
A new tri-nuclear Cu-carbonate cluster utilizing CO2 as a C1-building block – reactive intermediates, a probable mechanism, and EPR and magnetic studies†
Biswanath
Das
 *a,
Mohan
Bhadbhade
b,
Anders
Thapper
c,
Chris D.
Ling
d and
Stephen B.
Colbran
*a
*a,
Mohan
Bhadbhade
b,
Anders
Thapper
c,
Chris D.
Ling
d and
Stephen B.
Colbran
*a
aSchool of Chemistry, University of New South Wales, Sydney 2052, Australia. E-mail: das.biswanath85@gmail.com
bMark Wainwright Analytical Centre, University of New South Wales, Sydney 2052, Australia
cDepartment of Chemistry – Ångström Laboratory, Uppsala University, SE-75120, Uppsala, Sweden
dSchool of Chemistry, The University of Sydney, Sydney 2006, Australia
First published on 28th January 2019
Abstract
This work demonstrates that simple copper-bipyridine compounds and atmospheric CO2 react to produce useful/complex materials under appropriate conditions. Starting from a distorted square planar copper(II) complex, [(tbubpy)CuCl2](tbubpy = 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine), an air-sensitive, copper(I) complex, [(tbubpy)2CuI][BF4], which exhibits a distorted tetrahedral geometry, was synthesized and characterized. Reactions of [(tbubpy)2CuI][BF4] with CO2 inside a sealed tube and with air were carried out over a week and three weeks, respectively. A new tricopper(II)-carbonato cluster, [{(tbubpy)2Cu}3(μ-CO3)][PF6]4, was isolated with three distorted octahedral copper(II) centres bound by a carbonate-bridge formed from atmospheric CO2. NMR and UV-Vis spectroscopic analyses coupled with previous reports point to a multi-step process in the formation of a trinuclear CuII-carbonato cluster that includes the probable involvement of μ-hydroxo-bridged dicopper(II) type intermediates.
Introduction
Novel chemical features of first row transition metal complexes have attracted enormous attention with the aim of developing cost-effective, non-toxic and essential catalysts/materials.1–4 Copper is one of those metals which have shown significant promise.5–10 Being able to accommodate oxidation states from 0 to +4, its complexes have been studied as catalysts for a variety of metal-assisted synthetic, oxidation (e.g. of water, C–H bond) and reduction (e.g. of protons and CO2) reactions.11–16 CO2, a greenhouse gas, is being released from both natural processes (the respiration of living creatures) and anthropological processes (the burning of fossil fuels for industrial and household purposes of everyday life).17,18 The increasing amount of CO2 in the atmosphere is one of the major issues of the 21st century. In the current fast growing society, the requirement for energy from carbon-based (fossil) fuel is increasing every day as renewable carbon-free energy sources are yet to fulfil the demand to a significant degree. Therefore, one of the best possible ways to mitigate the concentration of CO2 in the atmosphere is to convert emitted CO2 to useful materials. This is an active and important field of modern-day research and the major focus is on converting CO2 to fuel and commodity chemicals. In this regard, direct air capture (DAC), more precisely direct extraction of CO2 from air, a concept introduced by Lackner in 1999, is highly relevant.19 The direct capture of CO2 from air and its conversion to useful materials (e.g. new chemicals, magnetic materials, metal organic frameworks etc) have been an active field of research in the last few decades.20Reduction and hydrogenation are the most common approaches for mitigating the amount of CO2 in the atmosphere as well as for producing fuels and manufacturing useful organic and inorganic compounds.21–23 In other words, the carbon atom that is trapped in waste CO2 is utilized as a C1-building block. In 2015, Beller et al. reviewed extensively such uses of CO2 as a building block in organic synthesis.24 Formation of organic carbonate from CO2 is one of the effective synthetic routes. CO2 reacts rapidly in basic medium. In the presence of strong nucleophiles, it can produce the corresponding carboxyl and carbamoyl compounds. These compounds upon reaction with organic electrophiles lead to the formation of organic carbonates and carbamates, respectively.24
In comparison, there are only a handful of examples of the formation of metal carbonates utilizing air.25–27 The reasons can be the difficulties in finding a suitable ligand–metal system and appropriate conditions to interact with air, which contains a low concentration of the substrate (such as 410 ppm for CO2).28 Here we report, starting from a simple air-stable bipyridine-CuII(Cl)2 complex {2: [(tbubpy)CuCl2] (tbubpy = 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine)}, in situ formation of an unstable bis(bipyridine)-CuI complex (3: [(tbubpy)2CuI][BF4]) which on slow reaction with air (over a period of 3 weeks) forms a CuII3-carbonate (4: [{(tbubpy)2Cu}3(μ-CO3)][PF6]4) cluster. Saturating the methanolic solution of 3 with CO2 speeds up the formation of 4 (to less than a week). These types of clusters can be interesting candidates for use as magnetic materials (e.g. single-molecule magnets) comprising first row transition metals. The utilization of the atmospheric CO2 as a C1-building block to form new materials and the understanding of the possible reactive intermediates are appealing topics.
Results and discussion
Freshly prepared 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine (tbubpy) (1) (1 mmol) was treated with one eq. of Cu(BF4)2 and two eq. of NaCl in methanol (Scheme S1†). The addition of 1.5 eq. of triethyl amine caused the pale blue solution to turn intense blue-green. A precipitate formed upon stirring for 90 min, which was successfully crystallized from acetonitrile solution to afford the complex-salt, [(tbubpy)CuCl2]·Et3NH+BF4−(2). Attempts made to crystallize [(tbubpy)CuCl2] using different combinations of Cu and Na salts, and without using Et3N, were unsuccessful. In the single crystal X-ray structure of 2, Fig. 1a, the CuII ion binds to two nitrogen and two chloride donors in a distorted square planar coordination geometry. CuII being a d9 system prefers the square planar, square pyramidal or trigonal bi-pyramidal geometry depending on the ligands over the tetrahedral geometry as the latter readily goes through ligand rearrangement/attack from solvent molecules.29 In 2, N1CuN2 has the smallest angle [80.14(9)°] out of the four angles around the Cu-center. The other angles are 93.90(7)°, 94.56(3)° and 95.63(6)° for N1CuCl1, Cl2CuCl1 and N2CuCl2, respectively.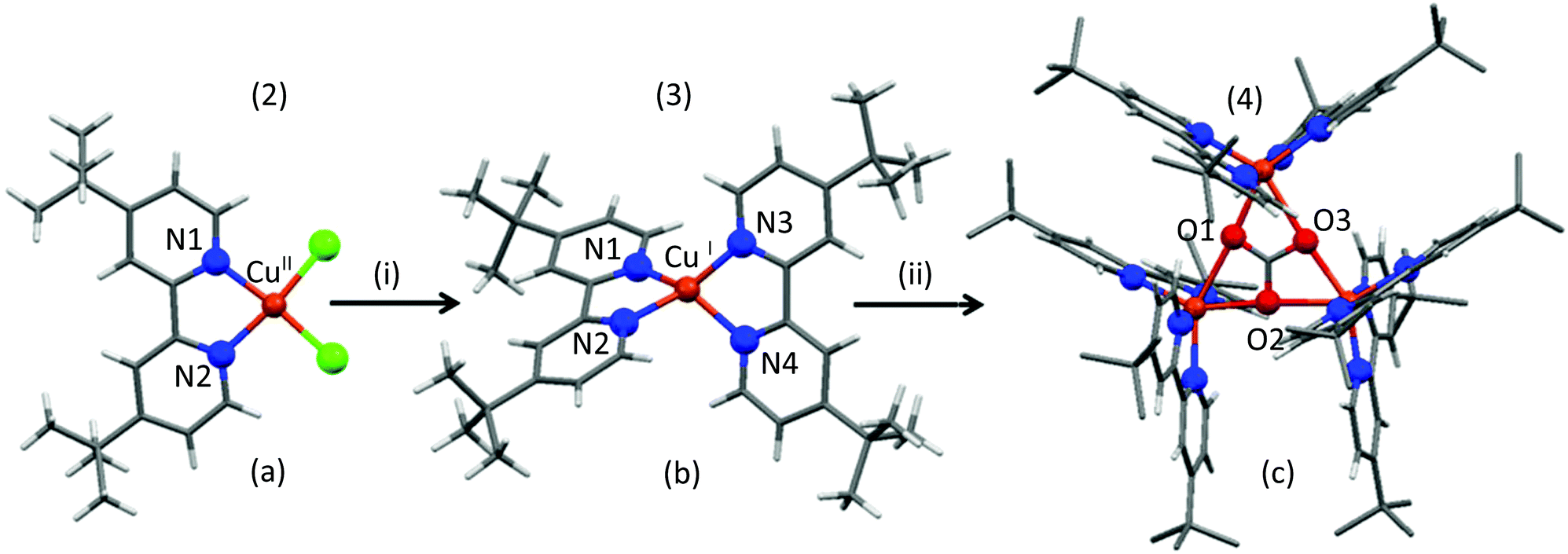 | ||
| Fig. 1 X-ray diffraction crystal structures of 2 (a), 3 (b) and 4 (c). A simplified depiction of the reaction pathway where (i) is NaBH4/d-methanol under N2 and (ii) is air. The details about the reaction procedure can be found in the Experimental section and Fig. S1.† The counter anions, methyl hydrogens of 4 and solvent molecules were removed for clarity. | ||
In order to perform the direct and easy analysis of the reactive intermediate/s by NMR spectroscopy, d-methanol was used as a solvent for all of the follow-up reactions. Treatment of a d-methanol solution of 2 (0.5 mM) with 2.5 eq. of NaBH4 under N2 shows an immediate colour change to black-brown, followed by a black precipitation within thirty minutes of continuous stirring. The precipitate was insoluble in water, methanol and acetonitrile. The filtrate was collected under N2. NMR spectra (1H and 13C) of the colourless solution showed peaks for a single diamagnetic species (3) containing tbubpy ligand(s). Air sensitive crystals were obtained by the slow diffusion of diethyl ether to the d-methanol solution of 3 inside a glovebox. Careful crystal mounting (at low temperature and in the absence of air) followed by X-ray diffraction crystallography shows that 3 is [(tbubpy)2CuI][BF4]. The CuI centre in 3 is surrounded by four N atoms from two tbubpy units in a distorted tetrahedral fashion (Fig. 1b). The instability of this complex can be explained by the soft nature of CuI and the structure. Being a d10 system, CuI prefers soft donors such as sulfur and iodide. With the intermediate donor capacity such as nitrogen (sp3 hybridized nitrogen is a harder donor, whereas sp2 hybridized nitrogen is a relatively soft donor) in the current study and the additional electron density from the tertiary butyl groups, CuI always tends to form CuII given a source of oxidation. Moreover, with N-donors CuI tends to have proper tetrahedral geometry which provides thermodynamic stability (CFSE, crystal field stabilization energy).29 Two tbubpy ligands make it impossible to achieve the tetrahedral geometry as N1CuN2 and N3CuN4 form an acute angle ∼80.4°. Two other angles of N1CuN3 and N2CuN4 are ∼107.9° and 122.3°, respectively.
NMR studies before (in a closed Young's NMR tube) and after careful purging of CO2 (1 atm) into a d-methanol solution of 3 in a Young's NMR tube (in the absence of air) show no immediate fast reaction, but over a period of 48 h a slow reaction took place. It results in an appreciable amount of new species with singlets at δ 2.03 ppm and δ 161 ppm in the 1H and 13C NMR spectra, respectively (Fig. S7 and S4†). The 13C chemical shift is characteristic of bicarbonate species (HCO3)−.30 Upon exposure to air, the solution starts changing colour slowly from yellow-orange to light brown to cyan over a period of four days. After a week, cyan coloured crystals of 4 (Fig. 1c) deposited from the reaction mixture. When another very similarly prepared d-methanol solution of 3 was directly exposed to air without prior exposure to CO2, it took 3 weeks before the cyan colored crystalline material of 4 came out from the solution.
The immediate changes that happen on exposing a methanol solution of 3 to air were followed by UV-Vis spectroscopy (Fig. 2) over two hours. A colourless solution of 3 under N2 turns light yellow-orange instantaneously upon the exposure to air. The formation of this new yellow-orange species was too fast to follow by room temperature UV-Vis spectroscopy. Previous reports by Stack et al. showed that a highly temperature sensitive bis(μ-oxo)dicopper(III) {[(TMED)2CuIII2(μ2-O)2]2+} (TMED = N,N,N′,N′-tetramethylethylenediamine) type intermediate formed.31 This intermediate was only possible to isolate/characterize at low temperature (183 K) using acetone as the solvent.31 The mononuclear counterpart {(TMED)CuI}+ reacts with O2 to form bis(μ-oxo)dicopper(III), which had a clear absorption maximum at around 390 nm with a shoulder at 470 nm. In the presence of an excess of CuI in the solution and with the limited steric demand from the ligand, {[(TMED)2CuIII2(μ2-O)2]2+} species reacts further, and forms a compact trinuclear [Cu3(μ3-O)2]3+ core.31
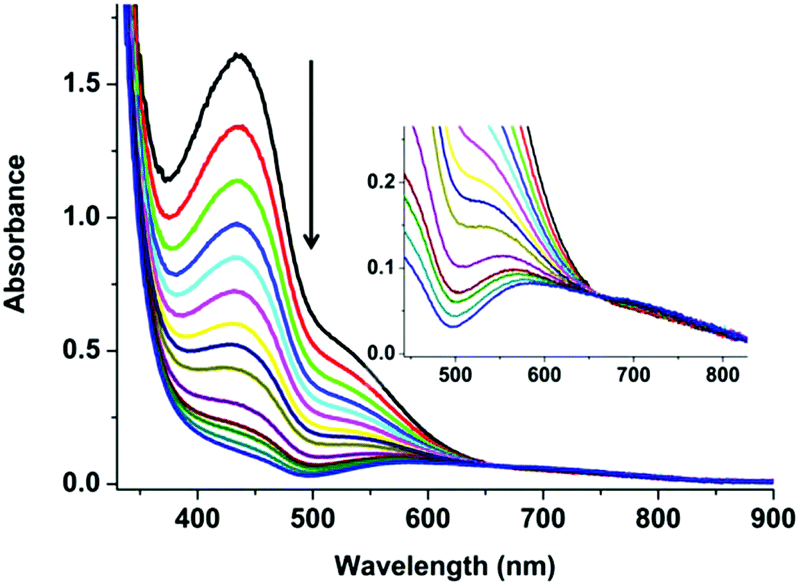 | ||
| Fig. 2 Room temperature UV-Vis spectra recorded for the reaction of 3 (0.5 mM) with air over a period of 120 minutes. The first nine spectra were recorded at 5 min intervals and the last 5 spectra were recorded at 15 min intervals. The inset shows a magnification of the spectra from 450–825 nm. | ||
In the current project, we carried out the reaction of the unstable CuI species (3) with air, instead of O2 only, to get an idea of the possible intermediates that might be involved in the construction of 4 (bluish green) in the basic medium. The reaction of 3 with air is a multistep process. The first and very fast process, i.e. the immediate development of yellow-orange species from the colourless solution of 3, can be attributed to the formation and transient presence of a high-valent Cu-intermediate in the presence of air oxygen as discussed above.31 The decay or the reaction of this new yellow orange species with air, being a slower process, can be carried out at room temperature. A UV-Vis study for a period of 2 h shows a steady decrease of the absorption band at 435 and 535 nm and the appearance of a new broad absorption band at 570 nm with a shoulder at 700 nm. The process becomes much slower after 2 h. This slower process can be attributed to slow ligand reorganization under air during the formation of 4 (Fig. 1c). Our claim is further supported by the very slow reaction of 3 in the presence of only CO2 in the absence of O2 (Fig. S4–9†). 13C NMR and 1H NMR studies over a period of two days reveal that 3 reacts very slowly with CO2 in d-methanol solution in the absence of O2. The clear formation of HCO3− was observed only after keeping 3 in the presence of 1 atm CO2 over a period of 48 h (Fig. S4–9†). No further reaction was observed in the absence of O2 (air). For the formation of 4, the presence of O2 (air) is mandatory. All these observations point towards the formation of a semi-stable CuII–O(H)–CuII intermediate with oxygen that reacts further and rather slowly with atmospheric CO2. The observations are consistent with the report from Mukherjee and co-workers, where the nucleophilic attack of the hydroxo group of an in situ generated, semi-stable di-μ-hydroxo-bridged dicopper(II) species, [L′Cu–OH–CuL′]2+ [L′ = methyl[2-(2-pyridyl)ethyl](2-pyridylmethy)amine], to the electrophilic carbon of the CO2 unit was proposed.32 From the cumulative results of ours and the previous reports we propose a reaction pathway to form 4 starting from 2 (Scheme 1).
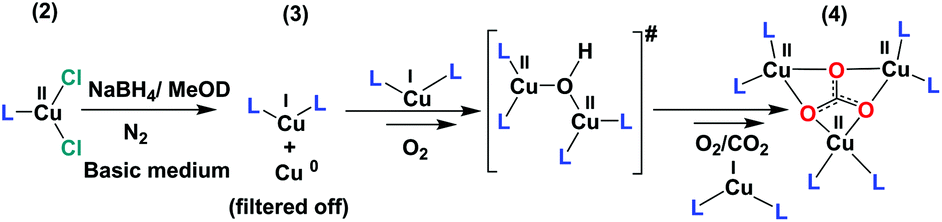 | ||
| Scheme 1 Proposed reaction pathway for the formation of 4 from 2. # represents the probable reactive intermediate. L stands for tbubpy. | ||
The X-ray diffraction structure reveals that 4 is a new trinuclear CuII-cluster {[(tbubpy)2CuII]3(CO3)(PF6)4} having a carbonato (CO32−) bridge as a central building block. Both O2 and CO2 from the air play important roles in forming the trinuclear CuII cluster (4). CO32− uses all its possible coordination and occupies six coordination sites (two per CuII units). Each CuII center is attached to two oxygen atoms of CO32− with O(Carbonato)–CuII bond distances of 2.31–2.35 Å. The CuII centers have distorted octahedral geometry and are surrounded by four nitrogen donors from two tbubpy units and share two of the six possible binding sites of the central CO32− (Fig. 1c). Although a few similar Cu-carbonate clusters are reported, to the best of our knowledge it is the first example where the formation of the cluster from the mononuclear counterpart in a reductive environment was followed up by NMR, UV-Vis spectroscopy and X-ray diffraction techniques.27,33,34 The structure makes it an interesting candidate for magnetic studies.
The crystal packing diagram of 4 (Fig. 3a and b) shows that it consists of trimeric complex cations having the carbonate carbon at the centre of the three-fold axis. Out of the four PF6− anions (per trimeric unit), one has 1/3 occupancy. The PF6− anions play an important role in the formation of the crystal packing through weak C–H⋯F (with H⋯F distance >2.75 Å) interactions between the F and C–H of the tert-butyl units. The presence of tert-butyl groups in the ligand periphery and PF6− ions makes the crystal-packing unique and unprecedented. The UV-Vis spectrum of 4 in d-methanol solution shows distinct broad absorption spectra at 640 nm, very different from those of 2 and 3 under similar conditions (Fig. S10†).
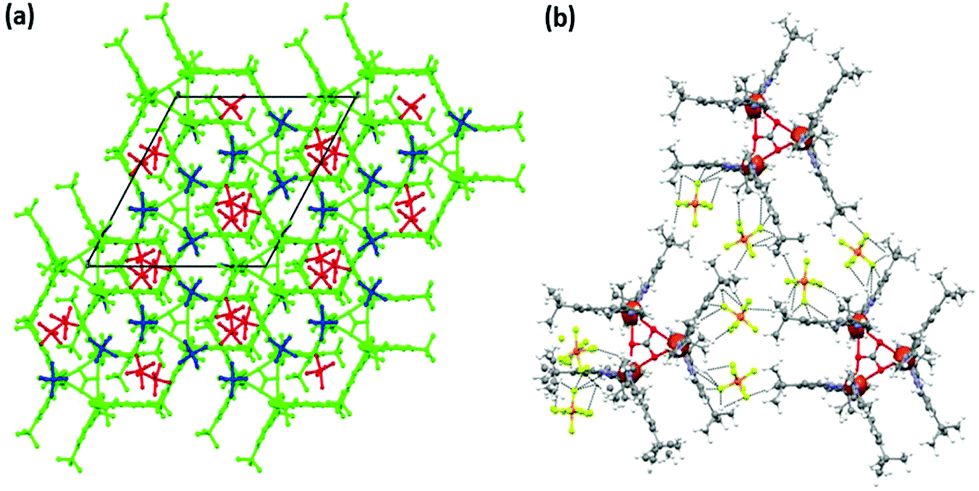 | ||
| Fig. 3 The packing diagram of 4·4PF6 (a) viewed along the C-axis and (b) showing intermolecular weak C–H⋯F interactions. In (a) blue and red colors are assigned to the PF6 anions where blue represents PF6 anions with single occupancy, whereas red represents PF6 anions with one third occupancy. | ||
Important bond distances in complexes 2–4 are summarized in Table 1. Cu–N distances vary from 1.99 Å to 2.115 Å in these three complexes. Interestingly, the longest and shortest distances are present in 4. In the central Cu3(CO3)4− unit, the Cu2+ centers are separated by 4.649 Å. Since both 2 and 4 are paramagnetic complexes, they were further characterized by EPR spectroscopy and magnetic measurements.
| Cu–N bond distances | Å |
|---|---|
| 2 | Cu–N1 [2.018(2)], Cu–N2 [2.003(2)] |
| 3 | Cu–N1 [2.029], Cu–N2 [2.005], Cu–N3 [2.029], Cu–N4 [2.005] |
| 4 | Cu–N1 [2.115(9)], Cu–N2 [1.99(1)], Cu–N3 [2.00(1)], Cu–N4 [2.11(2)] |
| Cu–O bond distances (4) | 2.21(1), 2.45(1) |
| Cu–Cu distance (4) | 4.649(3) |
The EPR spectrum of a frozen solution of 2 (1 mM in acetonitrile, 5 K) shows a rhombic EPR signal, consistent with the distorted square planar coordination geometry of 2, where the hyperfine interactions with the Cu-nucleus are not very well resolved (Fig. 4a). The EPR spectrum could be simulated as a rhombic S = 1/2 signal using the g- and A-values given in Table 2. An anisotropic line broadening (g-strain) was needed to obtain a good simulation. The rhombic EPR signal from 2 in acetonitrile differs from the previously reported axial EPR signal for (tbubpy)CuCl2·0.5CH3OH measured in methanol at 150 K where g‖ = 2.269, g⊥ = 2.01 and A‖ = 490 MHz, A⊥ ∼ 60 MHz.35 Another related complex, (tripbpy)CuCl2 (tripbpy = 6,6′-(2,4,6-triisopropylphenyl)-2,2′-bipyridine), is considerably more tetrahedral and has an EPR-signal (110 K) with g-values of ∼2.1 and ∼2.2 and unresolved hyperfine couplings.36 Varying the temperature from 5–20 K changed the intensity of the EPR signal from 2 but did not alter the shape (Fig. S11†).
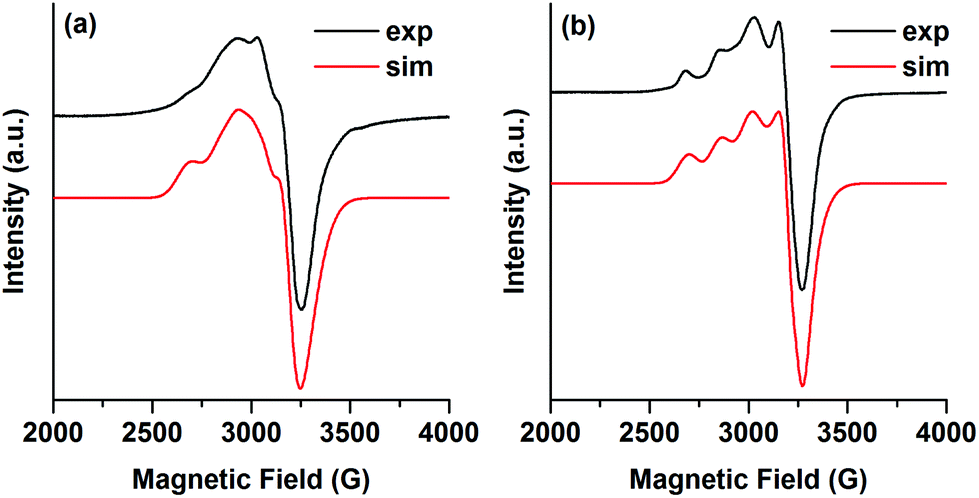 | ||
| Fig. 4 (a) Experimental (black) and simulated (red) EPR spectra of 2 (1 mM in acetonitrile). (b) Experimental (black) and simulated (red) EPR spectra of 4 (1 mM in acetonitrile). EPR conditions: T: 5 K, microwave power: (a) 200 μW (b) 20 μW, microwave frequency 9.287 GHz. The simulation was performed using EasySpin.37 | ||
| g 1 | g 2 | g 3 | A 1, MHz | A 2, MHz | A 3, MHz | g 1-strain | g 2-strain | g 3-strain | |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 2.26 | 2.16 | 2.06 | 510 | 200 | 10 | 0.11 | 0.06 | 0.15 |
| 4 | 2.256 | 2.10 | 2.075 | 500 | 225 | 10 | 0.09 | 0.15 | 0.03 |
The EPR-spectrum of a frozen solution of 4 (1 mM in acetonitrile, 5 K) has more resolved hyperfine interactions. Since the magnetic measurements indicate that the exchange interaction between the three Cu2+ is very small (see below), the EPR spectrum was treated as originating from a single S = 1/2 species. A satisfactory simulation was obtained with the g- and A-values given in Table 2. The EPR signal from 4 is more axial than the signal from 2 but the hyperfine interactions are of the same magnitude in both signals. Also for this signal an anisotropic line broadening was used for the simulations, probably reflecting the distorted octahedral coordination around the Cu ions. Varying the temperature from 5–20 K had only a very minor influence on the shape of the EPR signal from 4, in agreement with a very small exchange interaction between the Cu2+ ions (Fig. S12†).
The temperature dependence of the molar magnetic susceptibility, χm, of 2 and 4 is shown in Fig. 5(a) and (b). The right-hand axes show fits of the respective ZFC data to the Curie–Weiss (C–W) law χm = C/(T − Θ), where Θ is the characteristic Weiss temperature and C is the Curie constant, from 2–150 K. For 2, least-squares fitting gives: μeff = 1.62(5)μB/CuII, which agrees well with the theoretical spin-only magnetic moment (1.73μB) of the CuII (d9, S = 1/2) ion; and Θ = −1.3(2) K, a nearly insignificant deviation from 0 K.
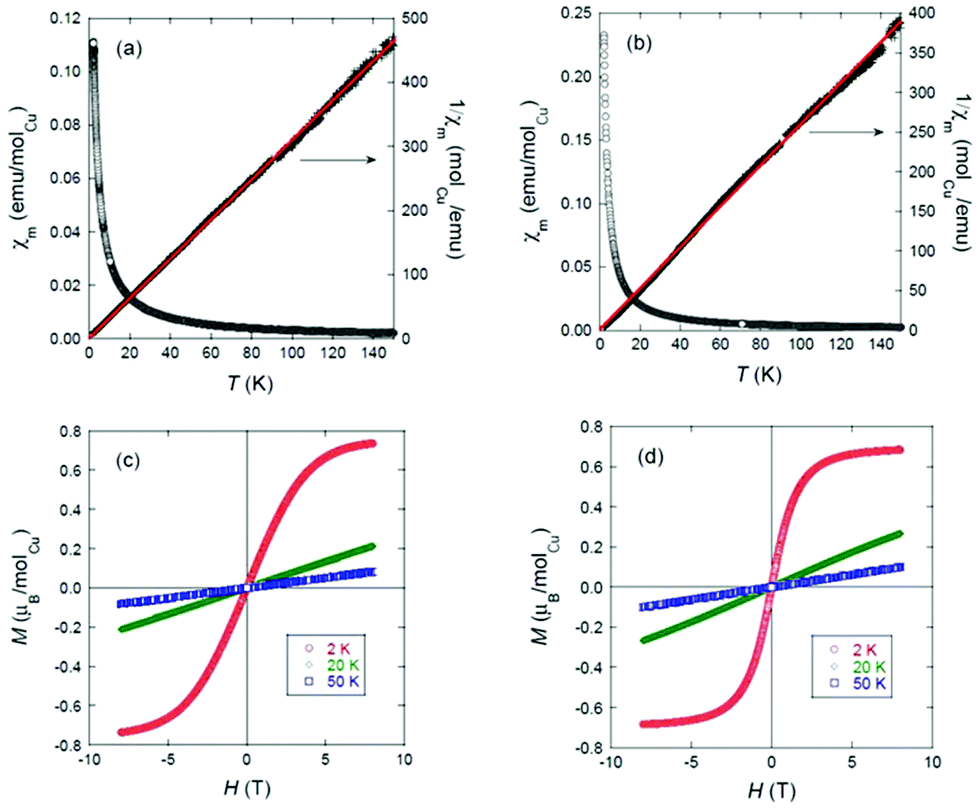 | ||
| Fig. 5 DC molar magnetic susceptibility χm and inverse susceptibility 1/χm as functions of temperature for (a) the mononuclear Cu(II) complex 2 and (b) the trinuclear complex 4 (red lines show linear fits to the Curie–Weiss equation); and field-dependent magnetisation of (c) 2 and (d) 4 at 2 K (red), 20 K (green) and 50 K (blue). | ||
No hysteresis is evident from the field-dependent magnetisation curves shown in Fig. 5. The mononuclear complex 2 is thus confirmed to contain the expected stoichiometric amount of CuII and to show purely paramagnetic behaviour, as expected. For 4, we observe a very small deviation from linear C–W behaviour. However, a fit over the 2–150 K range still yields μeff = 1.78(7)μB/Cu2+ and Θ = −0.8(3) K, and the field-dependent magnetisation behaviour is the same as for 2. These data are thus consistent with the purely paramagnetic behaviour of the expected stoichiometric amount of Cu2+ ions in the trinuclear complex, i.e., any exchange interactions among those ions through the central bridging carbonato unit must be very weak down to at least 2 K.
Conclusions
In this report, we have highlighted a new procedure that incorporates atmospheric CO2 to form new magnetic materials comprising copper(II) units. The reactive/air sensitive CuI intermediate was isolated and characterized by X-ray crystallography and NMR techniques (inert atmosphere). A reaction pathway was proposed based on the experimental findings and previous reports. Detailed EPR and temperature dependent magnetic studies were performed on a trinuclear CuII cluster and a mononuclear CuII-bipyridine complex.Experimental
General considerations
Commercially available reagents and deuterated solvents were used as received from Sigma Aldrich and Fisher chemicals. 1H and 13C NMR spectra were recorded using Bruker DPX 400 spectrometers. The UV-Vis-NIR spectra were recorded with a Varian Cary 50 UV/Vis spectrometer.Synthesis
EPR studies
EPR spectroscopic measurements were performed on 2 and 4 using a Bruker ESR-500 spectrometer, equipped with an ER 4122SHQ resonator, an ESR900 cryostat and an Oxford ITC503 temperature controller. The microwave frequency was 9.287 GHz and a modulation amplitude of 19.4 G and a modulation frequency of 100 kHz were used for all spectra. EPR signal simulations were performed with the EasySpin 5.2.21 software package.37Magnetic studies
DC magnetic susceptibility and magnetization data for the mononuclear Cu(II) complex 2 and the trinuclear complex 4 were collected on a Quantum Design Physical Property Measurement System (PPMS) using the vibrating sample magnetometer (VSM) method. Temperature-dependent susceptibility data were collected under zero field-cooled (ZFC) and field-cooled (FC) conditions in a coercive field of 1000 Oe between 2 and 300 K. Field-dependent magnetisation measurements were carried out at 2, 20 and 50 K up to 8 T. Molar magnetic parameters were calculated with respect to the magnetic Cu2+ ions [per Cu2+, i.e., 1/3 of a formula unit the trinuclear complex 4]. Diamagnetic corrections were applied using standard tabulated values of Pascal's constants.39X-ray crystallography
Crystals (2, 3 and 4) were mounted on a Bruker kappa-II CCD diffractometer at 150 K using an IμS Incoatec Microfocus Source with Mo-Kα radiation (λ = 0.710723 Å). Symmetry related absorption corrections using the program SADABS were applied and the data were corrected for Lorentz and polarisation effects using Bruker APEX3 software.40 The structure was solved by program SHELXT41(with intrinsic phasing) and full-matrix least-squares refinements were carried out using SHELXL-201441 through the Olex242 suite of software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BD and SBC would like to thank the Australian Research Council (Grant No. DP 160104383) for providing the funding for this research.References
- I. Roger and M. D. Symes, J. Mater. Chem. A, 2016, 4, 6724–6741 RSC.
- C. Liu, T. R. Cundari and A. K. Wilson, J. Phys. Chem. C, 2012, 116, 5681–5688 CrossRef CAS.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- F. Yu, F. Li, B. Zhang, H. Li and L. Sun, ACS Catal., 2015, 5, 627–630 CrossRef CAS.
- M. M. Najafpour, S. Mehrabani, Y. Mousazade and M. Hołyńska, Dalton Trans., 2018, 47, 9021–9029 RSC.
- T. Shinagawa, G. O. Larrazábal, A. J. Martín, F. Krumeich and J. Pérez-Ramírez, ACS Catal., 2018, 8, 837–844 CrossRef CAS.
- H. S. Jeon, S. Kunze, F. Scholten and B. Roldan Cuenya, ACS Catal., 2018, 8, 531–535 CrossRef CAS.
- Y. Lum and J. W. Ager, Angew. Chem., Int. Ed., 2017, 57, 551–554 CrossRef PubMed.
- J.-P. Costes, F. Dahan and W. Wernsdorfer, Inorg. Chem., 2006, 45, 5–7 CrossRef CAS PubMed.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS PubMed.
- M. K. Coggins, M.-T. Zhang, Z. Chen, N. Song and T. J. Meyer, Angew. Chem., Int. Ed., 2014, 53, 12226–12230 CrossRef CAS PubMed.
- Z. Weng, J. Jiang, Y. Wu, Z. Wu, X. Guo, K. L. Materna, W. Liu, V. S. Batista, G. W. Brudvig and H. Wang, J. Am. Chem. Soc., 2016, 138, 8076–8079 CrossRef CAS PubMed.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS PubMed.
- J. Zhang, H. Wang, S. Ren, W. Zhang and Y. Liu, Org. Lett., 2015, 17, 2920–2923 CrossRef CAS PubMed.
- X.-X. Guo, D.-W. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed.
- D. Schimel, B. B. Stephens and J. B. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 436 CrossRef CAS PubMed.
- K. Zickfeld, S. Solomon and D. M. Gilford, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 657 CrossRef CAS PubMed.
- K. S. Lackner, P. Grimes and H. J. Ziock, Proceedings of the 24th International Conference on Coal Utilization & Fuel Systems, 1999, pp. 885–896 Search PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- W. N. R. Wan Isahak, Z. A. Che Ramli, M. W. Mohamed Hisham and M. A. Yarmo, Renewable Sustainable Energy Rev., 2015, 47, 93–106 CrossRef CAS.
- T. D. Keene, M. J. Murphy, J. R. Price, N. F. Sciortino, P. D. Southon and C. J. Kepert, Dalton Trans., 2014, 43, 14766–14771 RSC.
- P. Kanoo, C. Madhu, G. Mostafa, T. K. Maji, A. Sundaresan, S. K. Pati and C. N. R. Rao, Dalton Trans., 2009, 5062–5064 RSC.
- E. A. Malinina, I. K. Kochneva, I. N. Polyakova, V. V. Avdeeva, G. A. Buzanov, N. N. Efimov, E. A. Ugolkova, V. V. Minin and N. T. Kuznetsov, Inorg. Chim. Acta, 2018, 479, 249–253 CrossRef CAS.
- I. Shimizu, Y. Morimoto, D. Faltermeier, M. Kerscher, S. Paria, T. Abe, H. Sugimoto, N. Fujieda, K. Asano, T. Suzuki, P. Comba and S. Itoh, Inorg. Chem., 2017, 56, 9634–9645 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- P. Kang, E. Bobyr, J. Dustman, K. O. Hodgson, B. Hedman, E. I. Solomon and T. D. P. Stack, Inorg. Chem., 2010, 49, 11030–11038 CrossRef CAS PubMed.
- J. Mukherjee, V. Balamurugan, M. S. Hundal and R. Mukherjee, J. Chem. Sci., 2005, 117, 111–116 CrossRef CAS.
- Y.-X. Peng, F. Xu, G. Yin, Q. Liu and W. Huang, J. Coord. Chem., 2012, 65, 3949–3959 CrossRef CAS.
- H.-X. Liu, X. Zhang, X.-J. Gao, C. Chen and D. Huang, Inorg. Chem. Commun., 2016, 68, 63–67 CrossRef CAS.
- D. J. Awad, U. Schilde and P. Strauch, Inorg. Chim. Acta, 2011, 365, 127–132 CrossRef CAS.
- E. E. Benson, A. L. Rheingold and C. P. Kubiak, Inorg. Chem., 2010, 49, 1458–1464 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- T. Ben Hadda and H. Le Bozec, Inorg. Chim. Acta, 1993, 204, 103–107 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef CAS.
- Bruker, APEX3, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880852–1880854. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04858d |
| This journal is © The Royal Society of Chemistry 2019 |