
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Broadband near-IR absorbing Au-dithiolene complexes bearing redox-active oligothiophene ligands†
Iain A.
Wright‡
 *a,
Claire
Wilson§
b,
Simon J.
Coles
b and
Peter J.
Skabara§
*a
*a,
Claire
Wilson§
b,
Simon J.
Coles
b and
Peter J.
Skabara§
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK. E-mail: i.a.wright@lboro.ac.uk; peter.skabara@glasgow.ac.uk
bUK National Crystallography Service. School of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
First published on 16th November 2018
Abstract
A series of three homoleptic, monoanionic gold dithiolene complexes of oligothiophene ligands which coordinate via a central thiophene-3,4-dithiolate chelate are presented. The oligomer chains are three, five and seven thiophenes long and the complexes display hybrid optoelectronic properties featuring characteristics of both the oligothiophene chains and the delocalised metal dithiolene centre. The properties of the complexes have been characterised using a variety of spectroscopic and electrochemical methods complemented by computational studies. Solid state spectroelectrochemistry has revealed that upon oxidation these complexes display intense and broad absorption across the visible spectrum. In attempting to produce nickel analogues of these materials a single crystal of a photo-oxidised nickel dithiolene complex has also been isolated.
Introduction
Binding two or more electronically delocalised substructures together by fusion across a shared aromatic bond presents an alternative and powerful method by which to facilitate electronic communication between redox-active sub-units.Bis(dithiolene) complexes have a non-innocent [C4S4M] chelate and have fascinated coordination and materials chemists alike for over 50 years.1–5 These complexes are often intensely coloured with a characteristic near-IR (NIR) absorption6,7 and display numerous reversible electron transfer processes.8 They are known to exhibit a range of fascinating and useful material properties including ferromagnetism,9 metallic conductivity and superconductivity,9–12 ambipolar charge transport,13–15 non-linear optical activity16,17 and catalytic water splitting.18–20
Conjugated polymers21–24 and oligomers25–27 of thiophene are archetypical organic p-type semiconductors that have been widely studied for use in optoelectronic devices. Combining the rich optoelectronic properties of the metal-containing non-innocent dithiolene core with the straightforward processing and easily tunable electronic properties of a conjugated polymer or oligomer presents an appealing approach in the search for new functional materials. A number of polymerisable complexes have been realised and subjected to electropolymerisation28–34 or electrodeposition35,36 to give low band gap metallopolymers, while discrete thiophene-containing dithiolene ligands have recently provided some novel photochromic complexes.37
Our interest in this area involves extending the conjugation length of the 34TDT ligand. Metallopolymers obtained by the electropolymerisation of complexes of the symmetrical 34TDT ligand 2,5-di(thien-2-yl)thiophene-3,4-dithiolate displayed broad absorption which is desirable for harvesting solar energy.33 An analogue of this ligand was synthesised with methyl groups capping the terminal α-positions of the oligothiophene chains to inhibit polymerisation; this material provided electrochemically stable complexes.34
Our family of ligands has grown to include methyl end-capped terthiophene (3T), quinquethiophene (5T) and septithiophene (7T) chains which we have previously exploited as precursors in the synthesis of new tetrathiafulvalenes57–59 and spirocyclic germanium complexes60,61 for applications in organic field effect transistor (OFET) and organic solar cell (OSC) devices, respectively. These ligands also bear hexyl groups to improve solubility. Here, we continue these studies by reporting the synthesis of monoanionic homoleptic Au complexes using 3T, 5T and 7T as 34TDT ligands.
The complexes have been characterised by electrochemical and spectroscopic methods and the optoelectronic properties of the neutral complexes have been probed using electronic absorption spectroelectrochemistry. Alongside this we attempted the synthesis of a complementary Ni complex which proved to be poorly stable, although we did obtain a single crystal of the complex [Ni(3T)2]2− in a partially oxidised state, the structure of which is also presented.
Results and discussion
Synthesis
All of the ligand precursors and complexes were synthesised by the same process (Scheme 1). Full experimental details can be found in our earlier publications57,58,60,62 and the ESI.†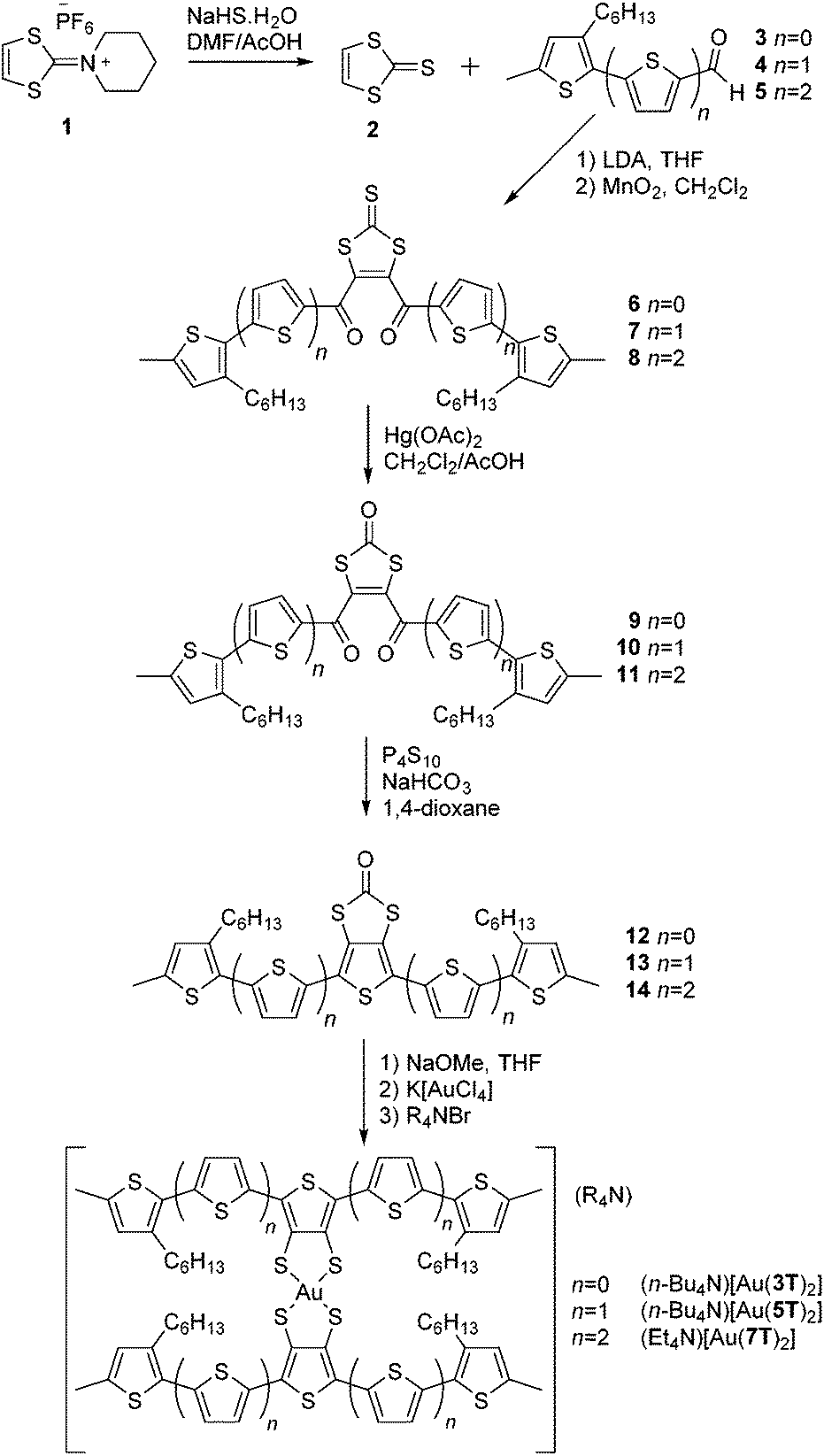 | ||
| Scheme 1 Synthesis of ligand precursors 12–14 and the Au complexes thereof. | ||
Hexafluorophosphate salt 1 was treated with NaHS·H2O in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMF/AcOH to produce the 1,3-dithiol-2-thione 2.63 Compound 2 was dissolved in tetrahydrofuran and treated twice, sequentially, at −78 °C with LDA followed by the corresponding 2-thienyl aldehyde 3–5. The temperature had to be carefully controlled throughout this process. This provided intermediate 1,4-diols which were then rapidly oxidised by MnO2 in CH2Cl2 to give the desired 1,4-diketone intermediates 6–8. Transchalcogenation of the 1,3-dithiol-2-thiones to 1,3-dithiol-2-ones 9–11 was achieved with Hg(OAc)2 in a mixture of CH2Cl2/AcOH. Finally, the 34TDT core was obtained by Paal–Knorr cyclisation using P4S10 and NaHCO3 in 1,4-dioxane to produce the ring-fused oligothiophenes 12–14.
:1 mixture of DMF/AcOH to produce the 1,3-dithiol-2-thione 2.63 Compound 2 was dissolved in tetrahydrofuran and treated twice, sequentially, at −78 °C with LDA followed by the corresponding 2-thienyl aldehyde 3–5. The temperature had to be carefully controlled throughout this process. This provided intermediate 1,4-diols which were then rapidly oxidised by MnO2 in CH2Cl2 to give the desired 1,4-diketone intermediates 6–8. Transchalcogenation of the 1,3-dithiol-2-thiones to 1,3-dithiol-2-ones 9–11 was achieved with Hg(OAc)2 in a mixture of CH2Cl2/AcOH. Finally, the 34TDT core was obtained by Paal–Knorr cyclisation using P4S10 and NaHCO3 in 1,4-dioxane to produce the ring-fused oligothiophenes 12–14.
The final complexes were obtained (Scheme 1) by treating a THF solution of the desired ligand precursor 12–14 with freshly made NaOMe prior to addition of K[AuCl4] followed immediately by a tetra(alkyl)ammonium bromide to provide the complexes in moderate to high yields. Purification was achieved by recrystallisation and verified by elemental analysis. Although the elemental analysis results for the largest complex [Au(7T)2]− showed some deviation from the predicted values, the results of optical and vibrational spectroscopy and its electrochemical properties are consistent with the other members of the series therefore we will include it within our discussion.
Electronic absorption spectroscopy
The absorption spectra of the complexes (Fig. 1 and Table 1) were recorded in CH2Cl2 solution and are dominated by intense bands in the visible region associated with π → π* transitions over the oligothiophene chains. As may be expected, the π → π* transitions become red-shifted as the conjugation length of the oligothiophene ligand increases. For [Au(3T)2]− this band has a λmax at 350 nm whereas complexes [Au(5T)2]− and [Au(7T)2]− have λmax values of 441 and 481 nm respectively. These are typical values for oligothiophenes of these chain lengths and are only slightly red-shifted in comparison with the ligands 3T–7T.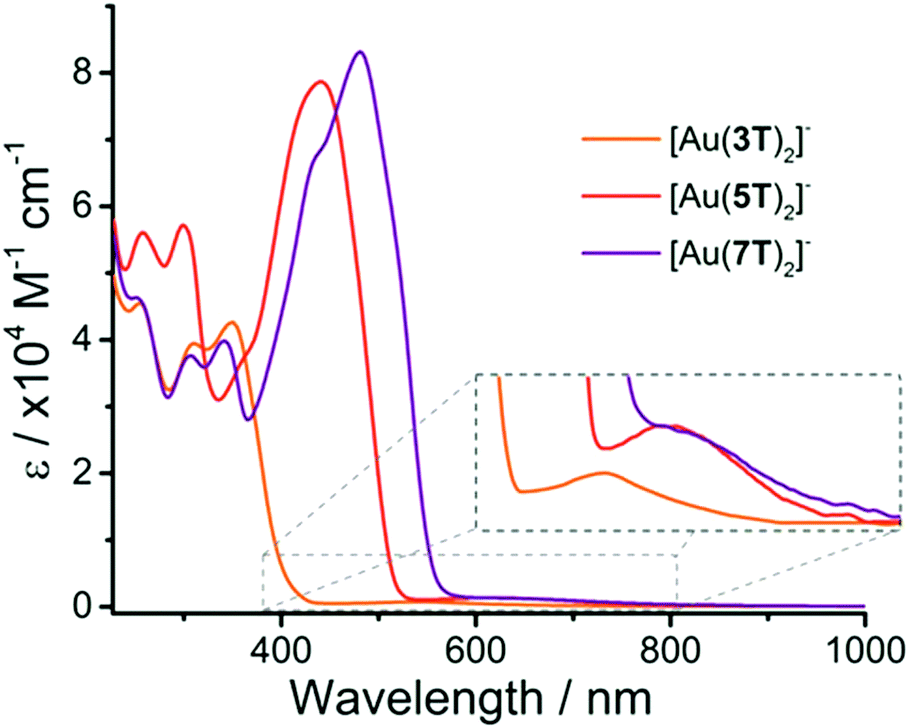 | ||
| Fig. 1 Electronic absorption spectra for complexes [Au(3T)2]−, [Au(5T)2]− and [Au(7T)2]− with low energy d–d transitions highlighted (inset). | ||
| λ max/nm | ε max × 103/mol−1 L cm−1 | π → π* energy gap/eV | d → d energy gap/eV | |
|---|---|---|---|---|
| 3T | 271(sh), 345 | 19.7 | 3.03 | — |
| 5T | 262, 302, 435 | 45.3 | 2.44 | — |
| 7T | 260(sh), 339, 467 | 61.2 | 2.29 | — |
| [Au(3T)2]− | 254, 310, 349, 543 | 42.6 | 2.99 | 1.64 |
| [Au(5T)2]− | 258, 299, 362(sh), 441, 616 | 78.6 | 2.37 | 1.42 |
| [Au(7T)2]− | 251, 307, 342, 438(sh), 481, 611 | 83.2 | 2.24 | 1.27 |
Common features include two high energy absorptions at ∼255 nm and ∼300 nm which are significantly more intense in complex [Au(5T)2]−. [Au(7T)2]− displays substructure featuring aspects of both of the smaller analogues, including a peak at ∼345 nm which coincides with the lowest energy π → π* band for [Au(3T)2]−; the peak maximum for [Au(5T)2]− correlates closely with a shoulder in the spectrum of [Au(7T)2]− indicating that some spectral characteristics of the smaller chains are maintained as the chain length increases.
The magnitude of the difference in extinction coefficients of the π → π* transitions between the free ligand and the complex decreases systematically as the chain length increases. It more than doubles in the case of 3T but increases by only a third in the case of 7T. Calculation of the energy of these π → π* transitions using the onset wavelengths reveals that they have only decreased by 0.04–0.07 eV with respect to their precursor ligands.
A characteristic low energy absorption which stretched towards the NIR region is also observed for each of the complexes and is associated with d → d transitions over the large Au atom (Fig. 1, inset).64–66 Increasing the ligand size from [Au(3T)2]− to [Au(5T)2]− results in a notable red-shift of this band however it does not shift further when the chain length increases to [Au(7T)2]−. The fact that this band is shifting to longer wavelengths as the oligothiophene chain increases in length serves to indicate that there is indeed some electronic communication with the delocalised dithiolene core.
Combined, these results indicate that while the Au dithiolene core itself does have interaction with the delocalised π-system of the ligands, it can facilitate only limited electronic communication between the π-systems of the two ligands, the extent of which decreases with increasing chain length.
Vibrational spectroscopy
Fourier transform infra-red (FTIR) spectroscopy of the ligands and complexes was performed using KBr pellets (Fig. 2 and Table S1†). There is good complementarity in the fingerprint regions of both the ligands and the complexes.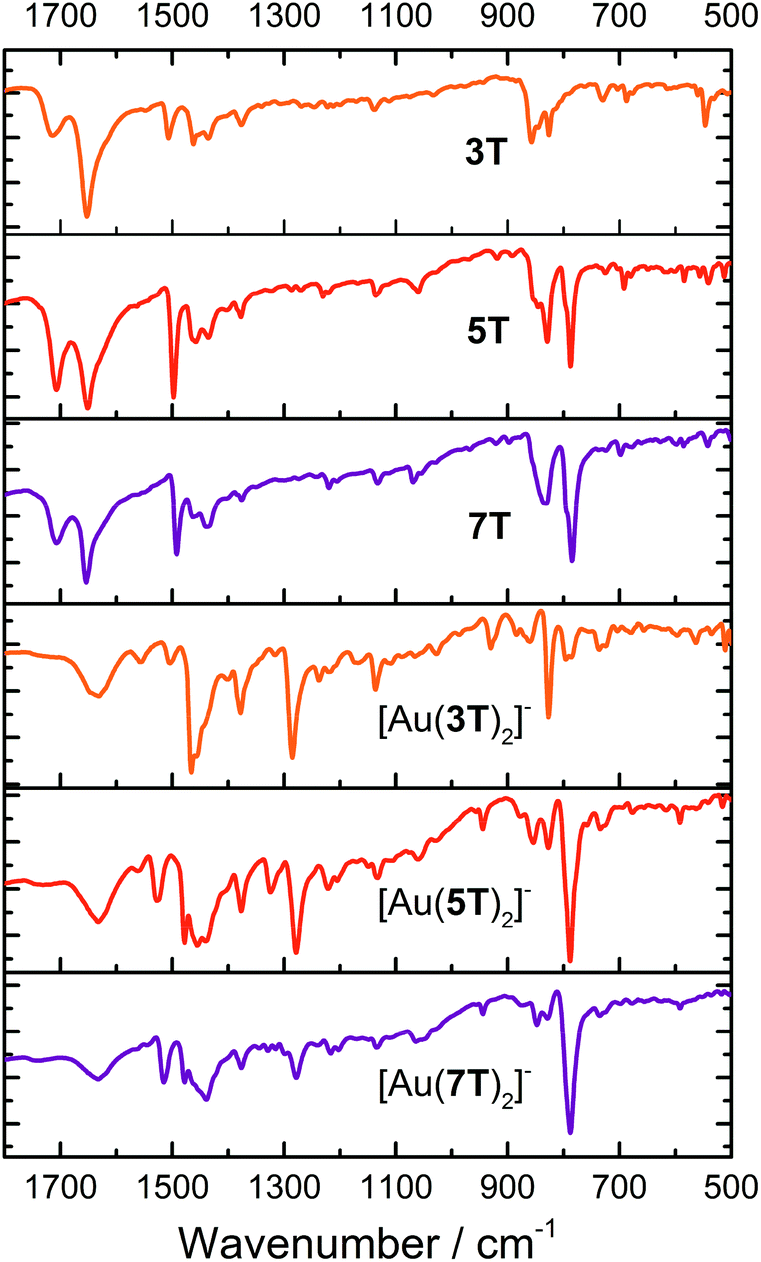 | ||
| Fig. 2 FTIR spectra for ligand precursors 3T, 5T and 7T and complexes [Au(3T)2]−, [Au(5T)2]− and [Au(7T)2]−. | ||
The ligands 3T–7T display peaks at ∼1650 and ∼1700 cm−1 due to the central carbonyl of the thieno-[3,4,d]-1,3-dithiol-2-one moiety. This correlates reasonably well with reported values, albeit at slightly higher wavenumber, and can be assigned confidently due to the absence of this peak in the FTIR spectra of the complexes.67,68 Aromatic overtones of the thiophene rings dominate the spectrum from 1650–1400 cm−1 (ref. 69) although a peak in this region between ∼1560 cm−1 is only observed in the metal complexes (hidden in [Au(7T)2]−) while a weak to medium intensity peak at ∼1377 cm−1 arises from deformations of the terminal –CH3 groups.69,70 A sharp peak at ∼1285 cm−1 is only present in the complexes. By comparing with Au complexes of 1,2-benzenedithiolate the absence of any intense bands around ∼1085 cm−1 indicates that the ligands have no significant radical character.64,66 The remaining notable characteristics are bands of moderate to strong intensity between 850–785 cm−1 assigned to aromatic C–H stretches of the remaining β-hydrogens on the thiophene backbone.69,70
Cyclic voltammetry
Cyclic voltammetry was used to examine the electrochemical properties of the complexes (Fig. 3 and Table 2). All of the complexes are significantly more easily oxidised than their ligand precursors57,58 and demonstrate a number of reversible or quasi-reversible processes. The complexity of the waveform increases and the reversibility decreases with increasing chain length.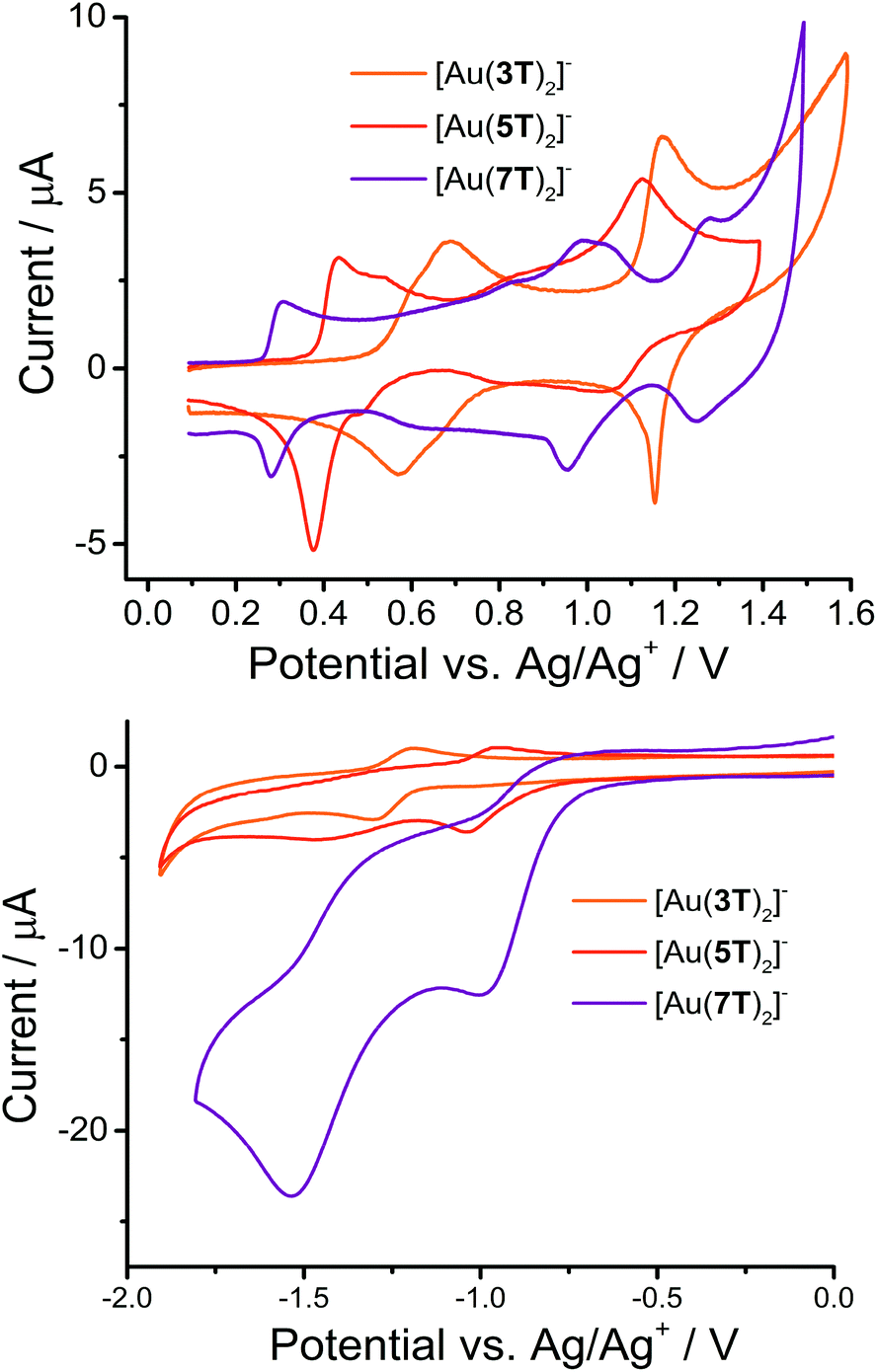 | ||
| Fig. 3 CV scans for the three gold complexes [Au(3T)2]−, [Au(5T)2]− and [Au(7T)2]− showing oxidation (upper) and reduction (lower) events. | ||
| [Au(3T)2]− | [Au(5T)2]− | [Au(7T)2]− | |
|---|---|---|---|
| a Electrochemical HOMO–LUMO gap calculated as the difference between the onset of the first oxidation and reduction waves. qQuasi-reversible peak. irrIrreversible peak. | |||
| E 1ox/V | +0.61/0.57 | +0.43/0.38 | +0.31/0.28 |
| E 2ox/V | +0.69/0.64 | +0.53/0.47 | +0.83/0.63 |
| E 3ox/V | +1.17/1.15 | +0.88/0.80 | +0.99/0.95 |
| E 4ox/V | — | +1.12/1.04 | +1.04irr |
| E 1red/V | −1.30/1.19q | −1.04/0.95q | −1.00irr |
| E 2red/V | — | — | −1.53irr |
| HOMO–LUMO gap/eVa | 1.68 | 1.19 | 1.04 |
The oxidative behaviour of aromatic Au dithiolene complexes is predominantly ligand based while the reductive behaviour is more metal centered.64,66 which can allow for great control to be exerted over the complexes’ oxidation potentials.65
In this series of compounds all major oxidative processes shift to lower potential as the oligothiophene chain lengthens, due to the increase in the number of π-electrons and extent of delocalisation, which result in stronger electron donating properties. Using the first oxidation potential as an example, the half-wave potential decreases steadily: E1/2 of +0.59 V for [Au(3T)2]−, +0.40 V for [Au(5T)2]− and +0.29 V for [Au(7T)2]−. On the other hand, reduction processes shift to a more positive potential in a similar fashion. The trend in reduction potentials coupled with the low onset of oxidation in all cases further indicates that some electronic coupling between the ligand π-system and the metal dithiolene core does indeed occur.
Electrochemical HOMO–LUMO gaps were calculated from the difference in the onsets of the first oxidation and reduction peaks and agree well with the d → d transition energies calculated from the absorption spectra.
Some specific comments about each complex follow:
[Au(3T)2]−
This complex displays greatly improved electrochemical reversibility when compared to the analogous non-capped complex which only had a single irreversible oxidation at +0.57 V.34 It has closely overlapping first and second oxidations with peak profiles and separations that indicate fairly good reversibility. In keeping with our previous observations we have assigned this to sequential radical cation formation on each terthiophene, which is localised primarily over the ligands. A third quasi-reversible oxidation occurs at E1/2 of +1.16 V, of which the narrow profile of the reverse peak may indicate some adsorption of the triply charged complex to the electrode surface. The complex undergoes a single quasi-reversible reduction at −1.26 V.
[Au(5T)2]−
In complex [Au(5T)2]−, the first oxidation peaks are similar to those observed for complex [Au(3T)2]−. The next region of the voltammogram shows no clearly defined peaks although there is evidence for a further oxidation occurring at approximately +0.83 V followed by a fourth, more easily discernible quasi-reversible peak centred at E1/2 = +1.08 V. This sequence of events is likely due to dication formation on each of the quinquethiophene chains. The complex undergoes a single quasi-reversible reduction at E1/2 = −1.00 V and an irreversible reduction at −1.48 V.
[Au(7T)2]−
Complex [Au(7T)2]− displays a voltammogram with a shape that is largely complementary to that of [Au(5T)2]−. A first reversible oxidation is centered at +0.28 V with another oxidation at +0.97 V. There appears to be at least one quasi-reversible oxidation in the broad waveform between these two oxidations. An irreversible shoulder can be identified at +1.04 V prior to the final quasi-reversible formation of a second dication at +1.27 V. The complex undergoes two irreversible reductions at −1.00 and −1.53 V. The magnitude of the current response of these reductions is very large and no reversibility is apparent.
Computational studies
DFT methods were used to gain a further insight into the electronic properties of [Au(3T)2]− and [Au(5T)2]−. The orbital contours for [Au(3T)2]− and [Au(5T)2]− are shown in Fig. 4 and 5 respectively. As it was desirable to see how the hexyl chains influence the geometry adopted by the ligands upon complexation they were included in the calculation. Similar observations can be made for both complexes:(1) In both cases steric hindrance results in significant twisting of the terminal thiophene rings of the oligothiophene chains
(2) The frontier orbitals of the HOMO manifold are delocalised over both the oligothiophene chains and metal dithiolene core. In [Au(5T)2]− it is not until the HOMO−4 that purely oligothiophene based contours begin to be observed while in [Au(3T)2]− even the HOMO−5 contains some influence from the coordinating sulfurs.
(3) The LUMO is localised almost exclusively over the gold-dithiolene centre for both complexes and is close in energy to the higher HOMO orbitals which correlates well with the low energy d → d band observed in the absorption spectra.
(4) The LUMO+1 and higher lie at significantly more positive energies than the LUMO and are delocalised over the conjugated π systems of the ligands with small contributions from the coordinating dithiolene sulfur atoms. This indicates that these orbitals make a much more substantial contribution to the shorter wavelength π → π* transitions.
(5) The HOMO in [Au(3T)2]− is quite similar to the HOMO−2 of [Au(5T)2]−. Noting that the energy differences between the HOMO, the HOMO−1 and the HOMO−2 are relatively small, it appears that the longer conjugation length of the 5T ligand leads to it having an increased influence on the frontier HOMOs than the less extended 3T ligand.
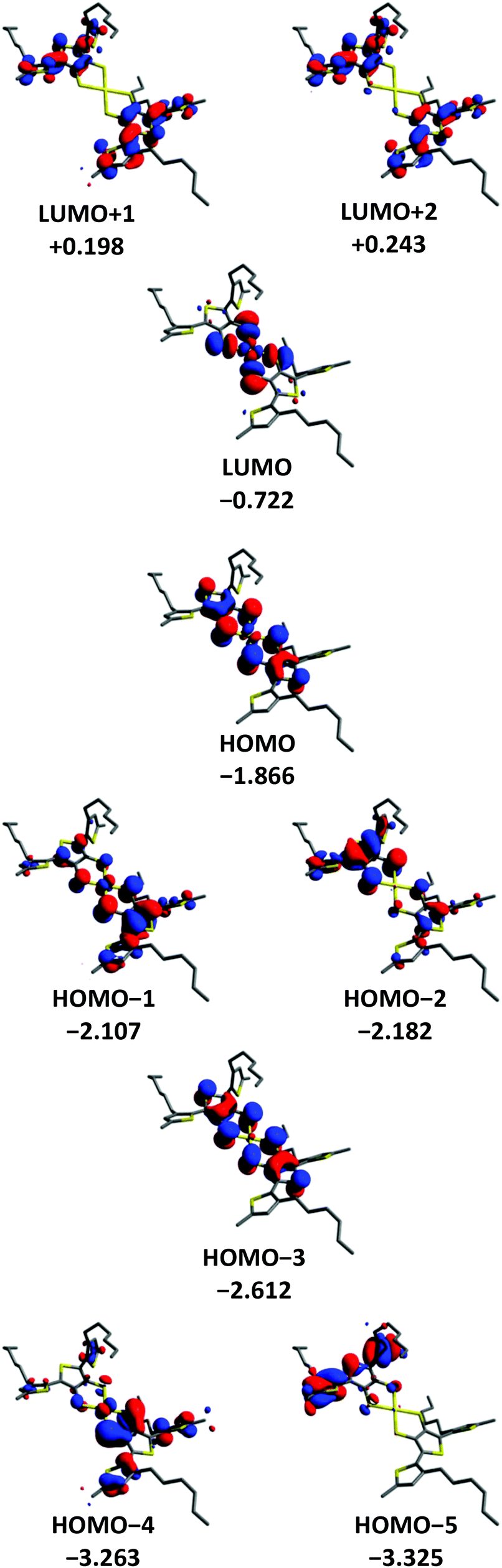 | ||
| Fig. 4 Frontier orbital profiles and energies for the [Au(3T)2]− anion. | ||
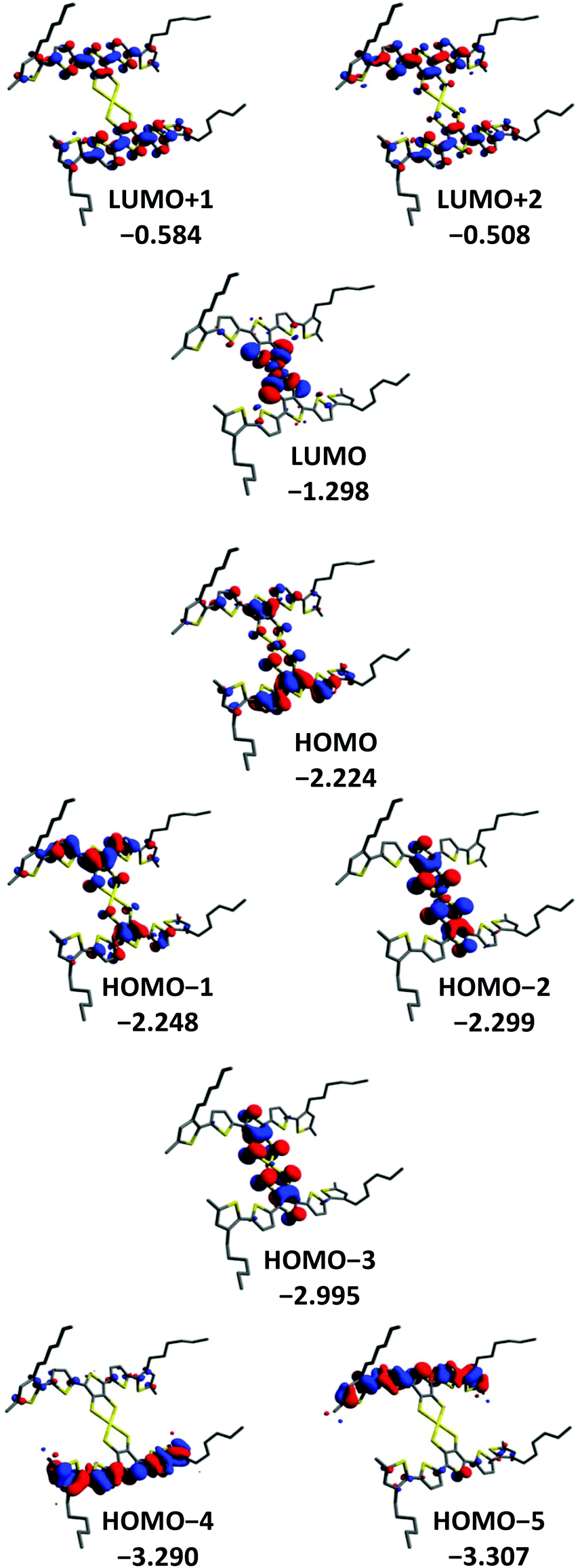 | ||
| Fig. 5 Frontier orbital profiles and energies for the [Au(5T)2]− anion. | ||
Paying particular attention to the mixed oligothiophene/dithiolene nature of the HOMO orbitals, these results clearly indicate the presence of constructive electronic interplay between the ligands and the gold-dithiolene core. Overall, the calculated electronic structure of the gold TDT core agrees fairly well with existing studies on both gold dithiolenes64,65,71 and other metal TDT complexes.56
Spectroelectrochemistry
Neutral Au 34TDT complexes have not as yet been isolated. Upon oxidation of the monoanionic complexes with iodine they yield poorly defined and insoluble polycrystalline precipitates which show some paramagnetic character.39,42 UV-vis spectroelectrochemical (SEC) measurements present one route by which to begin to observe their spectral properties. SEC allows changes in a molecule's spectroscopic properties upon oxidation or reduction to be monitored.Thin films of the complexes were deposited on ITO slides by drop casting. The slides were then suspended in an electrolytic solution and used as the working electrode of a standard three-electrode electrochemical cell held within a UV/vis spectrometer. A potential was applied to the film and its absorption spectrum measured at intervals of +0.10 V in a step-wise fashion. Upon changing potential the film was given approximately 60 seconds to equilibrate prior to recording the spectrum. The results of these experiments are presented in Fig. 6. Spectroelectrochemical studies on thin films of the ligands 5T and 7T can be found in our previous publications.57,58 The smallest ligand 3T is soluble in the electrolyte solution therefore thin film studies were not possible.
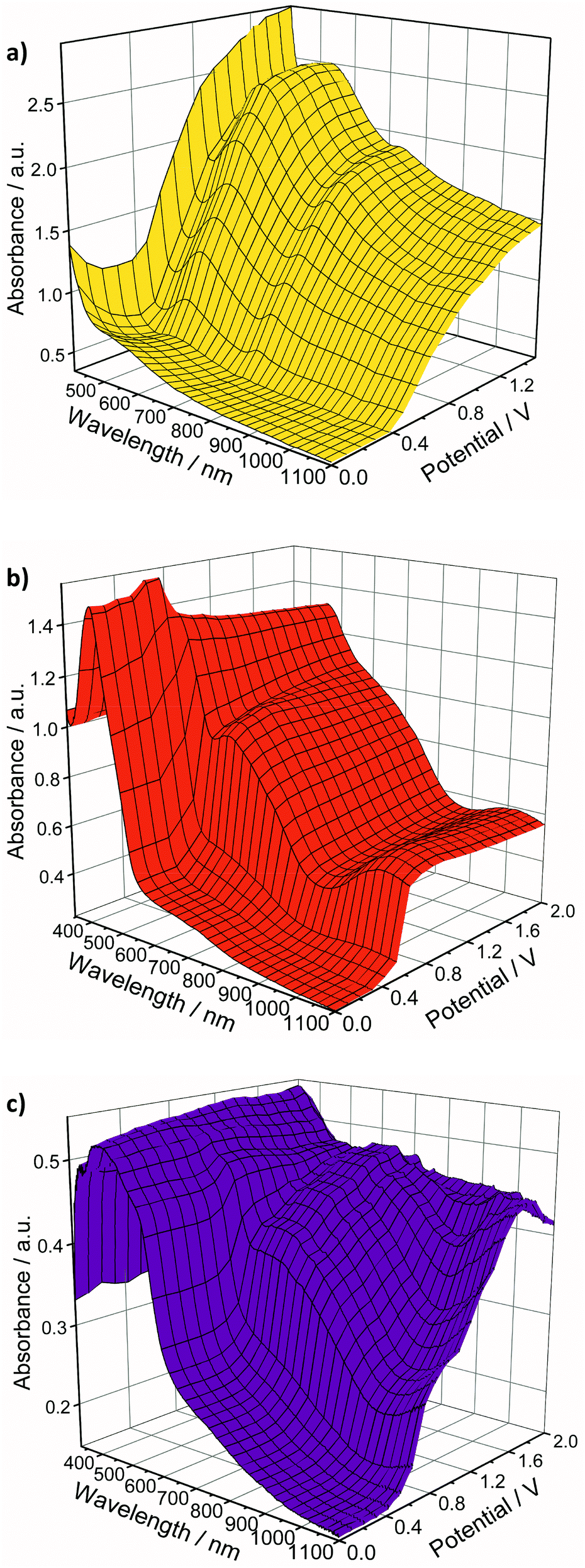 | ||
| Fig. 6 Spectroelectrochemistry plots for the oxidation of thin films of (a) [Au(3T)2]−, (b) [Au(5T)2]−, and (c) [Au(7T)2]−. | ||
Prior to oxidation, the thin film absorption spectra of all of the complexes are dominated by ligand based π → π* transitions with the weak d → d transition of the Au dithiolene core also evident. Upon oxidation a large increase in absorption across the entire visible spectrum is observed, featuring characteristics of both the conjugated ligand and the delocalised Au dithiolene centre. This is a somewhat gradual process for [Au(3T)2]− and [Au(7T)2]− but is very sharp for [Au(5T)2]− and remains remarkably stable at higher potentials.
The solid state SEC of bis(terthiophene) Au dithiolene complex [Au(3T)2]− is shown in Fig. 6a. Upon increasing the potential from 0.00 V to +0.40 V the intensity of the d → d absorption drops slightly then increases rapidly as at least one electron is lost with strong peaks emerging at 515 and 734 nm. The wavelength of the first of these peaks is due to the terthiophene based radical cation while the second will occur over the now neutral [AuC4S4]0 dithiolene centre. The intensity of the absorbance across the spectrum begins to stabilise at potentials >+0.70 V.
The SEC plot for [Au(5T)2]− (Fig. 6b) shows that upon oxidation at +0.30 V a large and sudden spectral response occurs: the π → π* absorption drops slightly in intensity and is redshifted from 429 to 459 nm and this is typical behaviour observed in conjugated thiophene oligomers and polymers.72 Concomitantly, the intensity of absorption across the rest of the spectrum is raised greatly with peaks at 602 nm and 1036 nm pushing deep into the NIR. The very limited change in the spectrum at higher potentials may indicate that it has become insulating.
The complex [Au(7T)2]− displays characteristics similar to those of the 5T analogue. At potentials above +0.30 V the generation of broad bands with peaks at 679 and >1100 nm respectively (Fig. 6c).
X-Ray crystallography
As previously mentioned, we attempted to synthesise nickel analogues of the gold complexes by using NiCl2·6H2O in place of K(AuCl)4 but found them poorly stable and therefore difficult to isolate. However we were able to isolate a small amount of the terthiophene nickel complex by diffusion of cyclohexane into a tetrahydrofuran solution and were surprised by our findings.A single crystal of (Et4N)2[Ni(3T)2] grew as an orange plate. X-ray crystallography revealed that the complex displayed varying amounts of oxidation of the coordinating sulfur atoms. We can only attribute this partial oxidation to the complexes having reacted with dissolved molecular oxygen. Similar spontaneous aerobic oxidation of dithiolenes has only been reported on a small number of occasions.73–75 This behaviour also leads us to postulate that oxidation of our Ni complexes could be an underlying factor of their poor stability. The asymmetric unit of the nickel complex contains two half complexes with the Ni lying on an inversion centre and two tetraethylammonium cations (Fig. 7). The coordinated S atoms are partially oxidised and have two oxygen sites modelled for each S which are refined with a common occupancy for each pair (for Ni(1) 0.19(3) and 0.50(2), for Ni(1a) 0.24(2) and 0.42(2)).
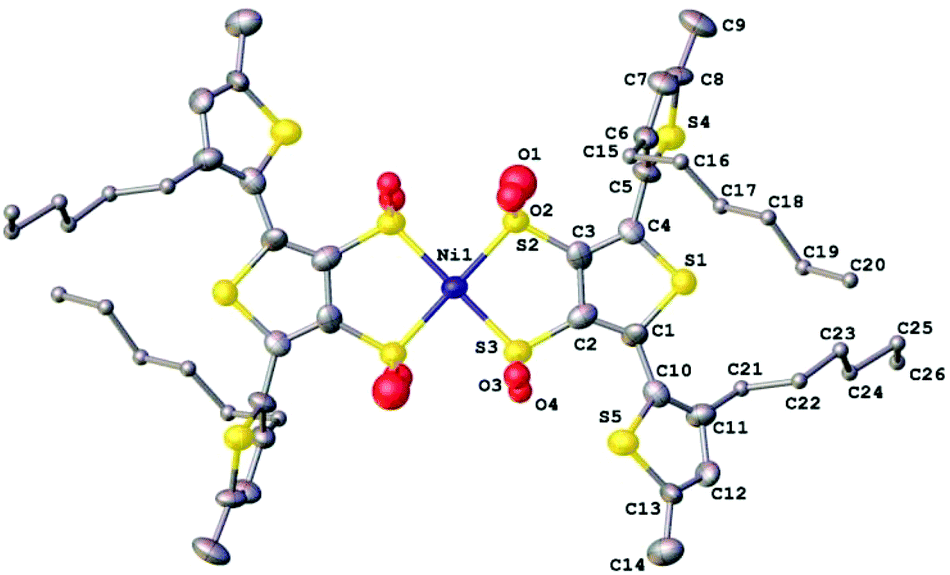 | ||
| Fig. 7 X-ray molecular structure determined for the partially oxidised [Ni(3T)2]2− dianion. | ||
The terthiophene chains do not adopt the all-anti conformation typical of short chain oligothiophenes, presumably due to steric hindrance between the hexyl chains and possible intermolecular forces between S(5)⋯O(3) (3.050(2)). The torsional angles between the terminal thiophenes are 72(2)° and 73(2)° for C3–C4–C5–C6 and C2a–C1a–C10a–C11a respectively and 150(2)° and 148(2) for C2–C1–C10–C11 and Ca–C4a–C5a–C6a respectively. The root mean square deviation from planarity for the central dithiolene rings, Ni–S2–C3–C2–S3 and Ni1a–S2a–C3a–C2a–S3a are 0.067 Å and 0.054 Å respectively.
Conclusions
In conclusion, six new homoleptic bis(thiophene-3,4-dithiolate) complexes featuring nickel or gold metal centres bound between end-capped oligothiophene ligands with chain lengths of three, five and seven thiophenes were synthesised. The Ni complexes proved to be poorly stable due to facile auto-oxidation as confirmed by the isolation of a crystal of a partially oxidised complex, the molecular structure of which was confirmed by X-ray crystallography.The monoanionic formally Au(III) complexes show hybrid optoelectronic properties resulting from interplay between the dithiolene core and the oligothiophene ligands including low oxidation potentials and a low energy metal centred absorption band, the position of which is sensitive to the oligothiophene chain length. This hybrid electronic behaviour has been confirmed using spectroscopic and electrochemical studies and corroborated with computational analysis of the two smaller complexes.
Spectroelectrochemical results for thin films of the Au(III) complexes in the solid state have shown that in all cases oxidation, ostensibly to the neutral Au(IV) complex, results in a large and sudden increase in the intensity of absorption across the entire visible window and into the NIR, which persists at higher potentials. Although their isolation remains challenging, the interesting optoelectronic and magnetic properties of neutral gold 34TDT complexes with extended conjugation means that they remain intriguing targets for further development. In particular, their broad and strong absorption across the visible and NIR regions of the spectrum make them excellent candidate materials for broadband light sensing and harvesting applications.76
Conflicts of interest
There are no conflicts to declare.Acknowledgements
PJS and IAW acknowledge the EPSRC for funding. IAW acknowledges Loughborough University and the Royal Society of Chemistry Research Fund (Grant No. RF18-5353) for funding.Notes and references
- G. N. Schrauzer and V. Mayweg, J. Am. Chem. Soc., 1962, 84, 3221 CrossRef CAS.
- H. B. Gray, R. Williams, I. Bernal and E. Billig, J. Am. Chem. Soc., 1962, 84, 3596–3597 CrossRef CAS.
- A. Davison, N. Edelstein, R. H. Holm and A. H. Maki, Inorg. Chem., 1963, 2, 1227–1232 CrossRef CAS.
- J. A. McCleverty, in Progress in Inorganic Chemistry, ed. F. A. Cotton, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1968, vol. 10, pp. 49–221 Search PubMed.
- R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed.
- M. L. Kirk, R. L. McNaughton and M. E. Helton, in Dithiolene Chemistry. Synthesis, Properties, and Applications. Progress in Inorganic Chemistry, ed. E. I. Stiefel and K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004, vol. 52, pp. 111–212 Search PubMed.
- S. D. Cummings and R. Eisenberg, in Dithiolene Chemistry: Synthesis, Properties, and Applications. Progress in Inorganic Chemistry, ed. E. I. Stiefel and K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004, vol. 52, pp. 315–367 Search PubMed.
- K. Wang, in Dithiolene Chemistry: Synthesis, Properties, and Applications. Progress in Inorganic Chemistry, ed. E. I. Stiefel and K. D. Karlin, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004, vol. 52, pp. 267–314 Search PubMed.
- N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93–127 CrossRef CAS.
- R. Kato, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed.
- M. Bousseau, L. Valade, J. P. Legros, P. Cassoux, M. Garbauskas and L. V. Interrante, J. Am. Chem. Soc., 1986, 108, 1908–1916 CrossRef CAS.
- D. G. Branzea, F. Pop, P. Auban-Senzier, R. Clérac, P. Alemany, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2016, 138, 6838–6851 CrossRef CAS PubMed.
- T. D. Anthopoulos, G. C. Anyfantis, G. C. Papavassiliou and D. M. De Leeuw, Appl. Phys. Lett., 2007, 90, 122105 CrossRef.
- T. D. Anthopoulos, S. Setayesh, E. Smits, M. Cölle, E. Cantatore, B. De Boer, P. W. M. Blom and D. M. De Leeuw, Adv. Mater., 2006, 18, 1900–1904 CrossRef CAS.
- S. Dalgleish, J. G. Labram, Z. Li, J. Wang, C. R. McNeill, T. D. Anthopoulos, N. C. Greenham and N. Robertson, J. Mater. Chem., 2011, 21, 15422–15430 RSC.
- P. Deplano, L. Pilia, D. Espa, M. L. Mercuri and A. Serpe, Coord. Chem. Rev., 2010, 254, 1434–1447 CrossRef CAS.
- D. Espa, L. Pilia, C. Makedonas, L. Marchiò, M. L. Mercuri, A. Serpe, A. Barsella, A. Fort, C. A. Mitsopoulou and P. Deplano, Inorg. Chem., 2014, 53, 1170–1183 CrossRef CAS PubMed.
- A. Zarkadoulas, E. Koutsouri and C. A. Mitsopoulou, Coord. Chem. Rev., 2012, 256, 2424–2434 CrossRef CAS.
- W. R. McNamara, Z. Han, C.-J. Yin, W. W. Brennessel, P. L. Holland and R. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15594–15599 CrossRef CAS PubMed.
- W. R. McNamara, Z. Han, P. J. Alperin, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2011, 133, 15368–15371 CrossRef CAS PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711–738 CrossRef CAS.
- T. P. Kaloni, P. K. Giesbrecht, G. Schreckenbach and M. S. Freund, Chem. Mater., 2017, 29, 10248–10283 CrossRef CAS.
- A. L. Kanibolotsky, N. J. Findlay and P. J. Skabara, Beilstein J. Org. Chem., 2015, 11, 1749–1766 CrossRef CAS PubMed.
- T. M. S. K. Pathiranage, D. S. Dissanayake, C. N. Niermann, Y. Ren, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3327–3346 CrossRef CAS.
- A. Mishra, C. Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1176 CrossRef CAS PubMed.
- L. Zhang, N. S. Colella, B. P. Cherniawski, S. C. B. Mannsfeld and A. L. Briseno, ACS Appl. Mater. Interfaces, 2014, 6, 5327–5343 CrossRef CAS PubMed.
- T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
- C. L. Kean and P. G. Pickup, Chem. Commun., 2001, 815–816 RSC.
- C. L. Kean, D. O. Miller and P. G. Pickup, J. Mater. Chem., 2002, 12, 2949–2956 RSC.
- T. Anjos, S. J. Roberts-Bleming, A. Charlton, N. Robertson, A. R. Mount, S. J. Coles, M. B. Hursthouse, M. Kalaji and P. J. Murphy, J. Mater. Chem., 2008, 18, 475–483 RSC.
- S. Dalgleish, C. A. Morrison, D. S. Middlemiss, A. R. Mount, A. Collins, L. Pilia, A. Serpe, M. L. Mercuri, S. J. Roberts-Bleming, A. Charlton, P. Deplano, P. J. Murphy and N. Robertson, J. Mater. Chem., 2009, 19, 6194–6200 RSC.
- S. Dalgleish and N. Robertson, Coord. Chem. Rev., 2010, 254, 1549–1558 CrossRef CAS.
- C. Pozo-Gonzalo, R. Berridge, P. J. Skabara, E. Cerrada, M. Laguna, S. J. Coles and M. B. Hursthouse, Chem. Commun., 2002, 2408–2409 RSC.
- P. J. Skabara, C. Pozo-Gonzalo, N. Lardiés Miazza, M. Laguna, E. Cerrada, A. Luquin, B. González, S. J. Coles, M. B. Hursthouse, R. W. Harrington and W. Clegg, Dalton Trans., 2008, 3070–3079 RSC.
- S. Dalgleish, H. Yoshikawa, M. M. Matsushita, K. Awaga and N. Robertson, Chem. Sci., 2011, 2, 316–320 RSC.
- E. Allwright, G. Silber, J. Crain, M. M. Matsushita, K. Awaga and N. Robertson, Dalton Trans., 2016, 45, 9363–9368 RSC.
- J. Wang, L.-X. Shi, J.-Y. Wang, J.-X. Chen, S.-H. Liu and Z.-N. Chen, Dalton Trans., 2017, 46, 2023–2029 RSC.
- D. Belo, H. Alves, E. B. Lopes, V. Gama, R. T. Henriques, M. T. Duarte, M. Almeida, A. Perez-Benitez, C. Rovira and J. Veciana, Synth. Met., 2001, 120, 699–702 CrossRef CAS.
- D. Belo, H. Alves, E. B. Lopes, M. T. Duarte, V. Gama, R. Teives Henriques, M. Almeida, A. Pérez-Benítez, C. Rovira and J. Veciana, Chem. – Eur. J., 2001, 7, 511–519 CrossRef CAS.
- D. Belo, M. J. Figueira, J. Mendonça, I. C. Santos, M. Almeida, R. T. Henriques, M. T. Duarte, C. Rovira and J. Veciana, Eur. J. Inorg. Chem., 2005, 3337–3345 CrossRef CAS.
- A. I. S. Neves, I. C. Santos, D. Belo and M. Almeida, CrystEngComm, 2009, 11, 1046–1053 RSC.
- D. Belo and M. Almeida, Coord. Chem. Rev., 2010, 254, 1479–1492 CrossRef CAS.
- D. Belo, H. Alves, S. Rabaça, L. C. Pereira, M. T. Duarte, V. Gama, R. T. Henriques, M. Almeida, E. Ribera, C. Rovira and J. Veciana, Eur. J. Inorg. Chem., 2001, 2001, 3127–3133 CrossRef.
- D. Belo, M. J. Figueira, J. P. M. Nunes, I. C. Santos, L. C. Pereira, V. Gama, M. Almeida and C. Rovira, J. Mater. Chem., 2006, 16, 2746–2756 RSC.
- J. P. M. Nunes, M. J. Figueira, D. Belo, I. C. Santos, B. Ribeiro, E. B. Lopes, R. T. Henriques, J. Vidal-Gancedo, J. Veciana, C. Rovira and M. Almeida, Chem. – Eur. J., 2007, 13, 9841–9849 CrossRef CAS PubMed.
- D. Belo, J. Mendonça, I. C. Santos, L. C. J. Pereira, M. Almeida, J. J. Novoa, C. Rovira, J. Veciana and V. Gama, Eur. J. Inorg. Chem., 2008, 5327–5337 CrossRef CAS.
- D. Belo, L. C. Jesus Pereira, M. Almeida, C. Rovira, J. Veciana and V. Gama, Dalton Trans., 2009, 4176–4180 RSC.
- A. I. S. Neves, J. C. Dias, B. J. C. Vieira, I. C. Santos, M. B. Castelo Branco, L. C. J. Pereira, J. C. Waerenborgh, M. Almeida, D. Belo and V. da Gama, CrystEngComm, 2009, 11, 2160–2168 RSC.
- A. I. S. Neves, I. C. Santos, L. C. J. Pereira, C. Rovira, E. Ruiz, D. Belo and M. Almeida, Eur. J. Inorg. Chem., 2011, 4807–4815 CrossRef CAS.
- D. Belo, A. I. S. Neves, L. C. J. Pereira and M. Almeida, Phys. Status Solidi, 2012, 9, 1199–1201 CrossRef CAS.
- E. Laukhina, V. Lebedev, V. Laukhin, A. P. del Pino, E. B. Lopes, A. I. S. Neves, D. Belo, M. Almeida, J. Veciana and C. Rovira, Org. Electron., 2012, 13, 894–898 CrossRef CAS.
- R. Silva, B. Vieira, M. Andrade, I. Santos, S. Rabaça, E. Lopes, J. Coutinho, L. Pereira, M. Almeida and D. Belo, Molecules, 2018, 23, 424 CrossRef PubMed.
- A. I. S. Neves, I. C. Santos, J. T. Coutinho, L. C. J. Pereira, R. T. Henriques, E. B. Lopes, H. Alves, M. Almeida and D. Belo, Eur. J. Inorg. Chem., 2014, 2014, 3989–3999 CrossRef CAS.
- T. Higashino, O. Jeannin, T. Kawamoto, D. Lorcy, T. Mori and M. Fourmigué, Inorg. Chem., 2015, 54, 9908–9913 CrossRef CAS PubMed.
- C. M. Amb and S. C. Rasmussen, Synth. Met., 2009, 159, 2390–2393 CrossRef CAS.
- C. M. Amb, C. L. Heth, S. J. Evenson, K. I. Pokhodnya and S. C. Rasmussen, Inorg. Chem., 2016, 55, 10978–10989 CrossRef CAS PubMed.
- I. A. Wright, P. J. Skabara, J. C. Forgie, A. L. Kanibolotsky, B. González, S. J. Coles, S. Gambino and I. D. W. Samuel, J. Mater. Chem., 2011, 21, 1462–1469 RSC.
- I. A. Wright, N. J. Findlay, S. Arumugam, A. R. Inigo, A. L. Kanibolotsky, P. Zassowski, W. Domagala and P. J. Skabara, J. Mater. Chem. C, 2014, 2, 2674–2683 RSC.
- R. G. D. Taylor, J. Cameron, I. A. Wright, N. Thomson, O. Avramchenko, A. L. Kanibolotsky, A. R. Inigo, T. Tuttle and P. J. Skabara, Beilstein J. Org. Chem., 2015, 11, 1148–1154 CrossRef CAS PubMed.
- I. A. Wright, A. L. Kanibolotsky, J. Cameron, T. Tuttle, P. J. Skabara, S. J. Coles, C. T. Howells, S. A. J. Thomson, S. Gambino and I. D. W. Samuel, Angew. Chem., Int. Ed., 2012, 51, 4562–4567 CrossRef CAS PubMed.
- S. Arumugam, I. A. Wright, A. R. Inigo, S. Gambino, C. T. Howells, A. L. Kanibolotsky, P. J. Skabara and I. D. W. Samuel, J. Mater. Chem. C, 2014, 2, 34–39 RSC.
- P. J. Skabara, I. M. Serebryakov, D. M. Roberts, I. F. Perepichka, S. J. Coles and M. B. Hursthouse, J. Org. Chem., 1999, 64, 6418–6424 CrossRef CAS.
- F. S. Guziec, J. M. Russo, F. F. Torres, G. C. Long and M. R. Tellez, J. Chem. Soc., Perkin Trans. 1, 1989, 1068–1070 RSC.
- K. Ray, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2005, 44, 5345–5360 CrossRef CAS PubMed.
- N. C. Schiodt, P. Sommer-Larsen, T. Bjornholm, M. F. Nielsen, J. Larsen and K. Bechgaard, Inorg. Chem., 1995, 34, 3688–3694 CrossRef CAS.
- K. Ray, T. Weyhermüller, A. Goossens, M. W. J. Crajé and K. Wieghardt, Inorg. Chem., 2003, 42, 4082–4087 CrossRef CAS PubMed.
- L. Y. Chiang, P. Shu, D. Holt and D. Cowan, J. Org. Chem., 1983, 48, 4713–4717 CrossRef CAS.
- X. Chen, N. R. de Tacconi and R. L. Elsenbaumer, J. Org. Chem., 2009, 74, 9188–9190 CrossRef CAS PubMed.
- D. Williams and I. Fleming, Spectroscopic Methods in Organic Chemistry, McGraw-Hill Publishing Company, London, 5th edn, 1995 Search PubMed.
- C. Wang, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, Chem. Mater., 1995, 7, 58–68 CrossRef CAS.
- S. Kokatam, K. Ray, J. Pap, E. Bill, W. E. Geiger, R. J. LeSuer, P. H. Rieger, T. Weyhermüller, F. Neese and K. Wieghardt, Inorg. Chem., 2007, 46, 1100–1111 CrossRef CAS PubMed.
- R. Berridge, S. P. Wright, P. J. Skabara, A. Dyer, T. Steckler, A. A. Argun, J. R. Reynolds, R. W. Harrington and W. Clegg, J. Mater. Chem., 2007, 17, 225–231 RSC.
- G. N. Schrauzer, C. Zhang and R. Chadha, Inorg. Chem., 1990, 29, 4104–4107 CrossRef CAS.
- R. Bolligarla and S. K. Das, Eur. J. Inorg. Chem., 2012, 2012, 2933–2939 CrossRef CAS.
- E. J. Uzelac and S. C. Rasmussen, Eur. J. Inorg. Chem., 2017, 2017, 3878–3883 CrossRef CAS.
- D. Cortizo-Lacalle, C. T. Howells, S. Gambino, F. Vilela, Z. Vobecka, N. J. Findlay, A. R. Inigo, S. A. J. Thomson, P. J. Skabara and I. D. W. Samuel, J. Mater. Chem., 2012, 22, 14119–14126 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental and synthetic and crystallography details and spectroscopic data. CCDC 1861791. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03915a |
| ‡ Present address: Department of Chemistry, Loughborough University, Loughborough LE11 3TU, UK. E-mail: i.a.wright@lboro.ac.uk. |
| § Present address: WestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. Email: peter.skabara@glasgow.ac.uk. |
| This journal is © The Royal Society of Chemistry 2019 |