
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-conventional synthesis and photophysical studies of platinum(II) complexes with methylene bridged 2,2′-dipyridylamine derivatives†
Evgeny
Bulatov
 and
Matti
Haukka
*
and
Matti
Haukka
*
Department of Chemistry, University of Jyväskylä, P.O. Box 35, FI-40014 Jyväskylä, Finland. E-mail: matti.o.haukka@jyu.fi
First published on 13th February 2019
Abstract
Methylene bridged 2,2′-dipyridylamine (dpa) derivatives and their metal complexes possess outstanding properties due to their inherent structural flexibility. Synthesis of such complexes typically involves derivatization of dpa followed by coordination on metals, and may not always be very efficient. In this work, an alternative synthetic approach, involving the derivatization step after – rather than prior to – coordination of dpa on metal center, is proposed and applied to synthesis of a number of platinum(II) complexes with substituted benzyldi(2-pyridyl)amines. Comparison with the more conventional synthetic route reveals greater efficiency and versatility of the proposed approach. The obtained complexes are not luminescent in solution at room temperature, but display blue phosphorescence emission (ca. 415 nm) with the lifetimes of μs order in glassy matrix at 77 K, with additional green (ca. 485 nm) and relatively long living (τ = 3.7 ms) emission in the case of iodine substituted derivative.
Introduction
Profound flexibility and chelating capabilities of 2,2′-dipyridylamine (di(2-pyridyl)amine, dpa) have stimulated excessive studies of its coordination chemistry,1 as well as incorporation of this moiety as a metal binding center into more complex organic materials.2 The latter is usually achieved by connecting dpa with an organic fragment via the amino nitrogen atom, and the resulting derivatives can be conjugated or non-conjugated (Scheme 1). Such organic molecules can be applicable in organic light emitting devices,3–6 while their ability to bind various transition metals through the dpa moiety allows their use as various luminescent sensors.7,8 Since dpa complexes of platinum(II) and palladium(II) have been found to possess cytotoxic activities comparable to or higher than cisplatin,9–11 complexes with dpa derivatives have been considered as potential antitumor agents.12–15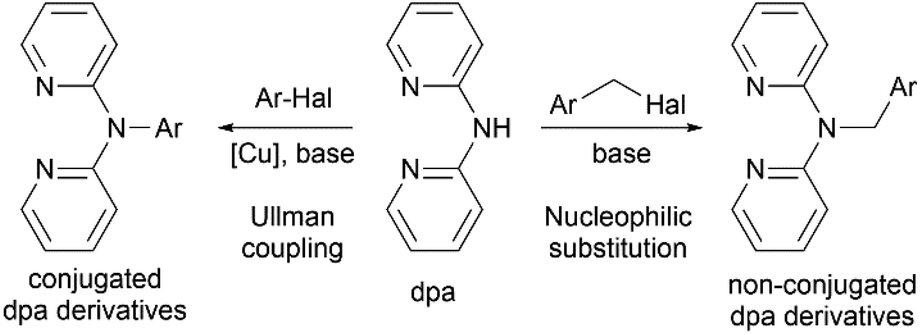 | ||
| Scheme 1 Conjugated and non-conjugated dpa derivatives and their syntheses. | ||
Variety of conjugated derivatives and their metal complexes, in which the amino nitrogen atom of dpa is directly connected with an aromatic system, have been synthesized and studied by the group of S. Wang6,16–18 and more recently by K.-J. Wei et al.19–21 Despite their attractive luminescent properties, real life application of such materials can be hindered by the complicated synthesis, which often involves transition metal catalyzed cross-coupling reactions (Scheme 1, left).
Non-conjugated derivatives, in which dpa moiety is connected with an aliphatic carbon atom, represent another family of dpa based materials. The number of studies on alkyl-substituted di(2-pyridyl)amines is rather limited,12,13 whereas methylene bridged aromatic derivatives receive more attention recently (Scheme 1, right). Inherent flexibility provided by the methylene linker not only expands the range of possibilities for various supramolecular motifs in solid state, as studied by L. Lindoy et al.,22–24 but also allows for non-trivial reactivity25 and luminescent properties26,27 of these compounds and their metal complexes. Compared to their conjugated counterparts, methylene bridged dpa derivatives are much easier to synthesize from dpa and corresponding halides via nucleophilic substitution reaction in presence of a base. Corresponding metal complexes can be then obtained by coordinating the dpa fragment on metals using conventional methods of coordination chemistry.
In this work we demonstrated, that the aforementioned conventional synthesis of methylene bridged dpa derivatives from dpa and corresponding halides does not always provide sufficient yields, and proposed an alternative synthetic approach, which involves the derivatization step after – rather than prior to – coordination of dpa on metal center. This approach makes the nucleophilic substitution reaction easier and more tolerant to various substituents in the halides, and allows more efficient synthesis of series of dpa based metal complexes with various organic backbones. Efficiency and versatility of the proposed synthetic approach were demonstrated on synthesis of series of substituted benzyldi(2-pyridyl)amines and their platinum(II) complexes. In addition, the products were structurally characterized and their photophysical properties were investigated.
Results and discussion
Synthesis
As mentioned above, the methylene bridged dpa derivatives are usually obtained by nucleophilic attack of the amino nitrogen atom of dpa on benzyl halides in presence of a base (Scheme 2a). However, several side processes can diminish the reaction yield; for example, the reported yields of benzyldi(2-pyridyl)amine vary from as high as 82%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 to as low as 16%.22 As we demonstrated in this work, the reaction becomes particularly problematic, when an electron withdrawing group is introduced in the phenyl ring (the same trend can also be found in another recent publication29). In such case, the methylene group can be easier deprotonated by the base due to stabilization of the resulting anion, leading to undesired side reactions. Additionally, benzylation of the pyridine nitrogen atoms instead of the amino nitrogen atom may take place (Scheme 2b).
28 to as low as 16%.22 As we demonstrated in this work, the reaction becomes particularly problematic, when an electron withdrawing group is introduced in the phenyl ring (the same trend can also be found in another recent publication29). In such case, the methylene group can be easier deprotonated by the base due to stabilization of the resulting anion, leading to undesired side reactions. Additionally, benzylation of the pyridine nitrogen atoms instead of the amino nitrogen atom may take place (Scheme 2b).
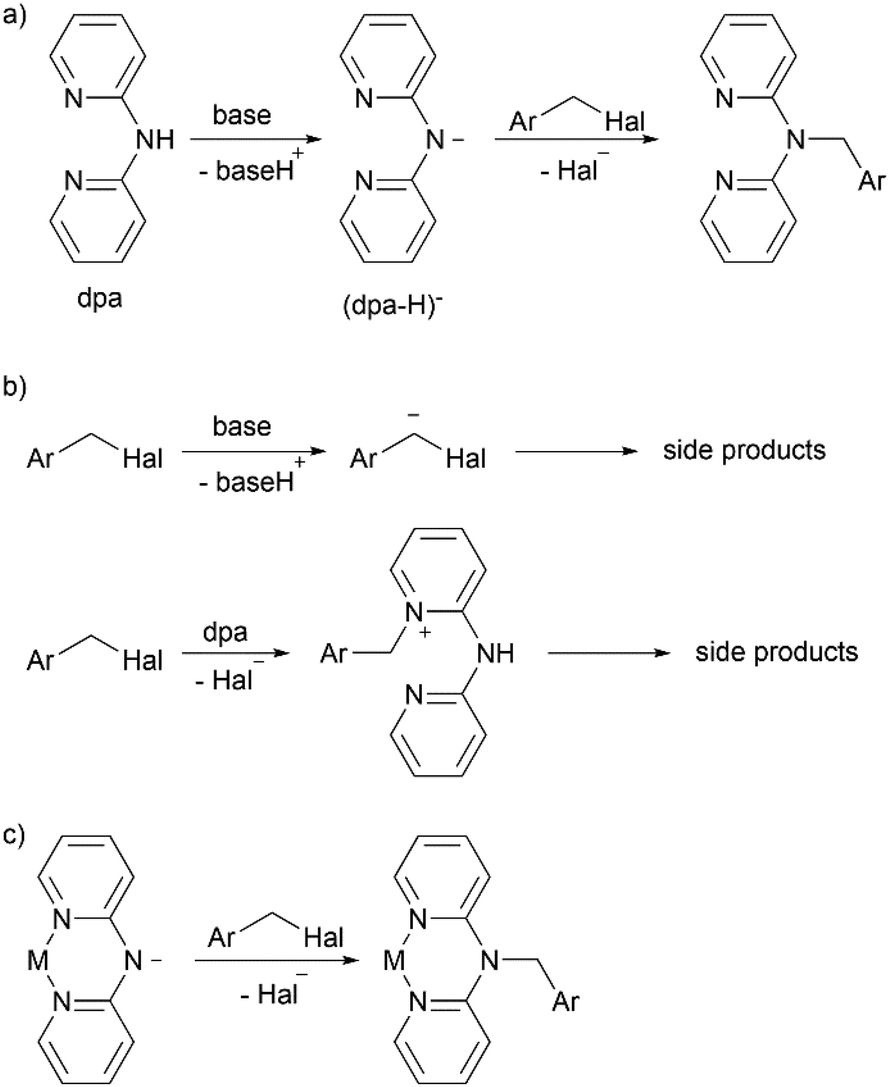 | ||
| Scheme 2 Reaction between dpa and benzyl halides: (a) target reaction, (b) undesired side reactions, (c) proposed reaction with (dpa-H)− complexes. | ||
Inspired by reports of various transition metal complexes containing deprotonated di(2-pyridyl)amide (dpa-H)− ligands,30–33 we suggested that use of such complexes in the benzylation reaction (Scheme 2c) could suppress the aforementioned undesired side processes. Thus, use of a base would be unnecessary in the reaction since the amino nitrogen atom of dpa is already deprotonated. Therefore, deprotonation of the methylene group of the benzyl halide would be avoided. The pyridine nitrogen atoms would be protected from benzylation by metal center. Moreover, such approach would allow more efficient synthesis of complexes of a particular metal with various non-conjugated dpa-derived ligands, which is a common research practice (for examples, see ref. 22, 23, 25, 27 and 29). Using the proposed method, such series of complexes could be easily obtained in one step from the (dpa-H)− containing complex and corresponding halides. The approach of derivatization of coordinated ligands has been applied, for example, to synthesis of various bipyridine and terpyridine complexes of ruthenium(II) and named as “Organic chemistry of coordination compounds”34 or “Chemistry on the complex”.35
We decided to test the proposed approach on synthesis of substituted benzyldi(2-pyridyl)amines 1–3 with substituents R = H, I, and NO2 in ortho-position of the phenyl ring as representatives of methylene bridged dpa derivatives with various electronic properties of the substituents. Metal center was chosen to be platinum(II), which is often used in development of phosphorescent materials due to its strong spin–orbit coupling;36 complexes of platinum(II) with dpa-based ligands are also considered as potential anticancer agents.12–15 In addition, synthesis of [Pt(dpa-H)2]33 and methylation of a structurally related bis(imidoylamidinate)platinum(II) complex [Pt{NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Ph)–NC(Ph)NPh}2],37 which can be seen as prototype for the dpa benzylation reaction, have been published previously (Scheme 3).
C(Ph)–NC(Ph)NPh}2],37 which can be seen as prototype for the dpa benzylation reaction, have been published previously (Scheme 3).
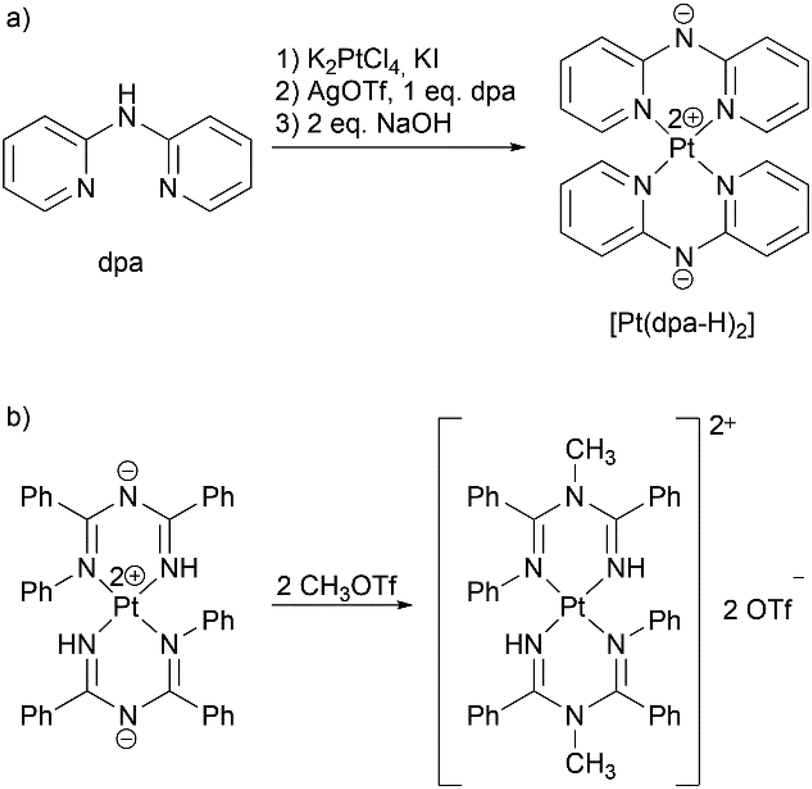 | ||
| Scheme 3 Known reactions of platinum(II) complexes, which inspired this work: (a) synthesis of [Pt(dpa-H)2],23 (b) methylation of [Pt{NHC(Ph)–NC(Ph)NPh}2].26 | ||
The target benzyldi(2-pyridyl)amines 1–3 and platinum(II) complexes 4–10 were synthesized using both conventional (benzylation prior to coordination) and alternative (benzylation after coordination) approaches (Scheme 4). Below particular details of syntheses are discussed in detail and comparison between the two approaches is presented.
 | ||
| Scheme 4 Synthesis of benzyldi(2-pyridyl)amines 1–3 and their platinum(II) complexes 4–10: the conventional (top) and alternative (bottom) approaches. | ||
The obtained compounds were then coordinated on platinum(II) by adapting known procedures33 in two steps. First, diiodide complexes 4–6 were obtained by reacting K2PtCl4 with one equivalent of 1–3 and excess of KI in water. The homoleptic complexes [7–9]·(OTf)2 were then obtained by reacting the diiodide complexes with another equivalent of the corresponding ligand 1–3 in acetonitrile in presence of silver(I) triflate (Scheme 4 top).
Yields of the two syntheses are compared in Table 1. Clearly, the alternative synthetic route provides higher overall yields of the homoleptic complexes 7–10 due to lower number and higher yields of the individual steps. In addition, introduction of the electron withdrawing groups in the benzyl moiety does not drastically affect the yields, in contrast with the conventional synthesis. Lesser reaction steps are also important for a practical synthesis. The conventional method required 9 steps to synthesize complexes [7–9]·(OTf)2 (3 reaction steps per complex), and many of them required individual approach due to variable properties (such as solubility and crystallizability) of the derivatives. On the other hand, starting from [Pt(dpa-H)2], the alternative method took only 3 steps to synthesize these complexes as bromides using very similar reaction conditions and workup. Moreover, any other derivative could be obtained just in one simple step using this method, as demonstrated by synthesis of [10]·Br2.
| Complex | Overall yield (yields of individual stages), % | |
|---|---|---|
| Conventional synthesisa | Alternative synthesisb | |
| a Overall yields were obtained by multiplication of the individual yields of the corresponding stages in the following order: dpa → ligands (1–3), ligands (1–3) → diiodide complexes (4–6), and diiodide complexes (4–6) → homoleptic complexes ([7–9]·(OTf)2). b Overall yields were obtained by multiplication of the yields of the corresponding individual stages in the following order: dpa → [Pt(dpa-H)2], [Pt(dpa-H)2] → homoleptic complexes ([7–10]·Br2). | ||
| 7 | 3 (26 × 82 × 14) | 37 (38 × 97) |
| 8 | 13 (44 × 48 × 61) | 24 (38 × 64) |
| 9 | 0.3 (9 × 20 × 15) | 32 (38 × 85) |
| 10 | — | 30 (38 × 79) |
These results demonstrate, that the alternative approach can be very convenient in synthesis of platinum(II) complexes with various methylene bridged dpa ligands. Moreover, even though separate tests were not performed, we believe that this approach can also be utilized for synthesis of platinum(II) complexes with only one methylene bridged dpa ligand. This suggestion is supported by the obtained side product dibromo(2-cyanobenzyldi(2-pyridyl)amine)platinum(II) (S4) in synthesis of [10]·Br2 (see section S2 in the ESI† for details).
Characterization of the products
In this section, crystal structures and general 1H NMR spectral features of all the obtained benzyldi(2-pyridyl)amines 1–3 and platinum(II) complexes 4–10 are discussed.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. In crystal structure of 1 pyridyl rings are in cis–trans conformation, whereas substituted derivatives 2 and 3 possess cis–cis conformation (using notation from ref. 1), and orientation of the phenyl ring in 2 and 3 is such that the substituents are turned outward from the pyridyl rings (Fig. 1). Halogen bond Npy⋯I (d(Npy⋯I) = 3.29 Å, ∠(Npy⋯I–C) = 166°) in crystal structure of 2 accounts for formation of the halogen bonded dimers (Fig. 2).
space group. In crystal structure of 1 pyridyl rings are in cis–trans conformation, whereas substituted derivatives 2 and 3 possess cis–cis conformation (using notation from ref. 1), and orientation of the phenyl ring in 2 and 3 is such that the substituents are turned outward from the pyridyl rings (Fig. 1). Halogen bond Npy⋯I (d(Npy⋯I) = 3.29 Å, ∠(Npy⋯I–C) = 166°) in crystal structure of 2 accounts for formation of the halogen bonded dimers (Fig. 2).
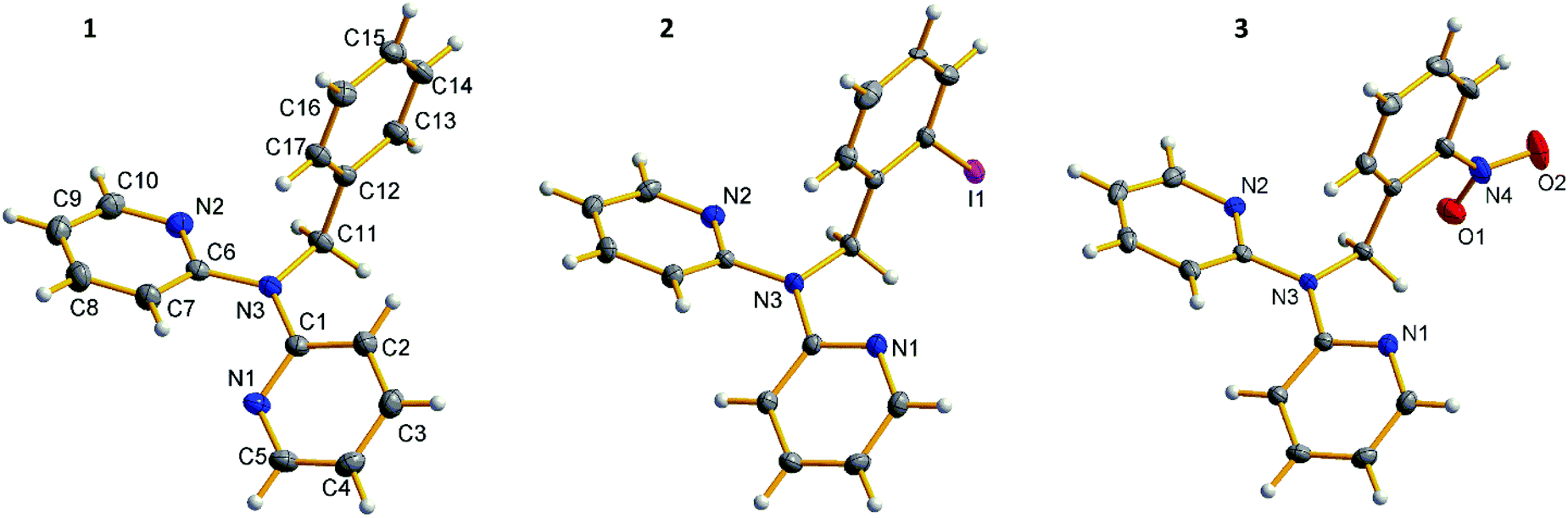 | ||
| Fig. 1 Crystal structures of 1–3. | ||
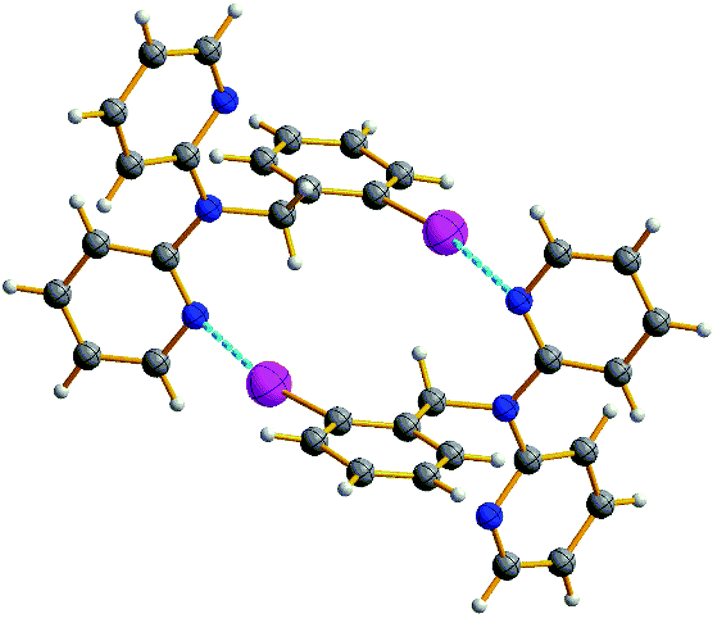 | ||
| Fig. 2 Halogen bonded dimers of 2. | ||
1H NMR spectra of 1–3 possess a number of multiplet signals in aromatic area (8.3–6.8 ppm) corresponding to the hydrogen atoms of pyridyl and phenyl rings, and one singlet signal at 5.8–5.4 ppm corresponding to the methylene hydrogen atoms.
space group, whereas 5 crystallizes in monoclinic space group P21/n. In all complexes, the metal center is coordinated by the dpa derived ligands via pyridine nitrogen atoms (d(Pt–N) = 2.03–2.06 Å, ∠(N–Pt–N) = 84.9–85.3°), forming six-membered chelate rings in boat conformation, with 50.6–53.5° dihedral angles between the pyridine planes. Such coordination is common for platinum(II) complexes with methylene bridged dpa derivatives.12,13 Two iodide ligands complete slightly distorted square planar configuration of the platinum metal centers with Pt–I bond lengths 2.58–2.60 Å and I–Pt–I angles 91.0–91.9°. In crystal structure of 5, the phenyl iodine atom I3 is oriented towards platinum center, with Pt⋯I distance of 3.94 Å. In 6, however, the nitro group is oriented away from platinum center (Fig. 3).
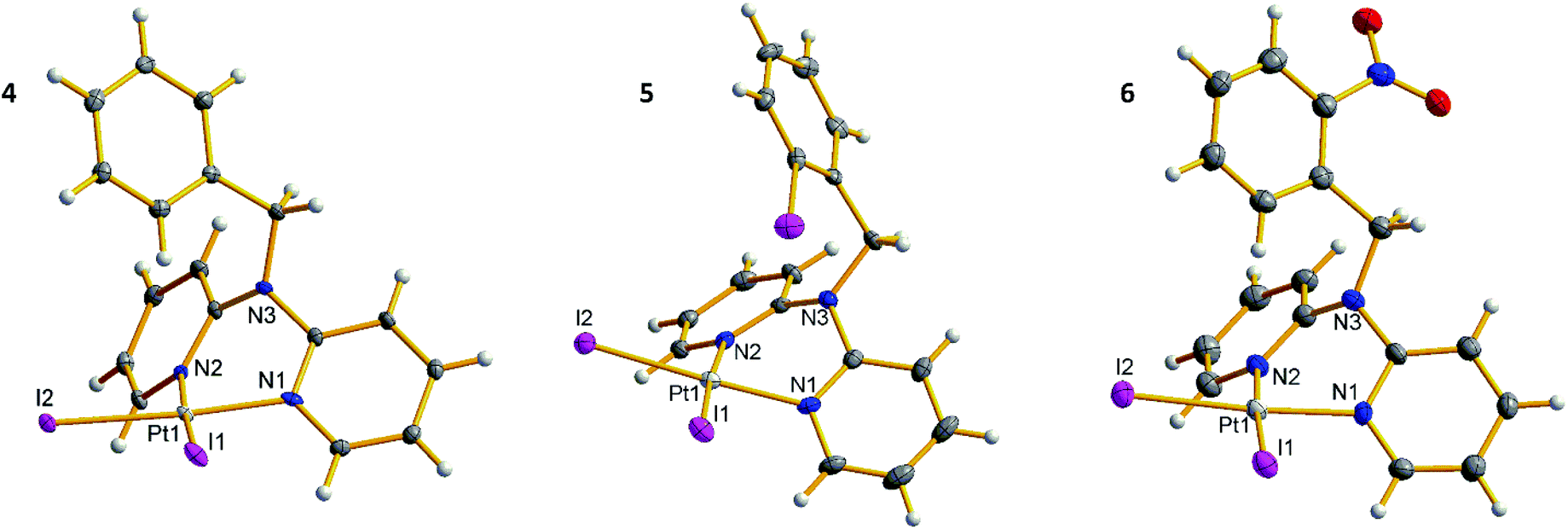 | ||
| Fig. 3 Crystal structures of 4–6. | ||
1H NMR spectra of 4–6 show downfielded (9.4–9.5 ppm) aromatic signal possessing characteristic broadened satellites due to splitting from 195Pt nucleus (3JH–Pt = 48–64 Hz), which can therefore be assigned to pyridine hydrogen atoms in 6th position. The other aromatic and methylene proton signals of the complexes resemble those of the corresponding ligands.
space group as dihydrate. Complexes 7–10 are centrosymmetric in solid state with platinum atoms at the inversion centers. Thus, asymmetric units in 7–10 contain platinum center with one ligand and one counter ion (and two and one water molecule in [9]·Br2 and [10]·(OTf)2 accordingly). Platinum centers possess square planar configuration with Pt–N bond lengths 2.01–2.02 Å and chelate N1–Pt–N2 angles 85.4–86.4°, and the six-membered chelate rings adopt boat conformation (dihedral angles between the pyridine planes 46.0–55.7°). Orientation of the phenyl ring in [8]·(OTf)2 is disordered between two positions: with iodine atom directed towards and away from platinum center. Nitro group in [9]·Br2 and cyano group in [10]·(OTf)2 are oriented towards platinum center with d(O2⋯Pt) = 3.27 Å in the former and d(N4⋯Pt) = 3.39 Å in the latter (Fig. 4).
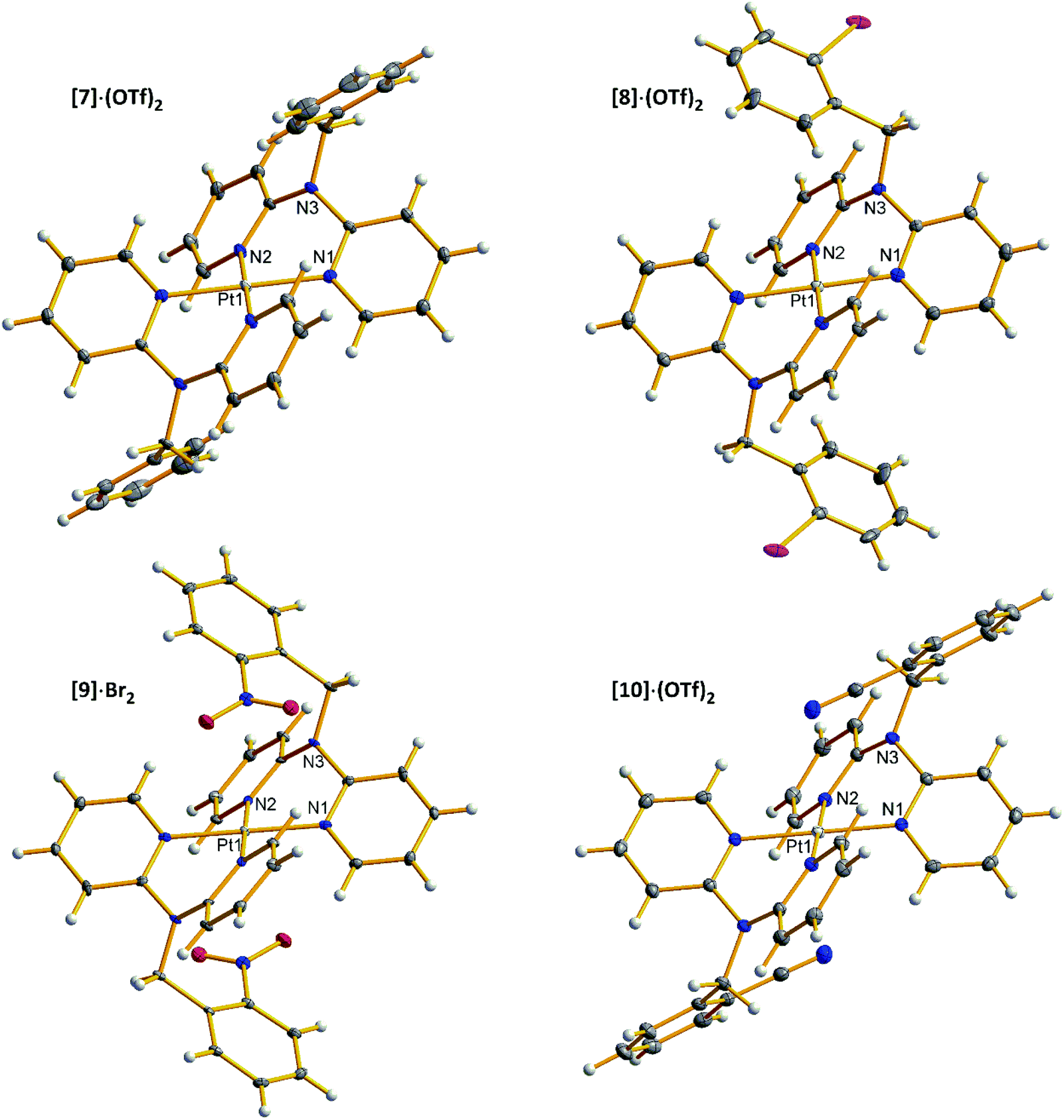 | ||
| Fig. 4 Crystal structures of 7–10. Anions and solvent molecules are omitted for clarity. | ||
1H NMR spectra of 7–10 show that hydrogen atoms of the two ligands are pairwise magnetically equivalent in all complexes. Signals of the pyridine hydrogen atoms in 6th position are shifted upfield compared with the precursor diiodide complexes, and the satellites due to splitting from 195Pt can barely be observed among the other aromatic signals.
Optical spectroscopy
Absorption spectra of 1–3 in methanol are presented in Fig. 5, and their features are summarized in Table S1 in the ESI.† The lowest energy absorption bands of the ligands can be assigned to the π–π* transitions within dpa moiety.27 Studies of photophysical properties of benzyldi(2-pyridyl)amines 1–3 are complicated due to their ability to undergo photocyclization in solution.39,40 While this process is slow enough to allow the measurement of absorption spectra, strong luminescence of the products and intermediates of the photocyclization interferes with the emission spectra of 1–3 in solution (for details, see section S3 in the ESI†). For this reason, studies of luminescent properties of the ligands were not included in this work. We have previously studied luminescent behavior of dpa hydrochloride, in which protonation of the pyridine nitrogen atoms suppresses the reaction of photocyclization in solution.41 Analogous studies on luminescence of ligands 1–3 and other methylene bridged dpa derivatives are underway in our research group.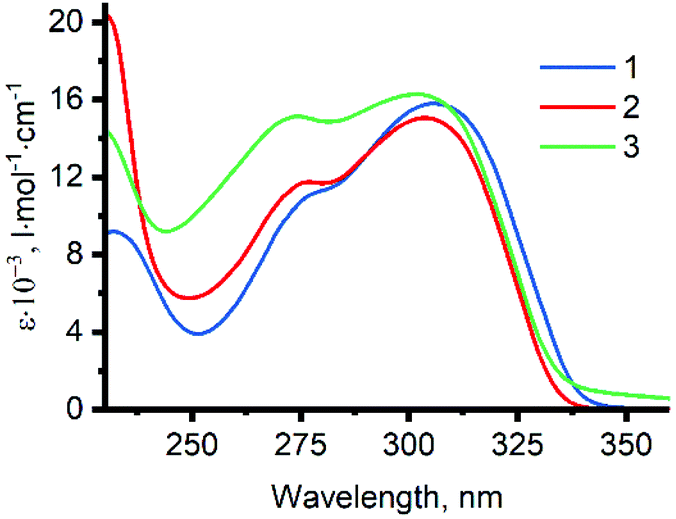 | ||
| Fig. 5 Absorption spectra of ligands 1–3 (C = 50 μM) in methanol. | ||
Absorption and photoluminescence emission spectra of [7–10]·Br2 are presented in Fig. 6 and summarized in Table 2. The lowest energy absorption bands of the complexes and the ligands possess similar wavelengths (λmax ≈ 310 nm and 305 nm accordingly) and extinction coefficients (ε ≈ (13–16) × 103 l mol−1 cm−1). The latter observation may indicate change in the nature of the lowest electronic transition upon coordination of the ligands to the platinum center. Complexes [7–10]·Br2 contain two ligands per molecule, and in the case of pure intraligand π–π* transitions approximately twice higher absorptivity would be expected for the complexes compared to the ligands.42 In addition, previously published computational studies on platinum(II) complexes with dpa derived ligands revealed a significant contribution of the metal center in the frontier molecular orbitals.15 Therefore, the low energy absorptions in [7–10]·Br2 are assigned to π–π* transitions within dpa moiety with an additional metal-to ligand charge transfer contribution.‡
 | ||
| Fig. 6 Absorption (methanol solution, RT, solid lines) and phosphorescence emission (methanol/ethanol matrix, 80 K, dashed lines) spectra of [7–10]·Br2 (C = 20 μM). Emission spectra were measured under excitation at λex = 320 nm, delay 20 ns, gate 10 μs (30 μs for [8]·Br2). | ||
| Complex | λ abs, nm | ε 10−3, l mol−1 cm−1 | λ em, nm | τ 1, μs | τ 2, μs |
|---|---|---|---|---|---|
| a Bands with multiple local maxima. b Lifetimes too short for reliable determination using the applied gate times. c Measured at an equilibrium state, since the sample was excited by the laser at 100 Hz pulse repetition rate. Therefore, the real value may be slightly lower. | |||||
| [7]·Br2 | 244 | 31.0 | 407 | 78 ± 4 | 200 ± 3 |
| 313 | 13.4 | ||||
| [8]·Br2 | 244 | 30.6 | 417 | ≤6b | 39 ± 1 |
| 312 | 13.3 | 470/500a | ≤90b | 3700 ± 60c | |
| [9]·Br2 | 243 | 35.8 | 425 | ≤5b | 45 ± 1 |
| 308 | 13.8 | ||||
| [10]·Br2 | 240 | 37.6 | 422/448/480a | 21 ± 1 | 160 ± 20 |
| 310 | 14.9 | ||||
Solutions of [7–10]·Br2 are not luminescent in methanol at room temperature. Quenching of luminescence of platinum(II) complexes is usually attributed to thermal population of relatively low lying metal centred excited states, leading to distortion of coordination geometry and consequent non-radiative relaxation.43 In order to observe the emission, solutions of the complexes in methanol/ethanol mixture (1:4, v/v)44 were cooled down to 80 K to form semi-rigid transparent glasses.
Emission properties of complexes [7–10]·Br2 share some common features. Thus, all complexes display emission bands with λmax = 407–425 nm, and a good match was observed between the absorption (Fig. 6) and excitation (Fig. S6 in the ESI†) spectra. Analysis of the time-resolved spectra (see section S4 in the ESI†) reveals biexponential decays of these emission bands. In all cases, the initial fast decay is followed by the slower one, characterized by lifetimes τ1 and τ2 accordingly in Table 2. The obtained values allow assignment of the observed emission to phosphorescence.
However, significant differences are apparent from the emission spectra of [8]·Br2, possessing additional shoulder at λmax ≈ 475 nm, and [10]·Br2, possessing additional peaks at λmax = 448 and 480 nm. While in the latter case the emission spectrum only slightly changes with time at 80 K (Fig. S7 in the ESI†), the two emission bands of [8]·Br2 at λmax = 417 and 470/500 nm possess strikingly different lifetimes of 39 μs and 3.7 ms accordingly. Time dependence of emission spectrum of [8]·Br2 is illustrated on Fig. 7 and Fig. S8 in the ESI.†
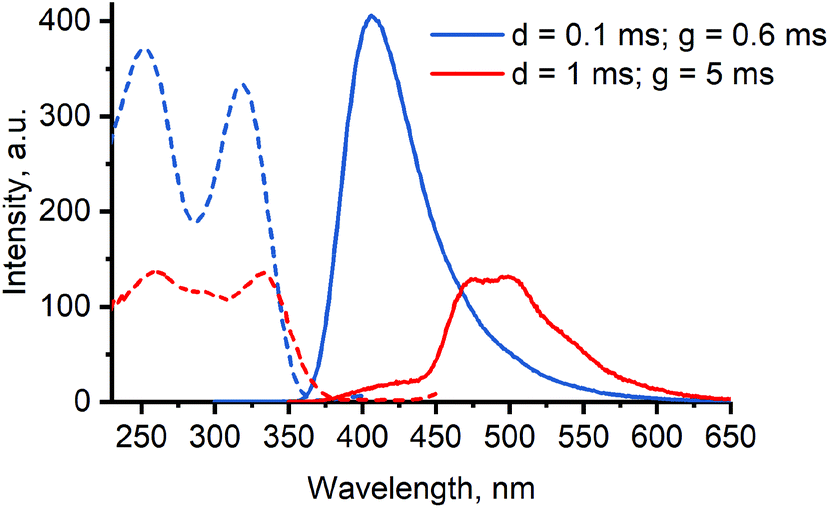 | ||
| Fig. 7 Phosphorescence excitation (dashed lines, monitored at emission maximum) and emission (solid lines, monitored at excitation maximum) spectra of [8]·Br2 in methanol/ethanol glass under two sets of timing parameters (77 K, C = 20 μM, d = delay, g = gate). | ||
At higher temperatures small red shift of all of the emission peaks is accompanied by decrease of lifetimes and emission intensity until complete disappearance at 140–150 K. Temperature dependencies of the observed rate constants reveal plateaus in most of cases at temperatures close to 80 K, indicating that the temperature-dependent non-radiative processes are suppressed at this point.45
In addition, [7,9]·Br2 display an additional longer wavelength emission band (λmax ≈ 500 nm) at temperatures of 120 K and higher, which strengthens upon continuous irradiation of the samples (Fig. S23 in the ESI†). Similar spectral changes may also take place for [8,10]·Br2, being less apparent due to the broad emission bands of these complexes. The appearance of the new emission bands can be tentatively attributed to the formation of excimers in viscous methanol/ethanol solution at higher temperatures, which is a common feature of platinum(II) complexes.43
Summarizing, the obtained complexes [7–10]·Br2 demonstrate generally similar absorption and phosphorescence spectra with excited state lifetimes of microsecond order, typical for platinum(II) complexes.43 On the other hand, the exceptionally long lifetime of the second emission band of [8]·Br2 – a unique feature of among the obtained complexes – is quite intriguing. This emission does not seem to originate either from excimer formation or from aggregation, since it is present in frozen matrix at 77 K, and does not depend on concentration (Fig. S11–18 in the ESI†). Thus, the additional green emission of [8]·Br2 can be supposedly associated with a specific conformation of the complex in the excited state, stabilized by an intramolecular interaction involving iodine atoms. Further studies of conformational flexibility and its impact on photophysical properties of 8 are currently being conducted in our research group.
Conclusions
Reaction between substituted benzyl bromides and [Pt(dpa-H)2] is demonstrated to be an efficient synthetic route towards benzyldi(2-pyridyl)amine complexes of platinum(II), which can serve as an example of the proposed non-conventional approach towards synthesis of metal complexes with methylene bridged dpa derivatives. This approach involves derivatization as the last step, when the dpa moiety is already coordinated on metal center, therefore allowing efficient one-step synthesis of multiple complexes and at the same time preventing undesired side reactions. The compounds obtained in this work possess conformational flexibility due to the methylene bridge, which results in various orientations of the substituents in solid state. The complexes are not emissive in solution at room temperature, but display blue and green phosphorescence in frozen methanol/ethanol matrix at 77 K with the lifetimes of μs order. The iodine substituted complex 8 possesses additional green emission with the lifetime of about 3.7 ms. Further studies on complex 8 and origin of its dual emission are underway.Experimental
All reagents and solvents for syntheses were purchased from Sigma-Aldrich, except for K2PtCl4 (Strem Chemicals and Alfa Aesar), and used as received without further purification. 1H NMR spectra were recorded using Bruker Avance III HD 300 MHz and Bruker Avance 400 MHz spectrometers. Chemical shifts are expressed in ppm using residual solvent peak as internal standard. All the obtained 1H NMR spectra are presented in section S5 in the ESI.† Elemental analyses (C, H, N) were preformed using Elementar Vario EL III elemental analyzer. Electrospray ionization mass spectra (ESI MS) were obtained with a Bruker micrOTOF or Agilent 6560 LC-IMMS TOF spectrometers equipped with an ESI source using methanol or acetonitrile as solvents.Crystallography
The crystals of 1–10 and S2–4 were immersed in cryo-oil, mounted in a MiTeGen loop and measured at 120 K on a Rigaku Oxford Diffraction Supernova diffractometer or at 170 K on a Bruker Axs KappaApexII diffractometer using Mo Kα (λ = 0.71073) radiation. The CrysAlisPro46 or Denzo-Scalepack47 program packages were used for cell refinements and data reductions. Multi-scan/Gaussian/Analytical absorption correction (CrysAlisPro,46 SADABS48) was applied to the intensities before structure solution. The structures were solved by intrinsic phasing method using the SHELXT49 software. Structural refinements were carried out using Olex250 graphical user interface. Intrinsic disorder was modelled over two positions for one of the nitro groups in 3 and for 2-iodobenzyl moiety in 5 and [8]·(OTf)2. C–H and O–H hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–1.00 Å, O–H = 0.85 Å, and Uiso = 1.2–1.5Ueq (parent atom), except for [10]·(OTf)2, in which the H2O hydrogen atoms were located from the difference Fourier and refined isotropically. The crystallographic details are summarized in section S6 in the ESI.†
Optical spectroscopy
HPLC grade methanol (J.T. Baker) and Uvasolv grade ethanol (Sigma-Aldrich) were used as solvents for optical measurements. Absorption spectra of 1–3 and [7–10]·Br2 were measured at ambient temperature in MeOH solution on Varian Cary 100 UV-visible spectrophotometer using quartz cuvettes with 1 cm path length in range of 220–700 nm. Steady state phosphorescence excitation and emission spectra of [7–10]·Br2 at 77 K in MeOH/EtOH (1:4, v/v) solution (presented in Fig. 7 and S6†) were measured on Varian Cary Eclipse Fluorescence spectrophotometer equipped with Oxford Optistat cryostat using cryogenic 1 cm quartz luminescence cuvette (FireflySci). Delay and gate times were adjusted in ranges of 0.1–1 ms and 0.6–5 ms accordingly using functions implemented in the spectrophotometer.
Phosphorescence emission decays were measured using the stroboscopic time resolution technique.51 Samples were excited at λex = 320 nm with 5 ns pulses (100 Hz repetition rate) using the diode pumped Q-switched Nd:YAG laser Ekspla NL230-100 with integrated optical parametric oscillator. The phosphorescence emission was detected at 90° angle using Oriel InstaSpec V ICCD detector with Acton SpectraPro sp-150i Spectrograph equipped with 300 grooves per mm grating blazed at 500 nm. Stanford Research Systems digital delay generator DG535 was used for controlling delay and gate timing parameters. Temperature of the samples was controlled within 80–150 K range using Oxford Optistat cryostat with nitrogen as static heat exchange gas. Sample solutions were placed in cryogenic 1 cm quartz luminescence cuvette (FireflySci) and deaerated by bubbling nitrogen gas for 10 minutes prior to measurements. A mechanical shutter was used to block the excitation beam between measurements to avoid photoinduced changes in the samples. The obtained spectra were processed using OriginPro software. The lifetime values were determined from the slopes of the fitted linear ln(I) vs. delay functions (see section S4 in the ESI†).
Synthetic procedures
:1, v/v) mixture as eluent. Product containing fractions were combined and evaporated to give yellow powder, which was recrystallized from acetone to obtain colorless crystals of the product (68 mg, 26%). Elemental analysis (%): C, 77.6; H, 5.4; N, 16.1. Calc. for C17H15N3: C, 78.1; H, 5.8; N, 16.1. 1H NMR (acetone-d6, 400 MHz): δ 8.29–8.25 (m, 2H), 7.63–7.57 (m, 2H), 7.38–7.34 (m, 2H), 7.27–7.20 (m, 4H), 7.16–7.12 (m, 1H), 6.92–6.87 (m, 2H), 5.53 (s, 2H, CH2). ESI MS (m/z): 262.14 (calcd for [M + H]+ 262.13), 545.26 (calcd for [2M + Na]+ 545.24). Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in acetone.
:1, v/v) solution to obtain the product as colorless crystals (345 mg, 44%). Elemental analysis (%): C, 52.6; H, 3.4; N, 10.9. Calcd for C17H14N3I: C, 52.7; H, 3.6; N, 10.9. 1H NMR (CDCl3, 300 MHz): δ 8.31–8.29 (m, 2H), 7.83 (dd, 1H, J = 1.0, 7.9 Hz), 7.58–7.50 (m, 2H), 7.25–7.15 (m, 4H), 6.93–6.83 (m, 3H), 5.41 (s, 2H, CH2). ESI MS (m/z): 387.99 (calcd for [M + H]+ 388.03). Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in chloroform.
:1, v/v) mixture as eluent. Product containing fractions were combined and subjected to second column chromatography purification using ethyl acetate/hexane (1:5 → 3:5, v/v) mixture as eluent. Product containing fractions were combined and slowly evaporated at room temperature to produce yellow crystals, which were recrystallized from ethyl acetate/pentane/diethyl ether (1:2:1, v/v) solution to get the product as yellowish crystals (137 mg, 9%). Elemental analysis (%): C, 66.3; H, 4.3; N, 18.05. Calcd for C17H14N4O2: C, 66.7; H, 4.6; N, 18.3. 1H NMR (CDCl3, 300 MHz): δ 8.29–8.27 (m, 2H), 8.04 (dd, 1H, J = 1.3, 8.0 Hz), 7.60–7.50 (m, 3H), 7.48–7.41 (m, 1H), 7.36–7.29 (m, 1H), 7.24–7.19 (m, 2H), 6.89–6.84 (m, 2H), 5.83 (s, 2H, CH2). ESI MS (m/z): 307.11 (calcd for [M + H]+ 307.12). Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in ethyl acetate/pentane/diethyl ether (1:2:1, v/v).
:1, v/v) was stirred for 20 min. Solid 2 (120 mg, 0.31 mmol) was added to the obtained dark-red solution and the resulting suspension was refluxed for 16 h at 85 °C. The solvent was partly evaporated on rotary evaporator and yellow-orange powder was filtered and washed with chloroform (5 × 3 ml) and THF (5 × 3 ml) to give the product as yellow powder (120.4 mg, 48%). Elemental analysis (%): C, 24.3; H, 1.7; N, 4.9. Calcd for C17H14N3I3Pt: C, 24.4; H, 1.7; N, 5.0. 1H NMR (THF-d8, 300 MHz): δ 9.49–9.29 (m, 2H, broad sidebands due to 3JPt,H = 46.3 Hz), 8.40 (d, 1H, J = 7.9 Hz), 7.99–7.92 (m, 2H), 7.84 (dd, 1H, J = 1.4, 7.9 Hz), 7.70–7.30 (m, 3H), 7.16–7.08 (m, 2H), 6.99–6.91 (m, 1H), 5.43 (s, 2H, CH2). ESI MS (m/z): 749.89 (calcd for [M − I + CH3CN]+ 749.91). Crystals suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in THF.
General procedure for conventional synthesis of bis(benzyldi(2-pyridyl)amine)platinum(II) triflate complexes [7–9]·(OTf)2
Suspension of precursor complex 4–6 (0.1 mmol) and corresponding ligand 1–3 (0.1 mmol) in acetonitrile (10 ml) was stirred at 60 °C for 10 min. Solution of silver triflate (55 mg, 0.21 mmol) in acetonitrile (5 ml) was added dropwise at this temperature within 5–7 min. Resulted mixture was stirred in darkness at 75 °C for 16 h. Yellow precipitate of silver iodide was filtered off and the filtrate was partially evaporated on rotary evaporator to volume of 2 ml and allowed to evaporate slowly at room temperature overnight. Resulted crystals were washed with THF (2 × 2 ml), recrystallized from methanol/acetonitrile mixture (1:1, v/v), and washed again with THF (3 × 2 ml) to give product as colorless crystals. Crystals of [7,8]·(OTf)2 suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in methanol. No high quality crystals could be obtained for [9]·(OTf)2, and crystals of [9]·Br2 were measured instead.
General procedure for alternative synthesis of bis(benzyldi(2-pyridyl)amine)platinum(II) bromide complexes [7–10]·Br2
[Pt(dpa-H)2] (53 mg, 0.1 mmol, prepared according to previously published procedure33 with 38% yield) and corresponding benzyl bromide (0.2 mmol) were suspended in acetonitrile (20 ml) and stirred at 75 °C for 24 h and refluxed for 2 h. Solvent was evaporated on rotary evaporator, and yellow residue was washed with chloroform (2 × 2 ml) and diethyl ether (2 × 2 ml). Resulting colorless solids were recrystallized from methanol to afford product as colorless crystalline material ([7]·Br2, 86 mg, 97%; [8]·Br2, 72 mg, 64%; [9]·Br2, 82 mg, 85%). 1H NMR spectra of [7–9]·Br2 matched those presented above. Crystals of [9]·Br2 suitable for X-ray diffraction analysis were obtained by slow evaporation of solution in methanol. In synthesis of [10]·Br2, additional side product S4 was isolated and identified (see section S2 in the ESI†).Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. B. and M. H. kindly acknowledge the financial support from the Academy of Finland (Proj. no. 295581). The authors would also like to acknowledge Elina Hautakangas for elemental analyses, Dr Elina Kalenius for mass spectrometry measurements, Dr Matti Tuikka for helping with X-ray analyses, and Dr Pasi Myllyperkiö and Dr Heikki Häkkänen for assisting with optical spectroscopy measurements.Notes and references
- D. W. Brogden and J. F. Berry, Comments Inorg. Chem., 2016, 36, 17–37 CrossRef CAS.
- C. J. Sumby, Coord. Chem. Rev., 2011, 255, 1937–1967 CrossRef CAS.
- B. Chen, G. Yu, X. Li, Y. Ding, C. Wang, Z. Liu and Y. Xie, J. Mater. Chem. C, 2013, 1, 7409–7417 RSC.
- R. Tan, Z.-B. Wang, Y. Li, D. J. Kozera, Z.-H. Lu and D. Song, Inorg. Chem., 2012, 51, 7039–7049 CrossRef CAS PubMed.
- W.-L. Jia, T. McCormick, Q.-D. Liu, H. Fukutani, M. Motala, R.-Y. Wang, Y. Tao and S. Wang, J. Mater. Chem., 2004, 14, 3344–3350 RSC.
- J. Pang, Y. Tao, S. Freiberg, X.-P. Yang, M. D'Iorio and S. Wang, J. Mater. Chem., 2002, 12, 206–212 RSC.
- J. Pang, E. J.-P. Marcotte, C. Seward, R. S. Brown and S. Wang, Angew. Chem., Int. Ed., 2001, 40, 4042–4045 CrossRef CAS PubMed.
- Y.-Q. Weng, F. Yue, Y.-R. Zhong and B.-H. Ye, Inorg. Chem., 2007, 46, 7749–7755 CrossRef CAS PubMed.
- A. K. Paul, H. Mansuri-Torshizi, T. S. Srivastava, S. J. Chavan and M. P. Chitnis, J. Inorg. Biochem., 1993, 50, 9–20 CrossRef CAS PubMed.
- A. K. Paul, T. S. Srivastava, S. J. Chavan, M. P. Chitnis, S. Desai and K. K. Rao, J. Inorg. Biochem., 1996, 61, 179–196 CrossRef CAS PubMed.
- C. Icsel, V. T. Yilmaz, Y. Kaya, H. Samli, W. T. A. Harrison and O. Buyukgungor, Dalton Trans., 2015, 44, 6880–6895 RSC.
- M. J. Rauterkus, S. Fakih, C. Mock, I. Puscasu and B. Krebs, Inorg. Chim. Acta, 2003, 350, 355–365 CrossRef CAS.
- S. Fakih, W. C. Tung, D. Eierhoff, C. Mock and B. Krebs, Z. Anorg. Allg. Chem., 2005, 631, 1397–1402 CrossRef CAS.
- I. Puscasu, C. Mock, M. Rauterkus, A. Röndigs, G. Tallen, S. Gangopadhyay, J. E. Wolff and B. Krebs, Z. Anorg. Allg. Chem., 2001, 627, 1292–1298 CrossRef CAS.
- P. W. Asman, Inorg. Chim. Acta, 2018, 469, 341–352 CrossRef CAS.
- C. Seward and S. Wang, Comments Inorg. Chem., 2005, 26, 103–125 CrossRef CAS.
- Y. Kang and S. Wang, Tetrahedron Lett., 2002, 43, 3711–3713 CrossRef CAS.
- W.-L. Jia, D. Song and S. Wang, J. Org. Chem., 2003, 68, 701–705 CrossRef CAS PubMed.
- K.-J. Wei, Y.-S. Xie, J. Ni, M. Zhang and Q.-L. Liu, Inorg. Chem. Commun., 2006, 9, 926–930 CrossRef CAS.
- K.-J. Wei, Y.-S. Xie, J. Ni, M. Zhang and Q.-L. Liu, Cryst. Growth Des., 2006, 6, 1341–1350 CrossRef CAS.
- J. Ni, K.-J. Wei, Y. Min, Y. Chen, S. Zhan, D. Li and Y. Liu, Dalton Trans., 2012, 41, 5280–5293 RSC.
- B. Antonioli, D. J. Bray, J. K. Clegg, K. Gloe, K. Gloe, O. Kataeva, L. F. Lindoy, J. C. McMurtrie, P. J. Steel, C. J. Sumby and M. Wenzel, Dalton Trans., 2006, 4783–4794 RSC.
- B. Antonioli, D. J. Bray, J. K. Clegg, K. Gloe, K. Gloe, A. Jäger, K. A. Jolliffe, O. Kataeva, L. F. Lindoy, P. J. Steel, C. J. Sumby and M. Wenzel, Polyhedron, 2008, 27, 2889–2898 CrossRef CAS.
- D. J. Bray, J. K. Clegg, K. A. Jolliffe and L. F. Lindoy, CrystEngComm, 2014, 16, 6476–6482 RSC.
- P. W. Asman and D. Jaganyi, Int. J. Chem. Kinet., 2017, 49, 545–561 CrossRef.
- J.-S. Yang, Y.-D. Lin, Y.-H. Chang and S.-S. Wang, J. Org. Chem., 2005, 70, 6066–6073 CrossRef CAS PubMed.
- M. A. Rohman, D. Sutradhar, S. Sangilipandi, K. Mohan Rao, A. K. Chandra and S. Mitra, J. Photochem. Photobiol., A, 2017, 341, 115–126 CrossRef CAS.
- A. J. Swarts and S. F. Mapolie, Dalton Trans., 2014, 43, 9892–9900 RSC.
- M. Vaquero, A. Ruiz-Riaguas, M. Martínez-Alonso, F. A. Jalón, B. R. Manzano, A. M. Rodríguez, G. García-Herbosa, A. Carbayo, B. García and G. Espino, Chem. – Eur. J., 2018, 24, 10662–10671 CrossRef CAS PubMed.
- J. F. Geldard and F. Lions, J. Am. Chem. Soc., 1962, 84, 2262–2263 CrossRef CAS.
- T. J. Hurley and M. A. Robinson, Inorg. Chem., 1968, 7, 33–38 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, G. T. Jordan and C. A. Murillo, Polyhedron, 1998, 17, 589–597 CrossRef CAS.
- Q. Wang, P. V. Gushchin, N. A. Bokach, M. Haukka and V. Yu. Kukushkin, Russ. Chem. Bull., 2012, 61, 828–835 CrossRef CAS.
- Y. Tor, Synlett, 2002, 1043–1054 CrossRef CAS.
- T. Mede, M. Jäger and U. S. Schubert, Chem. Soc. Rev., 2018, 47, 7577–7627 RSC.
- B. Minaev, G. Baryshnikov and H. Agren, Phys. Chem. Chem. Phys., 2014, 16, 1719–1758 RSC.
- G. H. Sarova, N. A. Bokach, A. A. Fedorov, M. N. Berberan-Santos, V. Yu. Kukushkin, M. Haukka, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Dalton Trans., 2006, 3798–3805 RSC.
- S. Licciulli, I. Thapa, K. Albahily, I. Korobkov, S. Gambarotta, R. Duchateau, R. Chevalier and K. Schuhen, Angew. Chem., Int. Ed., 2010, 49, 9225–9228 CrossRef CAS PubMed.
- V. M. Clark, A. Cox and E. J. Herbert, J. Chem. Soc. C, 1968, 831–833 RSC.
- J. D. Cocker, G. I. Gregory and G. B. Webb, Br. Pat., GB 1262864, 1972 Search PubMed.
- E. Bulatov and M. Haukka, ChemistrySelect, 2018, 3, 11535–11540 CrossRef CAS.
- K.-Y. Ho, W.-Y. Yu, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1999, 1581–1586 RSC.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- D. R. Scott and J. B. Allison, J. Phys. Chem., 1962, 66, 561–562 CrossRef CAS.
- T. O. Harju, J. E. I. Korppi-Tommola, A. H. Huizer and C. A. G. O. Varma, J. Phys. Chem., 1996, 100, 3592–3600 CrossRef CAS.
- Agilent, CrysAlisPro, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- Z. Otwinowski and E. Minor, in Methods in Enzymology, ed. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, vol. 276, Macromolecular Crystallography, Part A, pp. 307–326 Search PubMed.
- G. M. Sheldrick, SADABS - Bruker AXS, scaling and absorption correction, Bruker AXS, Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- G. M. Sheldrick, Acta Cryst. A, 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- D. R. James, A. Siemiarczuk and W. R. Ware, Rev. Sci. Instrum., 1992, 63, 1710–1716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details and discussion on syntheses and side products, photocyclization of 1, phosphorescence excitation and variable temperature time resolved emission spectra of [7–10]·Br2, 1H NMR spectra of all compounds, and crystallographic data. CCDC 1868987–1868999. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt03912g |
| ‡ This assignment is also supported by the preliminary computational DFT studies on [8]·Br2 (to be presented in the follow-up publication). |
| This journal is © The Royal Society of Chemistry 2019 |