DOI:
10.1039/C8DT04647F
(Paper)
Dalton Trans., 2019,
48, 3583-3592
Anilido-oxazoline-ligated rare-earth metal complexes: synthesis, characterization and highly cis-1,4-selective polymerization of isoprene†
Received
23rd November 2018
, Accepted 14th January 2019
First published on 16th January 2019
Abstract
Anilido-oxazoline-ligated rare-earth metal dialkyl complexes have been synthesized and structurally characterized. The complexes exhibited strong fluorescence emissions and good catalytic performance on isoprene polymerization with high cis-1,4-selectivity. The treatment of anilido-oxazoline precursors ortho-C6H4[NH(2,6-R12C6H3)][C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NC(R2,R3)CH2O] (R1 = R2 = R3 = Me (HL1); R1 = iPr, R2 = R3 = Me (HL2); R1 = R2 = iPr, R3 = H (HL3); and R1 = iPr, R2 = R3 = H (HL4)) with an equimolar amount of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) afforded rare-earth metal complexes L1–4-Ln(CH2SiMe3)2(THF)n (L1-Sc (1), n = 1; L2-Sc (2), n = 0; L1-Y (3), n = 1; L2-Y (4), n = 1; L3-Y (5), n = 1; and L4-Y (6), n = 1) in good yields. The complexes are stable in both the solid state and solution. Single crystal X-ray diffraction study showed that complexes 1, 3 and 4 exhibit a distorted trigonal bipyramidal configuration, while complex 2 is pseudo-tetrahedral without coordinated THF. The luminescence properties of complexes 1–4 were investigated and the emission maxima were found in the range of 465–477 nm. DFT and TD-DFT studies were carried out to explore their characteristic electronic structures and gain insight into their optical properties. Upon activation with organic borates, the reported complexes exhibited high activity and cis-1,4-selectivity for isoprene polymerization. The nature of the central metal and substituent groups in oxazoline have an influence on the cis-1,4-selectivity.
NC(R2,R3)CH2O] (R1 = R2 = R3 = Me (HL1); R1 = iPr, R2 = R3 = Me (HL2); R1 = R2 = iPr, R3 = H (HL3); and R1 = iPr, R2 = R3 = H (HL4)) with an equimolar amount of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) afforded rare-earth metal complexes L1–4-Ln(CH2SiMe3)2(THF)n (L1-Sc (1), n = 1; L2-Sc (2), n = 0; L1-Y (3), n = 1; L2-Y (4), n = 1; L3-Y (5), n = 1; and L4-Y (6), n = 1) in good yields. The complexes are stable in both the solid state and solution. Single crystal X-ray diffraction study showed that complexes 1, 3 and 4 exhibit a distorted trigonal bipyramidal configuration, while complex 2 is pseudo-tetrahedral without coordinated THF. The luminescence properties of complexes 1–4 were investigated and the emission maxima were found in the range of 465–477 nm. DFT and TD-DFT studies were carried out to explore their characteristic electronic structures and gain insight into their optical properties. Upon activation with organic borates, the reported complexes exhibited high activity and cis-1,4-selectivity for isoprene polymerization. The nature of the central metal and substituent groups in oxazoline have an influence on the cis-1,4-selectivity.
Introduction
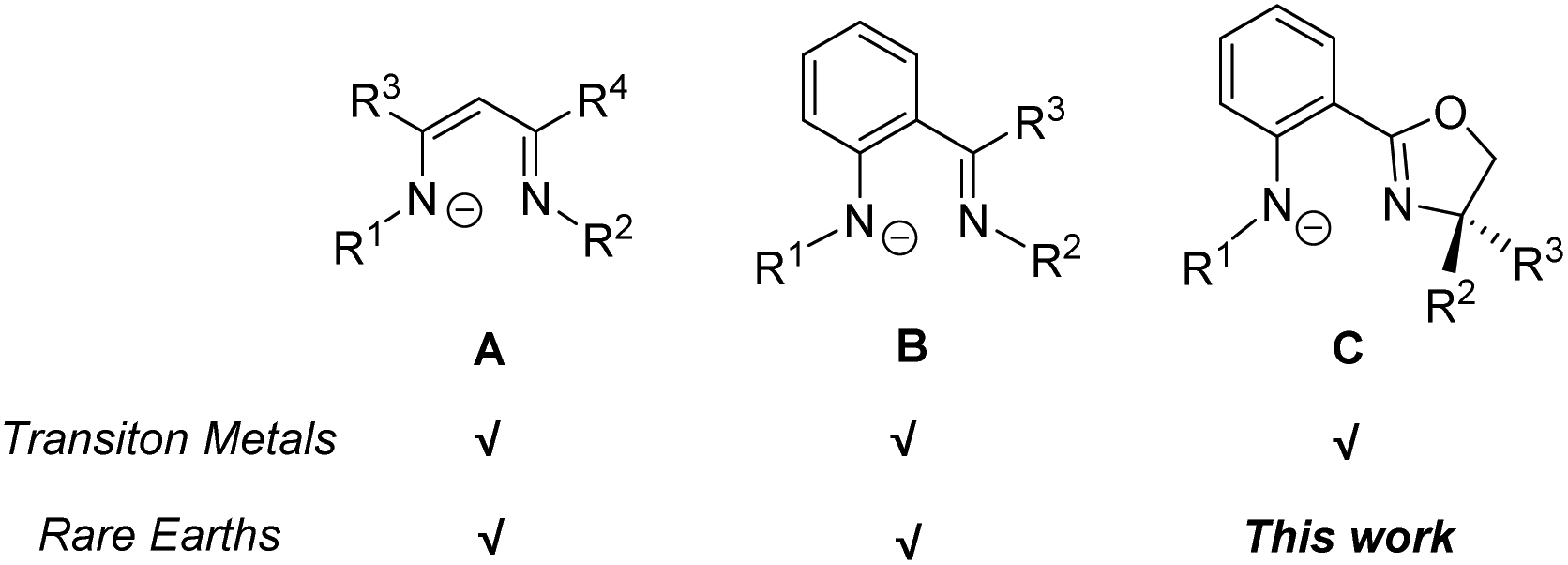
Owing to its wide variety of structural diversity for controlling the coordination environment around the metal center, the monoanionic bidentate β-diketiminate (Scheme 1, A) was shown to be one of the most versatile ligands for complexation with p-, d- and f-block elements to generate novel low-valent coordination-unsaturated complexes.1 Unlike its oxygen analogue, acetylacetonate, the R groups on the imine moieties of β-diketiminate can be easily manipulated by judicious choice of building blocks to fine tune its electronic and steric properties, hence resulting in effective and desirable catalyst design.2 Besides, efforts have also been devoted to the development of other β-diketiminato-type ligands, such as anilido-imine (Scheme 1, B) and anilido-oxazoline (Scheme 1, C).3 Both transition metal and rare earth (Ln) complexes containing β-diketiminate and anilido-imine ligands have been extensively studied and were demonstrated to exhibit outstanding catalytic reactivity in organic transformations, (co)polymerizations and small-molecule activations.4 For the anilido-oxazoline ligand, its particular merit is that, in addition to the presence of an easily derivatizable R group on the anilido moiety, there is an oxazoline that can efficiently incorporate designated chirality to metal catalysts.3d It has been successfully applied as a stereo-directing ligand in metal-based catalysis and polymerization.5 However, despite the employment of the anilido-oxazoline ligand being well documented for some main group and transition metal complexes, surprisingly, anilido-oxazoline supported Ln complexes are yet to be reported.
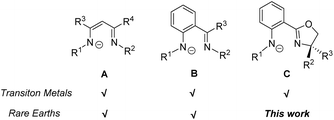 |
| | Scheme 1 Monoanionic N,N-bidentate ligands for transition metal and rare earth complexes. | |
As a kind of efficient ligand directed towards stereospecific catalysis, oxazoline-based ligands have been extensively used for developing both organometallic and organolanthanide catalysts.6 Ln complexes containing oxazoline-based ligands, such as bis(oxazolinyl)methane, 2,6-bis(oxazolinyl)pyridine and 1,1,1-tris(oxazolinyl)ethane, were found to be effective catalysts for a wide range of catalytic applications.7 Furthermore, oxazoline-based ligands have also been applied to Ln catalysts for polymerization and exert positive effects on stereochemistry controllability.8–10 For example, Ln complexes containing the chelating tris(oxazoline) ligand could catalyze isotactic polymerization of α-olefins upon activation with borate,8a and the Sc and Lu complexes with the bis(oxazolinylphenyl)amido ligand exhibited a high activity and trans-1,4-selectivity in the quasi-living polymerization of isoprene.9a We are particularly interested in Ln complexes supported by oxazoline-based ligands.10 The NCN-pincer Ln dichloride complexes supported by the monoanionic 2,6-bis(4′-(S)-isopropyl-2′-oxazolinyl)phenyl ligand were reported previously, which were found to be active in highly cis-1,4-selective polymerization of isoprene.10a Recently, we synthesized the first bis(oxazoline)-derived N-heterocyclic-carbene supported Ln complexes, that showed good activity in the (co)polymerization of 1-hexene.10c As our continuous effort in the exploration of new Ln complexes containing oxazoline-based ligands, herein, we report the synthesis and structural characterization of a series of anilido-oxazoline supported Ln dialkyl complexes, that display fluorescent properties. The good catalytic activity of these complexes for highly cis-1,4-selective polymerization of isoprene will also be described.
Results and discussion
Synthesis and characterization of complexes 1–6
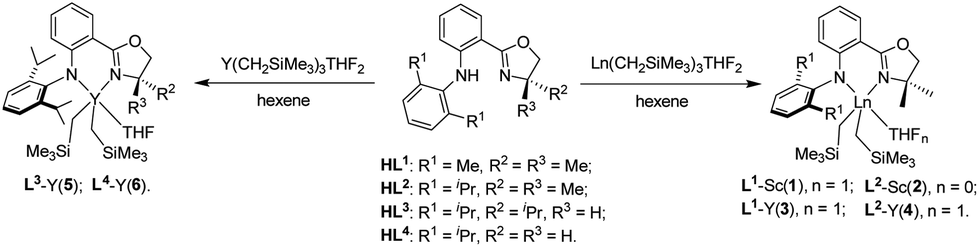
The anilido-oxazoline precursors (HL1–4) were prepared via the Pd-catalyzed C–N cross-coupling reaction using 1-bromo-2-oxazolinylbenzene and 2,6-dialkylaniline as the starting material.3d Subsequent treatment of these precursors with an equimolar amount of Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y) afforded the anilido-oxazoline Ln complexes 1–6 in good yields (Scheme 2). Complexes 1–6 are soluble in common organic solvents and stable in both the solid state and solution at room temperature.11 The good stability is in contrast to the tridentate bis(oxazolinylphenyl)amido supported Ln (Y, Tm) dialkyl complexes that were susceptible to the ring-opening of the oxazoline via alkyl migration,9a highlighting the good applicability of bidentate anilido-oxazoline for Ln complexation. The 1H NMR spectra of 1–6 revealed that the ratios of the anilido-oxazoline ligand to trimethylsilyl alkyls were approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The 1H NMR signals of Sc-CH2SiMe3 (0.07–0.20 ppm) and Y-CH2SiMe3 (−0.76 to −0.29 ppm) in 1–6 were similar to those of the reported Sc-silylalkyl and Y-silylalkyl complexes.12 Except 2, complexes 1 and 3–6 are each ligated with a THF molecule. Owing to the symmetrical dimethyl groups at 4-position of the oxazoline moiety, the resonance signals of protons at 5-position were displayed as singlets at δ = 3.52–3.68 ppm in 1–4. In comparison, the resonance signals of protons at 5-position showed splitting at δ = 4.01–4.03 and 4.88 ppm due to the influence of chiral groups in 5. The 13C NMR signals of sp2 carbon in the oxazoline moiety were observed at δC 168.86–169.51 ppm in 1–6, which moved to the downfield in comparison with those of free ligands (δC 162.25–164.03 ppm).
:2. The 1H NMR signals of Sc-CH2SiMe3 (0.07–0.20 ppm) and Y-CH2SiMe3 (−0.76 to −0.29 ppm) in 1–6 were similar to those of the reported Sc-silylalkyl and Y-silylalkyl complexes.12 Except 2, complexes 1 and 3–6 are each ligated with a THF molecule. Owing to the symmetrical dimethyl groups at 4-position of the oxazoline moiety, the resonance signals of protons at 5-position were displayed as singlets at δ = 3.52–3.68 ppm in 1–4. In comparison, the resonance signals of protons at 5-position showed splitting at δ = 4.01–4.03 and 4.88 ppm due to the influence of chiral groups in 5. The 13C NMR signals of sp2 carbon in the oxazoline moiety were observed at δC 168.86–169.51 ppm in 1–6, which moved to the downfield in comparison with those of free ligands (δC 162.25–164.03 ppm).
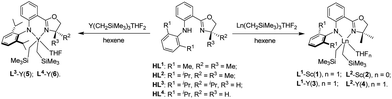 |
| | Scheme 2 Synthesis of complexes 1–6. | |
The IR spectra of 1–6 displayed an intense ν(C–N) band at 1607–1612 cm−1, which were red shifted compared with that of free anilido-oxazoline at 1626–1633 cm−1, indicating the coordination of nitrogen in the oxazoline moiety to the central Ln. Although the complexations of transition-metals with anilido-oxazoline ligands are well precedented,5 to the best of our knowledge, complexes 1–6 are the first reported Ln complexes supported by anilido-oxazoline ligands.
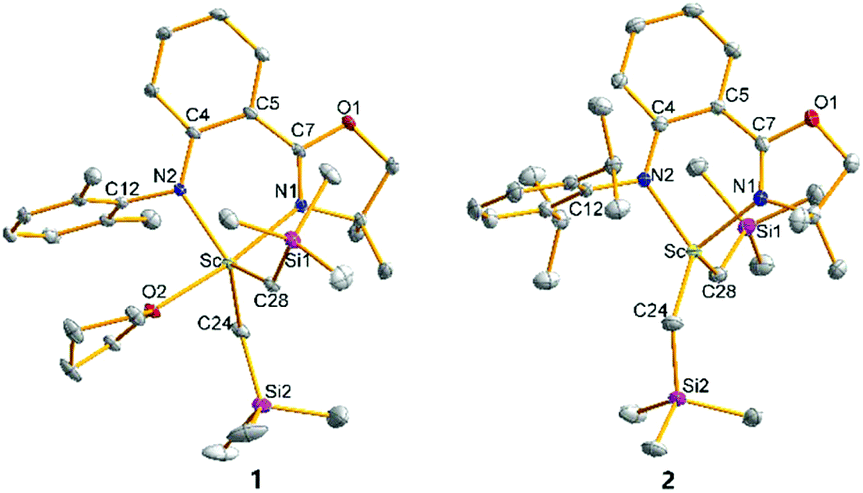
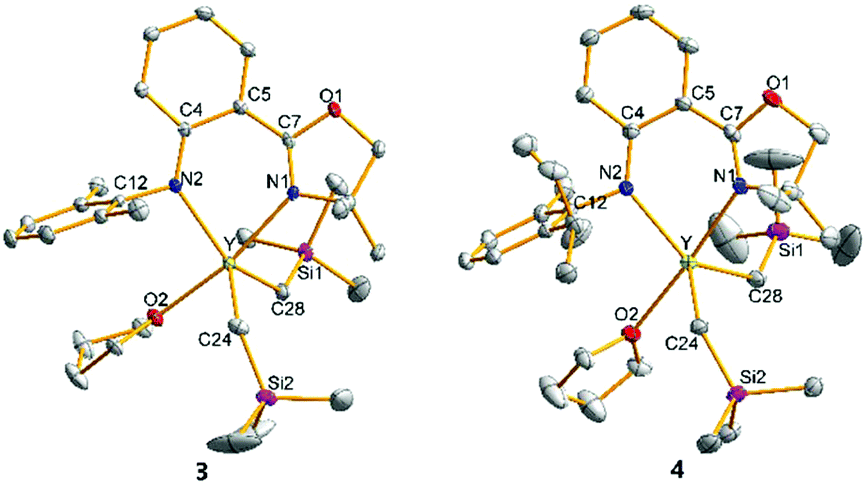
The crystal structures of 1–4 characterized by single crystal X-ray diffraction are shown in Fig. 1 and 2, respectively. The metal centres of 1, 3 and 4 were each coordinated with a bidentate anilido-oxazoline, two trimethylsilyl alkyls and one THF molecule, adopting a distorted trigonal bipyramidal configuration with an oxygen atom in THF and a nitrogen atom in oxazoline as vertexes. The respective bond distances between Ln and N2 of aniline for 1, 3 and 4 at 2.165, 2.315 and 2.322 Å are shorter than those between Ln and N1 of oxazoline (2.264, 2.406 and 2.427 Å, respectively). This indicates that the interactions between Ln and the two N atoms of anilido-oxazoline are inequivalent, suggesting a stronger interaction for the Ln–anilido bond. The bond distances of Sc–C bonds in 1 and Y–C bonds in 3 and 4 are in the range of 2.2378–2.2386, 2.3974–2.4027 and 2.3893–2.3923 Å, respectively. In comparison, the Sc–C bonds in 2 are in the range of 2.2042–2.2058 Å, which are shorter than those in 1. The five atoms of ligands around central metals are virtually coplanar, nevertheless, the central metals are out of this plane with distances in the range of 0.5538–0.6135 Å for 1, 3 and 4. The steric interaction between the N–aryl groups and the alkyls on the central metal can affect the out-of-plane distance. This phenomenon can be ascribed to the alleviation of steric interaction between the large central ion and the insufficient bite angle of the ligand.13 The out-of-plane distance in 1 was observed as 0.5538 Å, which is shorter than that of 0.815 Å in the penta-coordinated Sc complex [(2,6-iPr-C6H3)NC(Me)CHC(Me)N(2,6-iPr-C6H3)]ScMe2THF with the β-diketiminato ligand.13a This is presumably caused by a slight of rotation of the bond between phenyl and oxazolinyl derived from weak electron delocalization of the coordination five-ring and a relatively larger coordination space in 1, that do not require excessive bending of the Sc–N bond. Unlike 1, 3 and 4 that contain solvated THF molecules, the Sc center of 2 was only coordinated with the chelating anilido-oxazoline and two trimethylsilyl alkyls, adopting a pseudo tetrahedral geometry. The central Sc is out of the coordinated plane (NCPhCPhCN plane) with a distance of 0.7459 Å, which is significantly shorter than those distances (ca. 1.10–1.25 Å) in the tetra-coordinated scandium complexes with β-diketiminato ligands.13a The N1–Sc–N2 angle of 84.40° in 2 is larger than those in 1, 3 and 4 (74.55°–80.93°) (Table 1).
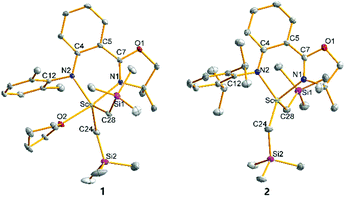 |
| | Fig. 1 Geometric structures of 1 and 2. Hydrogen atoms are omitted for clarity. The ellipsoids are drawn at 30% probability level. | |
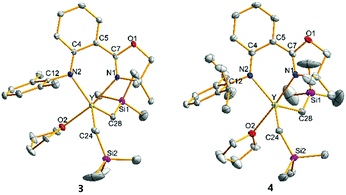 |
| | Fig. 2 Geometric structures of 3 and 4. Hydrogen atoms are omitted for clarity. The ellipsoids are drawn at 30% probability level. | |
Table 1 Selected bond lengths (Å) and angles (°) for complexes 1–4
| |
1 (Sc) |
2 (Sc) |
3 (Y) |
4 (Y) |
| Ln–N1 |
2.2641 |
2.1572 |
2.4064 |
2.4270 |
| Ln–N2 |
2.1650 |
2.0631 |
2.3148 |
2.3220 |
| Ln–O2 |
2.2624 |
— |
2.3774 |
2.4164 |
| Ln–C24 |
2.2378 |
2.2042 |
2.4027 |
2.3923 |
| Ln–C28 |
2.2386 |
2.2058 |
2.3974 |
2.3893 |
| N1–C7 |
1.2973 |
1.3026 |
1.2976 |
1.2794 |
| N2–C4 |
1.3776 |
1.3727 |
1.3682 |
1.3615 |
| N1–Ln–O2 |
172.66 |
— |
168.85 |
175.95 |
| N2–Ln–C24 |
116.68 |
115.44 |
117.85 |
121.39 |
| N2–Ln–C28 |
130.17 |
117.31 |
130.27 |
127.10 |
| C24–Ln–C28 |
113.15 |
114.63 |
111.86 |
111.09 |
UV-Vis and fluorescent properties
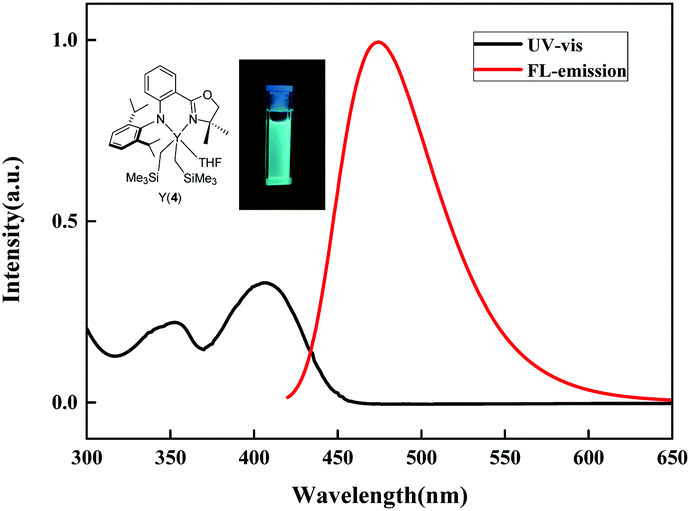
The UV-Vis and fluorescence spectra of complexes 1–4 were tested in n-hexane solution under an argon atmosphere and the results are summarized in Table 2. The UV-Vis spectra of 1–4 exhibited broad absorptions in the UV and violet region (341–359 and 396–403 nm) with strong absorptions (ε ∼ 104 L mol−1 cm−1) (Fig. S1–S4†). Upon excitation at 400 nm, complexes 1–4 displayed blue-green fluorescence with emission maxima of 477, 470, 465 and 474 nm, respectively (Fig. 3 and S5†). Small Stokes shifts (ca. 70 nm) were observed for these complexes. Complex 2 with the tetra-coordinated structure showed slightly blue-shifted fluorescence emission (470 nm) compared to 1 with the penta-coordinated structure (477 nm). For the Y complexes, the fluorescence emission peaks became slightly red-shifted with increase in the size of substituents at the ortho-positions of phenyl rings (465 nm for 3vs. 474 nm for 4).
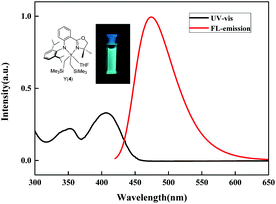 |
| | Fig. 3 UV-Vis and fluorescence emission spectra of complex 4. | |
Table 2 Optical properties of complexes 1–4 in n-hexane solution
| Complex |
UV-Vis abs. λ1 (nm) |
UV-Vis abs. λ2 (nm) |
Emission λ (nm) |
|
1
|
341 |
396 |
477 |
|
2
|
359 |
403 |
470 |
|
3
|
343 |
403 |
465 |
|
4
|
354 |
403 |
474 |
DFT and TD-DFT studies
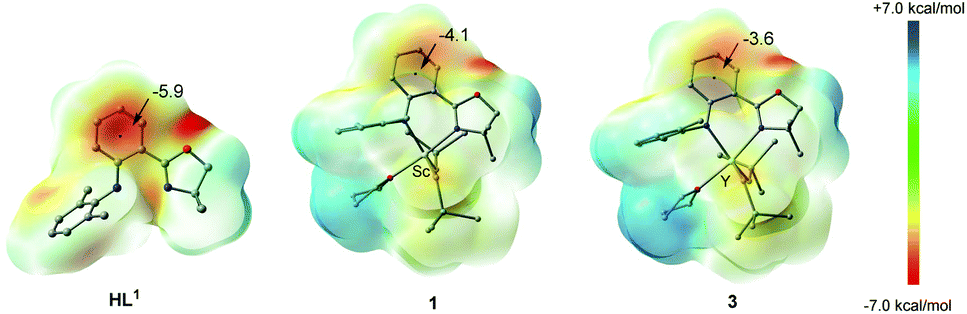
In order to understand the characteristic electronic structure of Ln complexes supported by anilido-oxazoline ligands, DFT calculations were performed and HL1, 1 and 3 were used as the model molecules. Based on the optimized geometric structures of HL1, 1 and 3, the nucleus-independent chemical shift (NICS), as a representative measure of aromaticity, was first calculated to evaluate the electron distribution of complexes.14 The NICS(0) distribution of the phenyl moiety center in the ligand skeleton was calculated to be −5.9 ppm for HL1, while the NICS(0) distributions of the same region decreased to −4.1 ppm for 1 and −3.6 ppm for 3, respectively (Fig. 4). These results mean that the phenyl moieties in 1 and 3 become less aromatic when the ligand is coordinated with the Ln center. Then, the electrostatic potential surfaces of HL1, 1 and 3 were calculated and shown in Fig. 4. The red regions indicate large negative charge distributions on the electrostatic potential surface and the blue regions indicate large positive charge distributions on the electrostatic potential surface. Despite the whole neutrality of the HL1, 1 and 3, negative electron density was largely populated at the phenyl moiety in the ligand skeleton. Compared with HL1, the light red color regions in 1 and 3 indicate a decrease in the electron density there. The imbalanced electron distribution is consistent with the NICS(0) distribution.
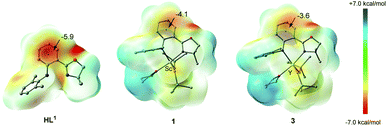 |
| | Fig. 4 The calculated NICS(0) distributions and electrostatic potential surface on the basis of the optimized geometry of HL1, 1 and 3. Hydrogen atoms have been omitted for clarity. | |
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of HL1, 1 and 3 were compared as well. The HOMO orbital energy in HL1 was calculated to be −0.189 eV, while the HOMO orbital energies were observed increasing to −0.181 eV in both 1 and 3. The HOMOs relevant to the π-bonding interactions of 1 and 3 locate at the phenyl moieties in the ligand skeleton. This result is consistent with the feature of the electrostatic potential surface, indicating a large negative charge distribution over the phenyl moieties.
In order to study the photophysical and photochemical properties of the complexes, the electronic transitions, frontier molecular orbitals (FMOs) and the emissions of complexes 1–4 were investigated by using time-dependent density functional theory (TDDFT) methods. After the light absorption, the four complexes are predominately excited to the S1 state, which corresponds to the absorption at 400 nm (Tables S1–S4†). In addition, the other absorptions observed at ca. 350 nm can be attributed to the partially electronic transition between the S0 state and the S2 state. In the S1 state, a significant π–π* transition can be observed as well as a slight metal to ligand charge transfer (MLCT) (Fig. 5). However, in the S2 state, the MLCT increases between the metal and the oxazoline moieties while the π–π* transition decreases. In addition, the 2,6-disubstituted phenyl twist occurs obviously in the S1 state compared to those in the S0 state. Thus a twisted intramolecular charge transfer (TICT) also occurs in the S0 → S1 transition. For the complexes 1–4, the calculated emissions are 431, 438, 508 and 461 nm, respectively, which are comparable to the experimental values (Table 2). It suggests that all the complexes experienced internal conversion and vibrational relaxation from the S2 state to the S1 state without any tautomerization before the fluorescence emission. Thus, there is only one emission even though two isosbestic points were found from both the electronic transition analysis and the experiments.
 |
| | Fig. 5 Frontier molecular orbitals (FMOs) of complexes 1–4. | |
Catalytic performance on isoprene polymerization
cis-1,4-Selective polymerization of isoprene is an important aspect in the synthetic rubber industry, which can produce the highly cis-1,4-regular PIPs with excellent rheological and mechanical properties.15 Towards this regard, rare-earth catalysts display good activity and cis-1,4-selectivity for isoprene polymerization with less gel formation and narrow molecular weight distribution.16–19 Therefore, it is of interest to study the catalytic performance of complexes 1–6 on isoprene polymerization, and the results are summarized in Table 3. All the neutral complexes were inactive towards isoprene polymerization even with a prolonged reaction time. On the other hand, upon activation with an equimolar amount of [Ph3C][B(C6F5)4], 1–6 could be employed as the initiators for isoprene polymerization. The Y complexes 3–6 achieved complete conversion of 1000 equivalent of isoprene within 30 min (Table 3, runs 3–6), while the isostructural complex 1 with the smaller Sc ion showed relatively lower activities (Table 3, run 1). The resultant poly(isoprene)s (PIPs) have high molecular weights and narrow molecular weight distributions (Mn = 7.53–13.82 × 104 and Mw/Mn = 1.27–2.40). In comparison, the THF-free complex 2 showed the highest activity for isoprene polymerization producing the PIP but with a relatively broader molecular weight distribution (Mw/Mn = 2.40) (Table 3, run 2). It might be ascribed to the higher Lewis acidity of 2 without coordinated THF that facilitates the coordination of the monomer. All the complexes 1–6 exhibited moderate to high cis-1,4-selectivity (75.7–95.9%) for isoprene polymerization. The Y complexes (3 and 4) displayed slightly higher cis-1,4-selectivity than the Sc complexes (1 and 2) with the same ligands, indicating the favourability of larger ionic radii (Table 3, runs 1–4). Besides the nature of the central metal, the substituents of the ligand were also found to have an impact on the cis-1,4 selectivity. The relatively higher cis-1,4-selectivities were observed by using 4 with a bulkier substituent at the ortho-positions of phenyl rings compared to 3. Observably, the substituent groups at 4-position of the oxazoline moiety displayed a significant influence on the cis-1,4-selectivity. The PIP with a cis-1,4 selectivity of 95.9% could be obtained by using 4 with dimethyl at 4-position of the oxazoline moiety as the catalyst, while the cis-1,4 selectivity of 75.7% was derived from 6 without any substituent groups in the oxazoline moiety (Table 3, run 6). The chiral groups in the oxazoline moieties of 5 did not result in prominent performances for catalytic activity or stereospecific selectivity obtaining the PIP with similar microstructures to those produced by 4 (Table 3, run 5).
Table 3 Polymerization of isoprene catalyzed by complexes 1–6a
| Run |
Cat. |
t (min) |
Yield (%) |
M
n
(×104) |
M
w/Mnb |
Microstructurec (%) |
|
cis-1,4 |
trans-1,4 |
3,4 |
|
Conditions: catalyst (10 μmol), [Ph3C][B(C6F5)4] (10 μmol), [IP]/[Ln] = 1000, toluene (5 mL), and T = 30 °C.
Determined by means of gel permeation chromatography (GPC) against polystyrene standards.
Determined by the 13C NMR spectrum of polyisoprene in CDCl3.
|
| 1 |
1
|
40 |
52.9 |
7.53 |
1.49 |
89.9 |
1.5 |
8.6 |
| 2 |
2
|
15 |
100 |
9.22 |
2.40 |
89.9 |
2.2 |
7.9 |
| 3 |
3
|
30 |
100 |
10.82 |
1.27 |
90.2 |
3 |
6.8 |
| 4 |
4
|
30 |
100 |
10.73 |
1.29 |
95.9 |
0 |
4.1 |
| 5 |
5
|
30 |
100 |
9.84 |
1.68 |
95.2 |
0.5 |
4.3 |
| 6 |
6
|
30 |
100 |
13.82 |
2.23 |
75.7 |
16.9 |
7.4 |
Complex 4 was chosen as a precursor to initiate isoprene polymerization under various conditions and the representative results are summarized in Table 4. When the feed ratio was increased from 500 to 4000, the molecular weights of the resultant PIPs increased linearly from 5.4 to 45.6 × 104 and the molecular weight distributions and cis-1,4-regularities were almost unchanged (Table 4, runs 1–4). Using [PhNHMe2][B(C6F5)4] as the activator, the polymerization was sluggish obviously as suggested by 85.3% conversion in 60 min (Table 4, run 5), while the system was almost inert under activation with B(C6F5)3 (Table 4, run 6). Using chlorobenzene or n-heptane instead of toluene as the solvent had a negligible effect on the selectivity, but resulted in a lower catalytic activity (Table 4, runs 7 and 8). Notably, the system 4/[Ph3C][B(C6F5)4] was found to be stable at an elevated temperature resulting in isoprene polymerization with enhanced activities but slightly decreased cis-1,4-selectivities (94.5% at 50 °C and 91.0% at 70 °C) (Table 4, runs 9 and 10).
Table 4 Polymerization of isoprene under various conditionsa
| Run |
[IP]:[Ln] |
T (°C) |
t (min) |
Yield (%) |
M
n
(×104) |
M
w/Mnb |
Microstructurec (%) |
| |
cis-1,4 |
trans-1,4 |
3,4 |
|
Conditions: complex 4 (10 μmol), [Ph3C][B(C6F5)4] (10 μmol), and toluene (5 mL).
Determined by means of gel permeation chromatography (GPC) against polystyrene standards.
Determined by the 13C NMR spectrum of polyisoprene in CDCl3.
Toluene = 6 mL.
Toluene = 12 mL.
Borate: [PhNHMe2][B(C6F5)4].
Borate: B(C6F5)3.
n.d.: not determined.
Chlorobenzene = 5 mL.
n-Heptane = 5 mL.
|
| 1 |
500 |
30 |
25 |
100 |
5.4 |
1.33 |
95.8 |
0 |
4.2 |
| 2 |
1000 |
30 |
30 |
100 |
10.7 |
1.29 |
95.9 |
0 |
4.1 |
| 3d |
2000 |
30 |
35 |
100 |
24.7 |
1.45 |
96.1 |
0 |
3.9 |
| 4e |
4000 |
30 |
45 |
83.8 |
45.6 |
1.32 |
95.6 |
0 |
4.4 |
| 5f |
1000 |
30 |
60 |
85.3 |
9.7 |
1.44 |
96.0 |
0 |
4.0 |
| 6g |
1000 |
30 |
180 |
Trace |
n.d.h |
n.d. |
n.d. |
n.d. |
n.d. |
| 7i |
1000 |
30 |
90 |
100 |
22.5 |
1.39 |
95.3 |
0 |
4.7 |
| 8j |
1000 |
30 |
240 |
89 |
19.0 |
1.28 |
91.9 |
4.2 |
3.9 |
| 9 |
1000 |
50 |
10 |
100 |
14.0 |
1.20 |
94.5 |
1.5 |
4.0 |
| 10 |
1000 |
70 |
5 |
100 |
11.8 |
1.35 |
91.0 |
4.3 |
4.7 |
Efforts have been made to identify the putative active species for the 4/[Ph3C][B(C6F5)4] system. The in situ reaction of 4 and an equimolar amount of [Ph3C][B(C6F5)4] were monitored by NMR spectroscopy and the cationic active species [L2YCH2SiMe3][B(C6F5)4] was speculated to be formed (Fig. S6–S8†). The 1H NMR spectrum features broad SiMe3 resonance at δ = −0.14 ppm and broad methylene resonance at δ = −0.34 ppm that can be attributed to the alkyl bound to Y. The characteristic resonance signals of the anilido-oxazoline ligand were observed as suggested by the resonance signals at δ = 7.92–7.91, 6.58–6.55, 5.97–5.95 ppm for the phenyl parts, δ = 3.94–3.92 ppm for the oxazoline moiety, δ = 1.41 1.14 ppm for the two methyl groups and δ = 3.21, 2.56, 1.15–1.05, 0.83 ppm for the two iPr groups. The resonance signals at δ = 3.41, 3.13, 1.41 ppm were attributed to the coordinated THF. At the same time, the liberation of two “Ph3CCH2SiMe3” species and Ph3CH was also observed according to the resonance signals at δ = 2.06, 1.97 ppm for methylene and −0.04, −0.26 ppm for SiMe3 of “Ph3CCH2SiMe3”, and δ = 5.45 ppm for Ph3CH.20 The 19F NMR spectrum of [L2YCH2SiMe3][B(C6F5)4] showed the absence of interaction between the fluorine atoms of [B(C6F5)4] and central Y3+. Unfortunately, attempts to determine the structural characteristics of the active species for 4 by using either [Ph3C][B(C6F5)4] or [Et3NH][B(C6H5)4] as an activator were unsuccessful, resulting in oily materials upon recrystallization. To indirectly prove the identity of the active species, a small quantity of CO2 (ca. 200 ppm) was introduced into the equimolar reaction of 4 with [Et3NH][B(C6H5)4] in THF, from which a cationic Y monoalkyl carboxylate complex (7) was isolated, as characterized by single crystal X-ray diffraction (Fig. S9†). The solid structure showed that complex 7 was an ionic pair complex and the cationic part was the Y3+ ion coordinated with the anilido-oxazoline ligand L2, carboxylate and three THF molecules. It was proposed that complex 7 was generated by insertion of one molecule CO2 into the Y–C bond of [L2-YCH2SiMe3THFn]+, which could be considered as the cationic part of the putative active species derived from 4.
Conclusions
In summary, we have prepared the first rare-earth metal dialkyl complexes bearing anilido-oxazoline ligands via the one-pot reaction of Ln(CH2SiMe3)3THF2 with the respective aniline-oxazolines. These complexes are mononuclear and their oxazoline moieties are stable in both the solid state and solution. The UV-Vis absorption spectra of 1–4 were recorded at 341–359 and 396–403 nm; and they exhibited blue-green fluorescence emissions with maxima of 465–477 nm. The TDDFT study indicated that there were π–π* transitions, MLCT and TICT in the S0 → S1 and the S0 → S2 transitions in the absorption process, and internal conversion and vibrational relaxation from the S2 state to the S1 state before fluorescence emission. Upon activation with organic borates, the complexes provided high cis-1,4-selectivity for the polymerization of isoprene (up to 96.1%). The yttrium complexes showed a higher catalytic activity and cis-1,4-selectivity compared to the Sc complexes; also, the substituent groups in the oxazoline ring played a role in cis-1,4-selectivity. The synthesis of other anilido-oxazoline-ligated Ln complexes containing various chiral substituents and the exploration of their application towards photochemical catalysis are underway.
Experimental section
General methods and materials
All air- and moisture-sensitive reactions were carried out using standard Schlenk line or glove box techniques under an argon atmosphere. Toluene, n-hexane, n-heptane and tetrahydrofuran (THF) were dried with sodium/benzophenone and n-hexane and n-pentane were dried with CaH2 under an argon atmosphere. Isoprene (IP) was purchased from TCI, dried with CaH2, degassed and stored in a glove box at −30 °C. Anhydrous LnCl3 (Ln = Sc, Y), [Ph3C][B(C6F5)4], [PhNHMe2][B(C6F5)4] and B(C6F5)3 were purchased from Jilin University and used as received. Ln(CH2SiMe3)3(THF)2 (Ln = Sc, Y)21 and anilido-oxazoline ligands (HL1–4)3d were synthesized according to the literature. Glassware and vials used for the polymerization were dried in an oven at 120 °C overnight and exposed to a vacuum–argon cycle three times. Deuterated solvents C6D6 (99.6 atom % D) and CDCl3 (99.8 atom % D) were purchased from the Cambridge Isotope Laboratory. All other solvents and reagents were purchased from commercial sources. Nuclear magnetic resonance (NMR) spectra were recorded by a Bruker AVANCE-500 NMR spectrometer and referenced to SiMe4 (TMS) or residual solvent resonances. Elemental analyses were performed on an Elementar Vario EL cube. UV-visible spectra were recorded using a Macy UV-1800A spectrophotometer at room temperature. Fluorescence measurements were carried out using a Lengguang Tech F97Pro fluorescence spectrophotometer. The molecular weight and molecular weight distribution of the polymers were tested with a Hitachi Chromaster 2000 (column: PL gel 5 μm MIXED-C) at 35 °C using THF as an eluent (flowing rate is 1.0 mL min−1) against polystyrene standards.
Geometry optimizations were performed with Gaussian 09 programs22 using the B3LYP level of theory. The Stuttgart–Dresden effective core potential (SDD) was employed for Lu, Sc, and Y. The 6-31G basis set was used for the other atoms. All of the structures were fully optimized and characterized by vibration frequency calculations.
Synthesis of L1-Sc(CH2SiMe3)2THF (1).
In a glovebox, a hexane solution (3 mL) of HL1 (131 mg, 0.444 mmol) was added dropwise to a hexane solution (3 mL) of Sc(CH2SiMe3)3THF2 (200 mg, 0.444 mmol) at low temperature (ca. −25 °C). The mixture was warmed to room temperature gradually and stirred for 2 h. After filtration, most of the solvents were removed under evacuation and 1 was obtained by crystallization from hexane solution with 65.0% yield (168.5 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 8.01 (dd, J = 8.2, 1.8 Hz, 1H, o-PhHN), 7.11 (d, J = 7.4 Hz, 2H, p-PhHN and m-PhHMe2N), 7.05 (dd, J = 8.3, 6.6 Hz, 1H, m-PhHMe2N), 6.91 (ddd, J = 8.7, 6.8, 1.8 Hz, 1H, m-PhHN), 6.47 (ddd, J = 8.1, 6.8, 1.2 Hz, 1H, p-PhHMe2N), 6.16 (dd, J = 8.7, 1.1 Hz, 1H, m-PhHN), 3.62–3.54 (m, 4H, THF), 3.52 (s, 2H, OCH2C), 2.24 (s, 6H, NPhMe2), 1.39 (td, J = 6.5, 5.7, 2.4 Hz, 4H, THF), 1.32 (s, 6H, NCMe2), 0.19–0.07 (m, 4H, CH2SiMe3), 0.13 (s, 18H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 169.34 (s, OCN), 154.98, 144.17, 136.63, 135.03, 132.07, 129.44, 125.61, 115.55, 115.24, 108.61, 78.31 (s, OCH2CN), 70.49 (s, THF), 67.67 (s, NCMe2), 42.71 (br, CH2SiMe3), 27.74 (s, NCMe2), 25.38 (s, THF), 19.14 (s, PhMe2), 3.98 (s, CH2SiMe3) ppm. IR (KBr): 2956 (m), 1608 (s), 1574 (m), 1534 (m), 1468 (m), 1432 (m), 1258 (m), 1209 (m), 858 (s). Anal. calcd for C31H51N2O2Si2Sc: C, 63.66; H, 8.79; N, 4.79. Found: C, 63.98; H, 8.87; N, 4.72.
Synthesis of L2-Sc(CH2SiMe3)2 (2).
Following the synthesis process of 1, 2 was synthesized using HL2 and Sc(CH2SiMe3)3THF2 with 68.5% yield (173 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 7.95 (dd, J = 8.2, 1.7 Hz, 1H, o-PhHN), 7.26 (s, 3H, p-PhHN and m-PhHMe2N), 6.88 (ddd, J = 8.7, 6.8, 1.8 Hz, 1H, m-PhHN), 6.44 (ddd, J = 8.1, 6.8, 1.1 Hz, 1H, p-PhHMe2N), 6.24 (dd, J = 8.7, 1.2 Hz, 1H, m-PhHN), 3.55 (s, 2H, OCH2C), 3.29 (p, J = 6.8 Hz, 2H, NPhCHMe2), 1.41 (d, J = 7.0 Hz, 6H, NPhCHMe2), 1.32 (s, 6H, NCMe2), 1.07 (d, J = 6.7 Hz, 6H, NPhCHMe2), 0.20–0.11 (m, 4H, CH2SiMe3), 0.09 (s, 18H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 170.34 (s, OCN), 156.91, 146.43, 139.22, 135.40, 131.80, 127.27, 125.19, 118.21, 116.24, 107.94, 78.55 (s, OCH2CN), 66.70 (s, NCMe2), 45.37 (br, CH2SiMe3), 28.60 (s, PhCHMe2), 27.96 (s, NCMe2), 25.31 (s, PhCHMe2), 25.29 (s, PhCHMe2), 3.43 (s, CH2SiMe3) ppm. IR (KBr): 2957 (s), 1607 (s), 1567 (s), 1531 (s), 1467 (m), 1430 (m), 1255 (s), 1239 (s), 1209 (s), 1163 (m), 1070 (m), 870 (s), 751 (m). Anal. calcd for C31H51N2OSi2Sc: C, 65.45; H, 9.04; N, 4.92. Found: C, 66.02; H, 9.13; N, 4.86.
Synthesis of L1-Y(CH2SiMe3)2THF (3).
Following the synthesis process of 1, 3 was synthesized using HL1 and Y(CH2SiMe3)3THF2 with 71.6% yield (184 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 8.24 (dd, J = 8.2, 1.8 Hz, 1H, o-PhHN), 6.98–6.90 (m, 3H, p-PhHN and m-PhHMe2N), 6.85 (dd, J = 8.1, 6.9 Hz, 1H, m-PhHN), 6.49 (ddd, J = 8.0, 6.7, 1.1 Hz, 1H, p-PhHMe2N), 6.08 (dd, J = 8.7, 1.1 Hz, 1H, m-PhHN), 3.66 (s, 2H, OCH2C), 3.31 (s, 4H, THF), 2.15 (s, 6H, NPhMe2), 1.54 (s, 6H, NCMe2), 1.22–1.08 (m, 4H, THF), 0.24 (s, 18H, CH2SiMe3), −0.29 (d, J = 11.7 Hz, 2H, CH2SiMe3), −0.65 (d, J = 11.9 Hz, 2H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 168.86 (s, OCN), 155.29, 145.28, 137.36, 134.41, 132.76, 129.29, 124.89, 115.09, 113.89, 108.88, 77.68 (s, OCH2CN), 70.98 (s, THF), 67.93 (s, NCMe2), 35.22 (s, CH2SiMe3), 34.90 (s, CH2SiMe3), 27.78 (s, NCMe2), 25.17 (s, THF), 18.99 (s, PhMe2), 4.71 (s, CH2SiMe3) ppm. IR (KBr): 2952 (m), 1608 (s), 1578 (m), 1533 (m), 1470 (m), 1432 (m), 1344 (w), 1270 (m), 1247 (m), 1205 (m), 1164 (w), 1101 (w), 1054 (w), 975 (w), 853 (s), 746 (m). Anal. calcd for C31H51N2O2Si2Y: C, 59.21; H, 8.18; N, 4.45. Found: C, 59.75; H, 8.25; N, 4.40.
Synthesis of L2-Y(CH2SiMe3)2THF (4).
Following the synthesis process of 1, 4 was synthesized using HL2 and Y(CH2SiMe3)3THF2 with 61.2% yield (169 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 8.22 (dd, J = 8.2, 1.8 Hz, 1H, o-PhHN), 7.16 (dt, J = 3.9, 1.8 Hz, 2H, p-PhHN and m-PhHMe2N), 7.14–7.07 (m, 1H, m-PhHMe2N), 6.89 (ddd, J = 8.7, 6.7, 1.8 Hz, 1H, m-PhHN), 6.45 (ddd, J = 8.0, 6.7, 1.2 Hz, 1H, p-PhHMe2N), 6.04 (dd, J = 8.7, 1.2 Hz, 1H, m-PhHN), 3.68 (s, 2H, OCH2C), 3.34–3.26 (m, 6H, NPhCHMe2 and THF), 1.58 (s, 6H, NCMe2), 1.23 (d, J = 6.9 Hz, 6H, NPhCHMe2), 1.20–1.14 (m, 4H, THF), 1.03 (d, J = 6.7 Hz, 6H, NPhCHMe2), 0.25 (s, 18H, CH2SiMe3), −0.33 (br, 2H, CH2SiMe3), −0.76 (br, 2H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 169.01 (s, OCN), 157.34, 148.01, 141.74, 133.58, 132.47, 126.42, 125.17, 117.75, 113.85, 108.56, 77.70 (s, OCH2CN), 70.45 (s, THF), 68.03 (s, NCMe2), 34.86 (s, CH2SiMe3), 34.55 (s, CH2SiMe3), 28.55 (s, PhCHMe2), 27.40 (s, NCMe2), 25.86 (s, PhCHMe2), 25.19 (s, THF), 24.34 (s, PhCHMe2), 4.80 (s, CH2SiMe3) ppm. IR (KBr): 2957 (s), 1608 (s), 1579 (m), 1534 (m), 1471 (m), 1430 (m), 1251 (m), 1206 (m), 1099 (w), 1056 (w), 855 (s). Anal. calcd for C35H59N2O2Si2Y: C, 61.38; H, 8.68; N, 4.09. Found: C, 61.81; H, 8.80; N, 4.04.
Synthesis of L3-Y(CH2SiMe3)2THF (5).
Following the synthesis process of 1, 5 was synthesized using HL3 and Y(CH2SiMe3)3THF2 with 54.7% yield (154 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 8.21 (dd, J = 8.2, 1.8 Hz, 1H, o-PhHN), 7.17 (d, J = 2.5 Hz, 1H, p-PhHN), 7.13–7.05 (m, 2H, m-PhHMe2N), 6.89 (ddd, J = 8.7, 6.7, 1.9 Hz, 1H, m-PhHN), 6.44 (ddd, J = 8.0, 6.7, 1.2 Hz, 1H, p-PhHMe2N), 6.04 (dd, J = 8.7, 1.2 Hz, 1H, m-PhHN), 4.88 (ddd, J = 7.2, 5.6, 3.3 Hz, 1H, OCH2CHN), 4.03–4.01 (m, 2H, OCH2CHN and NCHCH2), 3.52–3.48 (m, 2H, THF), 3.43 (p, J = 6.8 Hz, 1H, NPhCHMe2), 3.18 (p, J = 6.8 Hz, 1H, NPhCHMe2), 3.12–3.02 (m, 2H, THF), 2.70 (ddt, J = 10.6, 7.0, 3.0 Hz, 1H, NCHCHMe2), 1.29 (d, J = 6.8 Hz, 3H, NPhCHMe2), 1.14 (dd, J = 6.9, 3.6 Hz, 7H, NPhCHMe2 and THF), 1.10 (d, J = 6.7 Hz, 3H, NPhCHMe2), 0.96 (d, J = 6.6 Hz, 3H, NPhCHMe2), 0.80 (dd, J = 7.0, 2.1 Hz, 3H, NCHCHMe2), 0.76 (d, J = 6.8 Hz, 3H, NCHCHMe2), 0.26 (s, 18H, CH2SiMe3), −0.40 (br, 2H, CH2SiMe3), −0.58 (br, 2H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 169.51 (s, OCN), 157.68, 148.67, 147.36, 142.25, 133.53, 132.39, 126.32, 125.28, 125.03, 117.81, 113.65, 108.11, 70.56 (s, THF), 70.28 (s, OCH2CHN), 66.00 (s, OCH2CHN), 34.93 (br, CH2SiMe3), 31.31 (s, NCHCHMe2), 28.84 (s, PhCHMe2), 28.56 (s, PhCHMe2), 26.11 (s, PhCHMe2), 25.52 (s, PhCHMe2), 25.13 (s, THF), 24.94 (s, PhCHMe2), 24.20 (s, PhCHMe2), 19.08 (s, NCHCHMe2), 14.00 (s, NCHCHMe2), 4.60 (s, CH2SiMe3) ppm. IR (KBr): 2926 (s), 1608 (s), 1577 (m), 1537 (m), 1472 (m), 1450 (m), 1430 (m), 1381 (w), 1339 (w), 1258 (m), 1227 (s), 1166 (w), 1063 (m), 1017 (m), 854 (s), 747 (m). Anal. calcd for C36H61N2O2Si2Y: C, 61.86; H, 8.80; N, 4.01. Found: C, 62.13; H, 8.94; N, 4.09.
Synthesis of L4-Y(CH2SiMe3)2THF (6).
Following the synthesis process of 1, 6 was synthesized using HL4 and Y(CH2SiMe3)3THF2 with 47.5% yield (126 mg). 1H NMR (500 MHz, C6D6, 25 °C) δ 8.19 (dd, J = 8.2, 1.9 Hz, 1H, o-PhHN), 7.11 (d, J = 6.8 Hz, 2H, p-PhHN and m-PhHMe2N), 7.06 (dd, J = 8.8, 6.3 Hz, 1H, m-PhHMe2N), 6.89 (ddd, J = 8.7, 6.8, 1.8 Hz, 1H, m-PhHN), 6.47 (ddd, J = 8.0, 6.7, 1.2 Hz, 1H, p-PhHMe2N), 6.01 (dd, J = 8.7, 1.2 Hz, 1H, m-PhHN), 4.29 (t, J = 9.4 Hz, 2H, OCH2CH2N), 3.76 (t, J = 9.4 Hz, 2H, OCH2CH2N), 3.28 (tt, J = 11.3, 5.6 Hz, 6H, NPhCHMe2 and THF), 1.20 (d, J = 6.9 Hz, 6H, NPhCHMe2), 1.13 (s, 4H, THF), 1.01 (d, J = 6.7 Hz, 6H, NPhCHMe2), 0.28 (s, 18H, CH2SiMe3), −0.39 (br, 2H, CH2SiMe3), −0.69 (br, 2H, CH2SiMe3) ppm. 13C NMR (125 MHz, C6D6, 25 °C) δ 169.20 (s, OCN), 157.54, 148.35, 141.52, 133.40, 132.23, 126.48, 125.23, 117.80, 113.64, 108.75, 70.36 (s, THF), 66.23 (s, OCH2CH2N), 54.69 (s, OCH2CH2N), 34.29 (s, CH2SiMe3), 33.97 (s, CH2SiMe3), 28.51 (s, PhCHMe2), 25.75 (s, PhCHMe2), 25.13 (s, THF), 24.67 (s, PhCHMe2), 4.63 (s, CH2SiMe3) ppm. IR (KBr): 2958(m), 1612(s), 1534(w), 1469(m), 1434(m), 1260(m), 1228(m), 1095(m), 1021(m), 853(m), 800(m). Anal. calcd for C33H55N2O2Si2Y: C, 60.34; H, 8.44; N, 4.26. Found: C, 60.89; H, 8.51; N, 4.20.
General procedure for isoprene polymerization
A typical procedure (Table 3, run 4) was described as follows. In a glovebox, complex 4 (10 μmol) and [Ph3C][B(C6F5)4] (10 μmol) were mixed in 5 mL toluene, and then isoprene (0.68 g, 10 mmol) was added to the above-mentioned mixture in one portion. Polymerization was initiated and the mixture stirred vigorously for a desired reaction time. Afterward, the polymerization was quenched by ethanol and pouring into a large amount of ethanol (50 mL) containing hydrochloric acid (1%, v/v) and butylated hydroxytoluene (BHT, 0.1% w/w). The resultant polymer was precipitated, washed several times with ethanol and dried at 35 °C under vacuum to a constant weight.
Crystal structure determination
Single crystals suitable for X-ray single crystal analysis were generated from n-hexane solutions at −30 °C. Diffraction data were collected at 150 K on a Bruker D8 QUEST diffractometer equipped with a CPAD area detector, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The structures were solved by direct methods and refined by full-matrix least squares on F2. All non-hydrogen atoms were refined via anisotropic analysis and the hydrogen atoms were included in idealized positions. All calculations were performed using the SHELXTL crystallographic software packages.23 The details of the crystal data, data collections, and structure refinements are summarized in Table 5. CCDC 1880484 (for 1), 1880485 (for 2), 1880486 (for 3) and 1880487 (for 4)† contain the supplementary crystallographic data for this paper.
Table 5 Crystallographic data and refinement details for complexes 1–4
| |
1
|
2
|
3
|
4
|
| Formula |
C31H51N2O2Si2Sc |
C31H51N2OSi2Sc |
C31H51N2O2Si2Y |
C35H59N2O2Si2Y |
| Formula weight |
584.89 |
568.89 |
628.84 |
684.94 |
| Cryst. system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
P21/c |
P21/n |
|
a (Å) |
17.1880(8) |
10.3732(5) |
17.8753(5) |
10.4347(8) |
|
b (Å) |
9.4205(5) |
16.9019(7) |
9.5470(2) |
19.9397(15) |
|
c (Å) |
20.7058(9) |
19.2689(8) |
20.6698(5) |
18.8679(14) |
|
α (°) |
90 |
90 |
90 |
90 |
|
β (°) |
99.996(2) |
92.440(2) |
99.922(1) |
95.374(3) |
|
γ (°) |
90 |
90 |
90 |
90 |
|
V (Å3) |
3417.0(3) |
3375.3(3) |
3474.66(15) |
3908.5(5) |
|
Z
|
4 |
4 |
4 |
4 |
|
R
int
|
0.1317 |
0.0326 |
0.0510 |
0.0572 |
|
R
1
|
0.0720 |
0.0346 |
0.0640 |
0.0780 |
| wR2 |
0.1976 |
0.0864 |
0.1803 |
0.2201 |
| Goof |
1.0580 |
1.0290 |
1.0350 |
1.0430 |
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge Dr Bingyan Han for her help with the UV-Vis and fluorescence spectroscopy test, and Mr Guang-Jun Li and Ms Yajing Yang for the geometry optimizations. This work was supported by the National Natural Science Foundation of China (21404018, 21503030), the State Key Laboratory of Fine Chemicals (KF1711), the Fundamental Research Funds for the Central Universities (DUT18LK14) and the Education Department of the Liaoning Province (LT2015007). We extend our appreciation to the Supercomputing Center of the Dalian University of Technology for providing access to the supercomputer.
Notes and references
-
(a) L. Bourget-Merle, M. F. Lappert and J. R. Severn, Chem. Rev., 2002, 102, 3031 CrossRef CAS PubMed;
(b) C. Camp and J. Arnold, Dalton Trans., 2016, 45, 14462 RSC;
(c) S. Hohloch, B. M. Kriegel, R. G. Bergman and J. Arnold, Dalton Trans., 2016, 45, 15725 RSC;
(d) R. L. Webster, Dalton Trans., 2017, 46, 4483 RSC;
(e) W. Mao, L. Xiang and Y. Chen, Coord. Chem. Rev., 2017, 346, 77 CrossRef CAS.
- C. Chen, S. M. Bellows and P. L. Holland, Dalton Trans., 2015, 44, 16654 RSC.
- See some examples:
(a) P. G. Hayes, G. C. Welch, D. J. H. Emslie, C. L. Noack, W. E. Piers and M. Parvez, Organometallics, 2003, 22, 1577 CrossRef CAS;
(b) X. Liu, W. Gao, Y. Mu, G. Li, L. Ye, H. Xia, Y. Ren and S. Feng, Organometallics, 2005, 24, 1616 Search PubMed;
(c) C.-T. Chen, C.-Y. Chan, C.-A. Huang, M.-T. Chen and K.-F. Peng, Dalton Trans., 2007, 36, 4073 RSC;
(d) P. I. Binda, S. Abbina and G. Du, Synthesis, 2011, 2609 CAS.
-
(a) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 250, 602 CrossRef CAS;
(b) S. P. Sarish, S. Nembenna, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2011, 44, 157 CrossRef CAS PubMed;
(c) Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722 CrossRef CAS;
(d) S. Yao and M. Driess, Acc. Chem. Res., 2012, 45, 276 CrossRef CAS PubMed;
(e) V. T. Annibale and D. Song, RSC Adv., 2013, 3, 11432 RSC;
(f) C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC;
(g) Y. S. Liu, J. Li, X. L. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387 CrossRef CAS.
-
(a) C.-T. Chen, C.-Y. Chan, C.-A. Huang, M.-T. Chen and K.-F. Peng, Dalton Trans., 2009, 4073 Search PubMed;
(b) M.-T. Chen, P.-J. Chang, C.-A. Huang, K.-F. Peng and C.-T. Chen, Dalton Trans., 2009, 9068 RSC;
(c) M.-T. Chen and C.-T. Chen, Dalton Trans., 2011, 40, 12886 RSC;
(d) K.-F. Peng and C.-T. Chen, Eur. J. Inorg. Chem., 2011, 5182 CrossRef CAS;
(e) T. V. Truong, E. A. Kastl and G. Du, Tetrahedron Lett., 2011, 52, 1670 CrossRef CAS;
(f) S. Abbina and G. Du, Organometallics, 2012, 31, 7394 CrossRef CAS;
(g) V. K. Chidara and G. Du, Organometallics, 2013, 32, 5034 CrossRef CAS;
(h) Z. Lu, S. Abbina, J. R. Sabin, V. N. Nemykin and G. Du, Inorg. Chem., 2013, 52, 1454 CrossRef CAS PubMed;
(i) Z. Lu, S. Abbina, J. R. Sabin, V. N. Nemykin and G. Du, Inorg. Chem., 2013, 52, 1454 CrossRef CAS PubMed;
(j) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689 CrossRef CAS PubMed;
(k) S. Bian, S. Abbina, Z. Lu, E. Kolodka and G. Du, Organometallics, 2014, 33, 2489 CrossRef CAS PubMed.
-
(a) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef CAS;
(b) J. Zhou and Y. Tang, Chem. Soc. Rev., 2005, 34, 664 RSC;
(c) Q.-H. Deng, R. L. Melen and L. H. Gade, Acc. Chem. Res., 2014, 47, 3162 CrossRef CAS PubMed;
(d) S. H. Liao, X. L. Sun and Y. Tang, Acc. Chem. Res., 2014, 47, 2260 CrossRef CAS PubMed;
(e) J. S. Cannon and L. E. Overman, Acc. Chem. Res., 2016, 49, 2220 CrossRef CAS PubMed;
(f) S. Bellemin-Laponnaz, T. Achard, D. Bissessar, Y. Geiger and A. Maisse-Francois, Coord. Chem. Rev., 2017, 332, 38 CrossRef CAS;
(g) Y.-F. Yang, X. Hong, J.-Q. Yu and K. N. Houk, Acc. Chem. Res., 2017, 50, 2853 CrossRef CAS PubMed;
(h) G. Yang and W. Zhang, Chem. Soc. Rev., 2018, 47, 1783 RSC;
(i) K. Zheng, X. Liu and X. Feng, Chem. Rev., 2018, 118, 7586 CrossRef CAS PubMed.
-
(a) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327 RSC;
(b) B. D. Ward and L. H. Gade, Chem. Commun., 2012, 48, 10587 RSC.
-
(a) B. D. Ward, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 1668 CrossRef CAS PubMed;
(b) L. Lukesova, B. D. Ward, S. Bellemin-Laponnaz, H. Wadepohl and L. H. Gade, Dalton Trans., 2007, 920 RSC;
(c) B. D. Ward, L. Lukesova, H. Wadepohl, S. Bellemin-Laponnaz and L. H. Gade, Eur. J. Inorg. Chem., 2009, 866 CrossRef CAS.
-
(a) H. Liu, J. He, Z. Liu, Z. Lin, G. Du, S. Zhang and X. Li, Macromolecules, 2013, 46, 3257 CrossRef CAS;
(b) C. Yu, D. Zhou, X. Yan, F. Gao, L. Zhang, S. Zhang and X. Li, Polymers, 2017, 9, 531 CrossRef.
-
(a) Y. Pan, T.-Q. Xu, G.-W. Yang, K. Jin and X.-B. Lu, Inorg. Chem., 2013, 52, 2802 CrossRef CAS PubMed;
(b) Y. Pan, T.-Q. Xu, S. Fu, G.-W. Yang and X.-B. Lu, Macromolecules, 2013, 46, 8790 CrossRef CAS;
(c) Y. Pan, A. Zhao, Y. Li, W. Li, Y.-M. So, X. Yan and G. He, Dalton Trans., 2018, 47, 13815 RSC.
- The thermostability of anilido-oxazoline Ln complexes was investigated by taking complex 4 as an example, which showed stability in C6D6 solution at an elevated temperature (50 °C) for more than 4 h.
-
(a) W. P. Kretschmer, A. Meetsma, B. Hessen, T. Schmalz, S. Qayyum and R. Kempe, Chem. – Eur. J., 2006, 12, 8969 CrossRef CAS PubMed;
(b) G. Du, Y. Wei, W. Zhang, Y. Dong, Z. Lin, H. He, S. Zhang and X. Li, Dalton Trans., 2013, 42, 1278 RSC;
(c) X. Zhu, Y. Li, D. Guo, S. Wang, Y. Wei and S. Zhou, Dalton Trans., 2018, 47, 3947 RSC.
-
(a) P. G. Hayes, W. E. Piers, L. W. M. Lee, L. K. Knight, M. Parvez, M. R. J. Elsegood and W. Clegg, Organometallics, 2001, 20, 2533 CrossRef CAS;
(b) X. Liu, W. Gao, Y. Mu, G. Li, L. Ye, H. Xia, Y. Ren and S. Feng, Organometallics, 2005, 24, 1614 CrossRef CAS.
-
(a) P. von Ragué Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef PubMed;
(b) A. D. Allen and T. T. Tidwell, Chem. Rev., 2001, 101, 1333 CrossRef CAS PubMed;
(c) P. von Ragué Schleyer, Chem. Rev., 2001, 101, 1115 CrossRef.
-
(a)
W. Kuran, Principle of Coordination Polymerization, John Wiley and Sons Ltd., New York, 2001, p. 275 CrossRef;
(b)
L. S. Baugh and J. A. M. Canich, Stereoselective Polymerization with Single-Site Catalysts, Taylor & Francis, New York, 2008, p. 447 Search PubMed;
(c) D. Wilson, Makromol. Chem., Macromol. Symp., 1993, 66, 273 CrossRef CAS;
(d) J. Zhao and G. N. Ghebremeskel, Rubber Chem. Technol., 2001, 74, 409 CrossRef CAS.
-
(a) Z. Hou and Y. Wakatsuki, Coord. Chem. Rev., 2002, 231, 1 CrossRef CAS;
(b) P. M. Zeimentz, S. Arndt, B. R. Elvidge and J. Okuda, Chem. Rev., 2006, 106, 2404 CrossRef CAS PubMed;
(c) L. Friebe, O. Nuyken and W. Obrecht, Adv. Polym. Sci., 2006, 204, 1 CrossRef CAS;
(d) A. Fischbach and R. Anwander, Adv. Polym. Sci., 2006, 204, 155 CrossRef CAS;
(e) Z. Zhang, D. Cui, B. Wang, B. Liu and Y. Yang, Struct. Bonding, 2010, 137, 49 CrossRef CAS.
-
(a) A. Fischbach, C. Meermann, G. Eickerling, W. Scherer and R. Anwander, Macromolecules, 2006, 39, 6811 CrossRef CAS;
(b) C. Meermann, K. W. Törnroos, W. Nerdal and R. Anwander, Angew. Chem., Int. Ed., 2007, 46, 6508 CrossRef CAS PubMed;
(c) X. Li, M. Nishiura, L. Hu, K. Mori and Z. Hou, J. Am. Chem. Soc., 2009, 131, 13870 CrossRef CAS PubMed;
(d) Z. Jian, D. Cui, Z. Hou and X. Li, Chem. Commun., 2010, 46, 3022 RSC;
(e) G. Zhang, Y. Wei, L. Guo, X. Zhu, S. Wang, S. Zhou and X. Mu, Chem. – Eur. J., 2015, 21, 2519 CrossRef CAS PubMed;
(f) G. Zhang, B. Deng, S. Wang, Y. Wei, S. Zhou, X. Zhu, Z. Huang and X. Mu, Dalton Trans., 2016, 45, 15445 RSC;
(g) G. Zhang, S. Wang, X. Zhu, S. Zhou, Y. Wei, Z. Huang and X. Mu, Organometallics, 2017, 36, 3812 CrossRef CAS.
-
(a) L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909 CrossRef CAS PubMed;
(b) W. Gao and D. Cui, J. Am. Chem. Soc., 2008, 130, 4984 CrossRef CAS PubMed;
(c) K. Lv and D. Cui, Organometallics, 2010, 29, 2987 CrossRef CAS;
(d) L. Wang, D. Cui, Z. Hou, W. Li and Y. Li, Organometallics, 2011, 30, 760 CrossRef CAS;
(e) P. Zhang, H. Liao, H. Wang, X. Li, F. Yang and S. Zhang, Organometallics, 2017, 36, 2446 CrossRef CAS.
-
(a) D. Li, S. Li, D. Cui and X. Zhang, Organometallics, 2010, 29, 2186 CrossRef CAS;
(b) L. Li, C. Wu, D. Liu, S. Li and D. Cui, Organometallics, 2013, 32, 3203 CrossRef CAS;
(c) L. Guo, X. Zhu, G. Zhang, Y. Wei, L. Ning, S. Zhou, Z. Feng, S. Wang, X. Mu, J. Chen and Y. Jiang, Inorg. Chem., 2015, 54, 5725 CrossRef CAS PubMed;
(d) G. Zhang, S. Wang, S. Zhou, Y. Wei, L. Guo, X. Zhu, L. Zhang, X. Gu and X. Mu, Organometallics, 2015, 34, 4251 CrossRef CAS.
- Similar organic by-products were observed in the activations of rare-earth metal or titanium alkyls with [Ph3C][B(C6F5)4] previously. However, the generation of Ph3CH still remains unclear, see:
(a) P. D. Bolton, E. Clot, N. Adams, S. R. Dubberley, A. R. Cowley and P. Mountford, Organometallics, 2006, 25, 2806 CrossRef CAS;
(b) M. Zimmermann, K. W. Törnroos, R. M. Waymouth and R. Anwander, Organometallics, 2008, 27, 4310 CrossRef CAS;
(c) D. M. Lyubov, L. Luconi, A. Rossin, G. Tuci, A. V. Cherkasov, G. K. Fukin, G. Giambastiani and A. A. Trifonov, Chem. – Eur. J., 2014, 20, 3487 CrossRef CAS PubMed;
(d) L. Luconi, D. M. Lyubov, A. Rossin, T. A. Glukhova, A. V. Cherkasov, G. Tuci, G. K. Fukin, A. A. Trifonov and G. Giambastiani, Organometallics, 2014, 33, 7125 CrossRef CAS.
- M. F. Lappert and R. Pearce, J. Chem. Soc., Chem. Commun., 1973, 126 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, A64, 112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880484–1880487. For ESI and crystallographic data in CIF or other electronic format, see DOI: 10.1039/c8dt04647f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Wenqiang
Li
a,
Ning-Ning
Wei
a,
Wenqiang
Li
a,
Ning-Ning
Wei