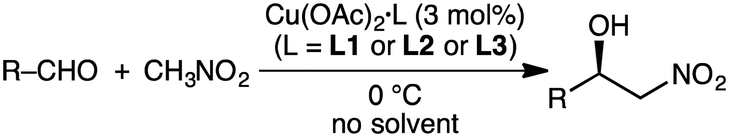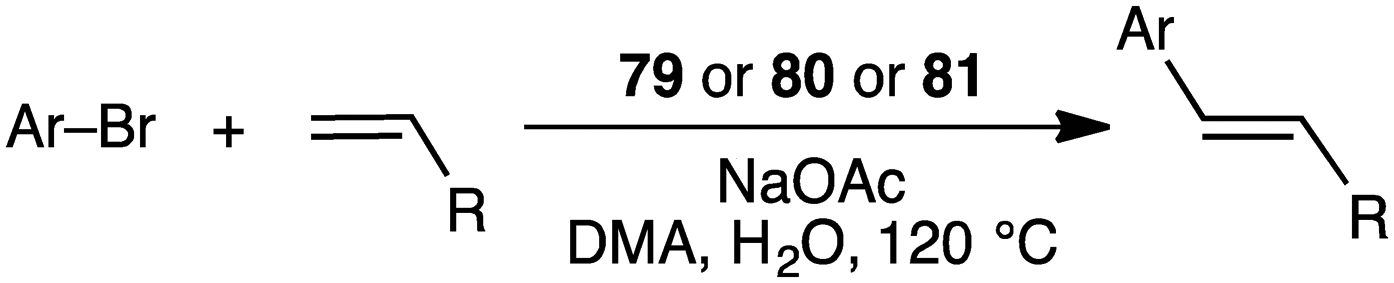Macrocyclic multinuclear metal complexes acting as catalysts for organic synthesis
Received
19th September 2019
, Accepted 21st November 2019
First published on 21st November 2019
Abstract
Metal clusters in nature and artificial systems are known to exhibit excellent catalytic activities. Among various coordination systems, macrocyclic multinuclear metal complexes have unique structural features and great potential in organic synthesis. The multiple metal centers in a macrocyclic framework act as specific sites for the binding and activation of substrates, showing high catalytic activity and selectivity as a result of the cooperative effect of the multiple metal centers and robustness originating from macrocyclic skeletons. Most of them are precisely designed and synthesized, while occasionally, nice complexes also emerge from a fortuitous combination of metal ions and ligands. Here we overview the recent achievements of (i) multinuclear metal complexes with a covalently-linked macrocyclic ligand (category A), (ii) multinuclear metal complexes forming a macrocyclic skeleton (category B), and (iii) self-assembled supramolecular coordination complexes (category C). Various organic reactions catalyzed by these coordination systems are summarized here.
 Bikash Dev Nath | Bikash Dev Nath was born in Barisal, Bangladesh in 1984. He received his bachelor's degree in 2010 and master's degree in 2011 from Jahangirnagar University. He worked as a synthesis engineer at Pilarquim (Shanghai) Co., Ltd. in China from 2011 to 2016. He completed his doctor's degree in 2019 at Okayama University under the supervision of Professor Tadashi Ema. His subject during doctoral study was “Novel Macrocyclic Multinuclear Ni(II) and Zn(II) Complexes for Catalytic CO2 Conversions”. |
 Kazuto Takaishi | Kazuto Takaishi was born in Ehime, Japan in 1980. He received his bachelor's degree in 2003 and master's degree in 2005 from Okayama University, and he obtained his doctor's degree in 2008 from Kyoto University under the supervision of Professor Takeo Kawabata. He then moved to RIKEN as a postdoctoral fellow. He was appointed as an Assistant Professor at Seikei University in 2012. He moved to Okayama University to join the Tadashi Ema research group as a Senior Assistant Professor in 2015. His main research topic is the synthesis of chiral functional compounds. |
 Tadashi Ema | Tadashi Ema was born in Gifu, Japan in 1966. He received his bachelor's degree from Kyoto University in 1989, and he obtained his master's degree in 1991 and doctor's degree in 1994 from Kyoto University under the supervision of Professor Hisanobu Ogoshi. He was appointed as an Assistant Professor at Okayama University in 1994 and was promoted to an Associate Professor in 2002 and a Full Professor in 2012. His research interest is the design and synthesis of unique molecular catalysts. |
1. Introduction
1.1 Classification of macrocyclic multinuclear metal complexes
Multinuclear metal complexes have attracted much attention of chemists because of their fascinating structures and functions, and some of them act as catalysts for organic synthesis and enzyme model reactions.1–5 Metal clusters can be created in the cavity of a covalently-bonded macrocyclic polydentate ligand,6 or they can be synthesized by the self-assembly of metal ions and non-macrocyclic polydentate ligands. Simultaneous coordinations of multiple metal ions can lead to the effective recognition and activation of substrates.7,8
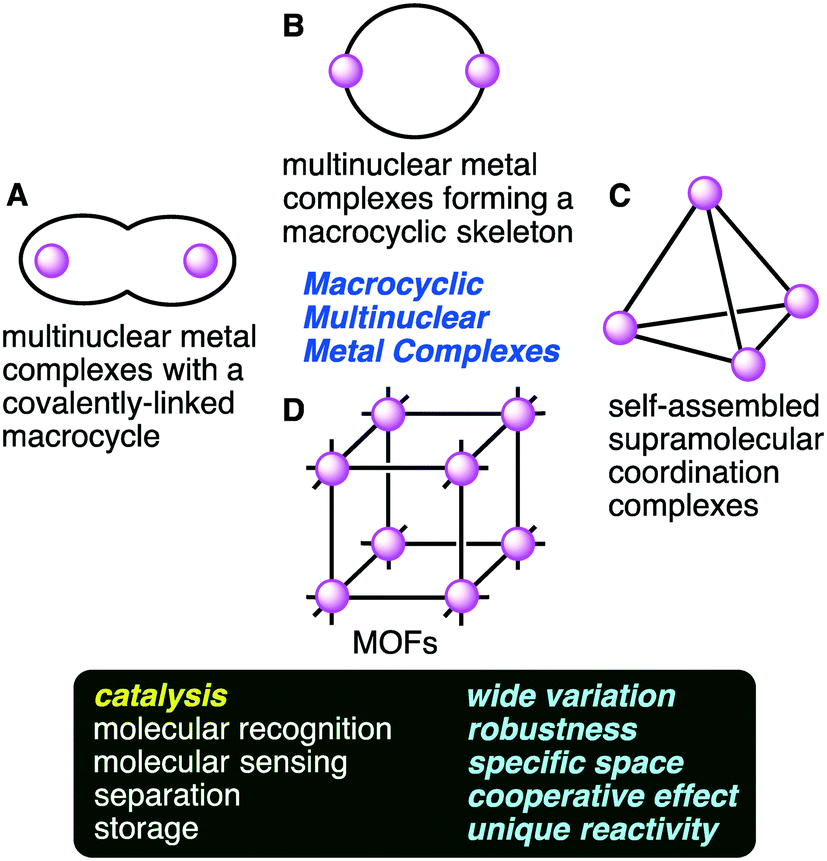
Multinuclear metal complexes can be classified into two types: macrocyclic and non-macrocyclic ones. The former possesses a macrocyclic framework useful for the spatial disposition of not only metal ions but also catalytic functional groups. The precise preorganization of metal ions and catalytic functional groups is a great advantage of the macrocyclic ones over the non-macrocyclic ones. Macrocyclic multinuclear metal complexes can be subdivided into four categories (Fig. 1): (A) multinuclear metal complexes with a covalently-linked macrocyclic ligand, (B) multinuclear metal complexes forming a macrocyclic skeleton, (C) self-assembled supramolecular coordination complexes, and (D) metal–organic frameworks (MOFs).
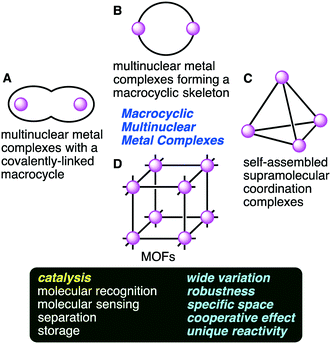 |
| | Fig. 1 Macrocyclic multinuclear metal complexes classified into four categories (A–D) and their functions and features. | |
Multinuclear metal complexes with a covalently-linked macrocyclic ligand (category A) are created by combining an elaborately designed macrocyclic ligand with metal ions. The coordination of multiple metal ions to different chelating sites in the macrocyclic cavity gives homo- or heteronuclear metal complexes. Multinuclear metal complexes forming a macrocyclic skeleton (category B) are created by the self-assembly of metal ions and polydentate ligands. The macrocyclic skeletons are constructed through multiple metal–ligand coordination bonds. Specific structures characterized by multiple metal centers and macrocyclic frameworks lead to excellent catalytic activities. Heterometallic complexes that are similar to those in nature are also created.9–12 Self-assembled supramolecular coordination complexes (category C) are molecular cages with a vacant inner space, and some of them are excellent supramolecular homogeneous catalysts.13–19 On the other hand, metal–organic frameworks (MOFs) (category D) provide another effective approach to create a sophisticated coordination space. MOFs are three-dimensional lattices composed of metal ions and bridging organic ligands, having high porosity, a large surface area, dense active sites, high adsorption capacity, and good thermal stability.20–39 MOFs can work as excellent heterogeneous catalysts. There may be room for improvement of the above classification; for example, molecular knots with multiple metal ions,40–44 which might belong to category A, are not considered here because the use of molecular knots as catalysts has not been reported.
1.2 Advantages of macrocyclic multinuclear metal complexes
Macrocyclic multinuclear metal complexes have both promising advantages and challenging aspects as follows: (i) synthetic challenges for the construction of macrocyclic structures, (ii) specific sites for substrate binding and catalysis, (iii) synergetic effects based on the metal–metal or metal–ligand cooperation, (iv) variations of (mixed) metal elements, (v) good reactivity, selectivity, and robustness resulting from macrocyclic structures, and (vi) unlimited combinations of metals and ligands.
Macrocyclic multinuclear metal complexes are sometimes difficult to prepare because of the lack of versatile synthetic methods. The macrocyclization step sometimes requires rigorous tuning of reaction conditions and suffers from low yields. The careful design of organic ligands is key to the successful construction of specific molecular structures with good synthetic accessibility.
The binding of a substrate molecule to a metal center is a crucial step in catalysis. Therefore, the catalytic activity can be significantly improved if the complex offers a specific coordination site to bind and activate the substrate molecule. The cooperative effect of multiple metal centers is important for high catalytic performance.45–51 Cooperative catalysis may originate from two or more metal ions in close proximity. The presence of different metals is beneficial for excellent catalytic activities. Macrocyclic ligands with multiple chelating sites should be well designed to systematically introduce different metal elements.
Another significant feature of macrocyclic multinuclear metal complexes is robustness, which may come from the sophisticated and tight assembly of metal ions and ligands with multiple coordinations. Although macrocyclic metal complexes can be created by designing macrocyclic ligands, the self-assembly of metal ions and ligands may be assisted by more complicated and subtle metal–ligand and ligand–ligand interactions. Combinations of metal ions and ligands sometimes give rise to unusual robustness.
Macrocyclic multinuclear metal complexes show a variety of physical and chemical properties based on structural diversity.52,53 In the area of host–guest chemistry, they can act as host molecules for the multipoint recognition of guest molecules.7,8 They play significant roles in catalysis,14–19,27–39,54–56 and they can mimic the active site of metalloenzymes.57 They also find applications in molecular sensing,58–60 separation,61–68 and ionic/proton conductivity.69–71 They can be used in biochemical applications72 and drug delivery.73,74 Taking advantage of high porosity and vacant inner space, they can store,75–78 separate, and purify a gas.63–67
1.3 Scope of this perspective
Despite the wide range of catalytic applications of macrocyclic multinuclear metal complexes in organic synthesis, only a limited number of reviews have been published so far, all of which are related to self-assembled supramolecular coordination complexes or MOFs (categories C and D in Fig. 1).14–19,27–39 In contrast, there are few reviews on categories A and/or B in Fig. 1. Here we provide, for the first time, an overview of homogeneous catalysis with macrocyclic multinuclear metal complexes in categories A–C in Fig. 1, illuminating the characteristics and advantages of each category. For example, it can be seen that the catalysts in categories B and C show quite different activities, selectivities, and mechanisms although the latter is structurally defined as a three-dimensional version of the former. In addition, it is also shown that all of them catalyze a wide range of useful organic reactions. In most cases, it is impossible to clearly compare catalytic performances between macrocyclic/multinuclear and non-macrocyclic/mononuclear systems because of the lack of data although structural features and proposed mechanisms strongly suggest the advantages and benefits of macrocyclic multinuclear metal complexes. Each achievement is concisely summarized one by one using reaction schemes with representative catalyst structures to help the readers easily understand the recent progress and great potential of this research field.
2. Multinuclear metal complexes with a covalently-linked macrocyclic ligand (category A)
2.1 CO2 fixation
Carbon dioxide (CO2) is a renewable raw material for the production of value-added chemicals, and a number of important catalytic reactions have been developed.79–86 A typical example can be seen in the copolymerization of epoxides and CO2 to form polycarbonates,87–95 and several multinuclear metal complexes were developed for this purpose.
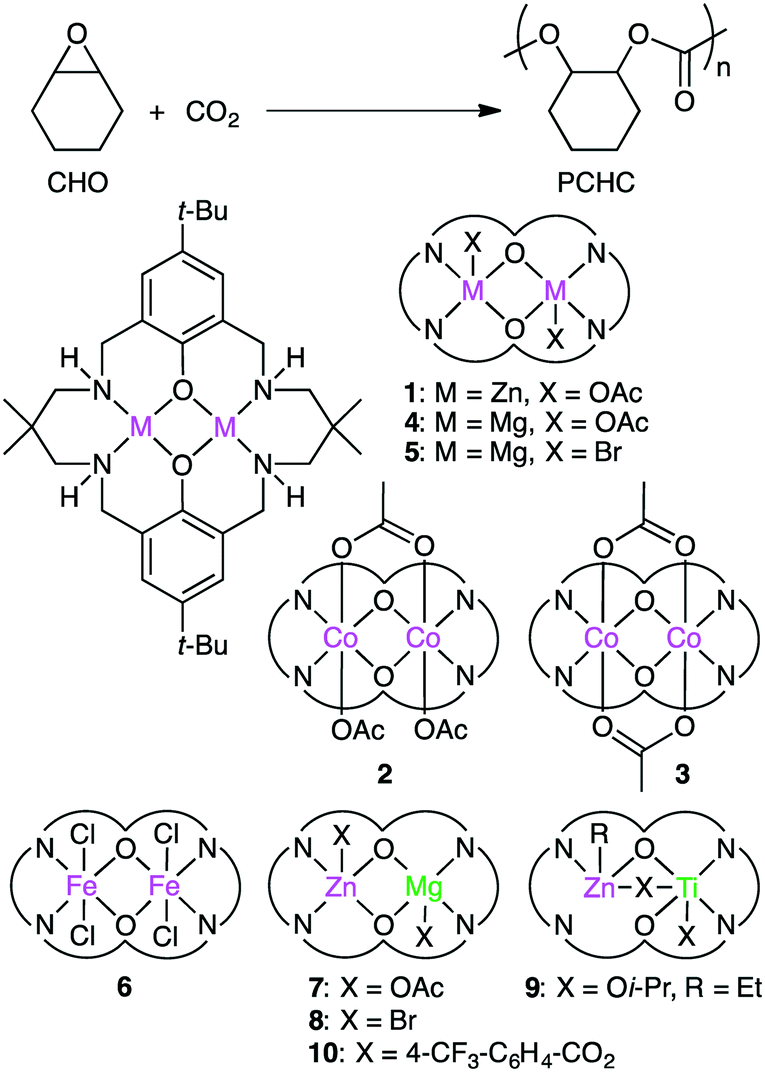
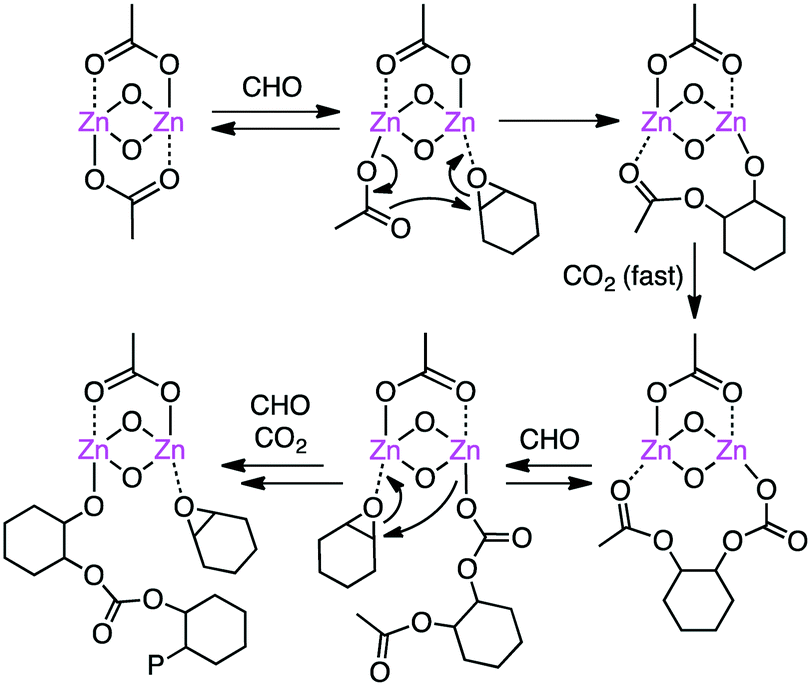
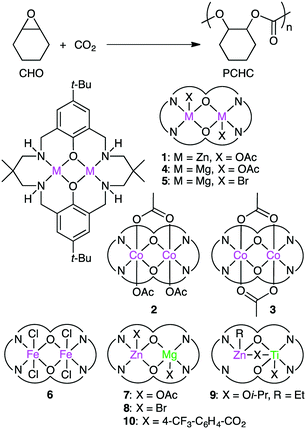
Williams and co-workers developed several macrocyclic multimetallic complexes showing high catalytic activity for the copolymerization of cyclohexene oxide (CHO) and CO2 (Scheme 1). A robust catalyst 1 catalyzed the copolymerization of CHO and CO2 (1 atm) with a catalyst loading of 0.1 mol% at 80–100 °C to form poly(cyclohexene carbonate), PCHC.96 The turnover number (TON) and turnover frequency (TOF) values reached 430–530 and 18–25 h−1, respectively. Increasing the CO2 pressure to 10 atm at 100 °C increased both the TON (838) and TOF (38 h−1). When the catalyst loading was reduced to 0.01 mol%, the TON and TOF values increased up to 3350 and 140 h−1, respectively. A mechanism was proposed on the basis of kinetic studies (Scheme 2).97 DFT calculations suggested that the rate-determining step is the epoxide-ring opening of CHO by the nucleophilic attack of the carbonate anion in the propagation step.98
 |
| | Scheme 1 Copolymerization of CO2 and CHO. | |
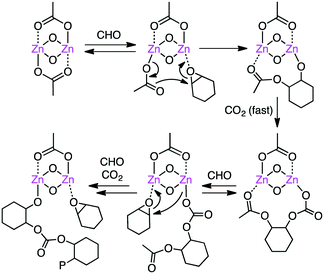 |
| | Scheme 2 Mechanism of copolymerization. | |
Dinuclear Co complexes 2 and 3 exhibited comparable catalytic activity at 1 atm CO2 pressure.99,100 Complex 2 with a mixed valence Co(II)/Co(III) core showed 20 times higher TOF (500 h−1) at 100 °C than complex 1. At a higher CO2 pressure (10 atm), both 2 and 3 showed much higher catalytic activities, and complex 2 displayed a TOF of 3700 h−1 at 100 °C. Mg complex 4 with a catalyst loading of 0.01 mol% showed a TOF of 730 h−1 at 100 °C at 12 atm CO2 pressure.101 The Mg–carbonate bond may be more nucleophilic because of the decreased Lewis acidity of Mg(II) as compared with Zn(II), which can facilitate the ring opening of the epoxide. Fe(III) complex 6 exhibited 8 times higher activity at 10 atm CO2 pressure than at 1 atm CO2 pressure.102
Williams' group also reported the first heterodinuclear Mg/Zn complex 7,103 and complex 8 was the first isolated heterometallic complex synthesized from a macrocyclic ligand.104 Heterodinuclear Ti/Zn complex 9 was also synthesized.105 Importantly, mixed metal catalyst 7 was remarkably more effective than either homodinuclear Zn complex 1 or Mg complex 4. The catalytic activity of heterodinuclear complex 8 was about 5 times higher than that of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of homodinuclear Zn and Mg complexes. 8 was more than twice as active as Mg complex 5, while a Zn analog of 5 showed no activity at all. Clearly, the enhanced catalytic activity of heterodinuclear Mg/Zn complexes 7 and 8 is due to the synergistic effect of the two different metal ions. In addition, 8 formed the polycarbonate linkages highly selectively (>99%), and 8 with a catalyst loading of 0.01 mol% showed a TOF of 624 h−1. More recently, when the axial ligand (Br) in 8 was replaced with p-trifluoromethylbenzoate to prepare 10, a maximal TOF of 8830 h−1 was obtained under optimized reaction conditions (20 bar CO2, 120 °C).106 A series of dimetallic complexes were characterized by X-ray crystal structural analysis.107
:1 mixture of homodinuclear Zn and Mg complexes. 8 was more than twice as active as Mg complex 5, while a Zn analog of 5 showed no activity at all. Clearly, the enhanced catalytic activity of heterodinuclear Mg/Zn complexes 7 and 8 is due to the synergistic effect of the two different metal ions. In addition, 8 formed the polycarbonate linkages highly selectively (>99%), and 8 with a catalyst loading of 0.01 mol% showed a TOF of 624 h−1. More recently, when the axial ligand (Br) in 8 was replaced with p-trifluoromethylbenzoate to prepare 10, a maximal TOF of 8830 h−1 was obtained under optimized reaction conditions (20 bar CO2, 120 °C).106 A series of dimetallic complexes were characterized by X-ray crystal structural analysis.107
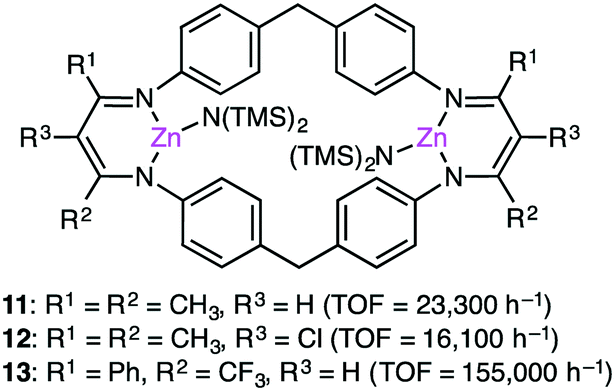
Based on DFT calculations on the copolymerization of CHO and CO2 with 1, showing the importance of the shuttling mechanism (Scheme 2),98,108 Rieger and co-workers synthesized 11 containing two β-diketiminate Zn complexes (Fig. 2).109,110 Complex 11 showed a TOF of 9130 h−1 at 100 °C and 40 bar CO2 pressure. The catalytic activity decreased at higher temperature (120 °C) probably due to decomposition of the catalyst. Several other catalysts with modified structures were also synthesized, and their catalytic activities for the copolymerization of CHO and CO2 were studied by online ATR-IR measurements.111 Surprisingly, the in situ IR spectroscopic measurements revealed that the TOF values reached up to 23300 h−1 for complex 11 at 30 bar CO2 pressure and 100 °C. Complexes 12 and 13 containing Cl and CF3 groups, respectively, were expected to have metal centers with enhanced Lewis acidity. Indeed, complex 13 exhibited exceptionally high catalytic activity for the copolymerization of CHO and CO2 with a TOF of up to 155000 h−1.
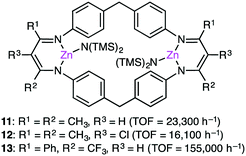 |
| | Fig. 2 Structures of 11–13. | |
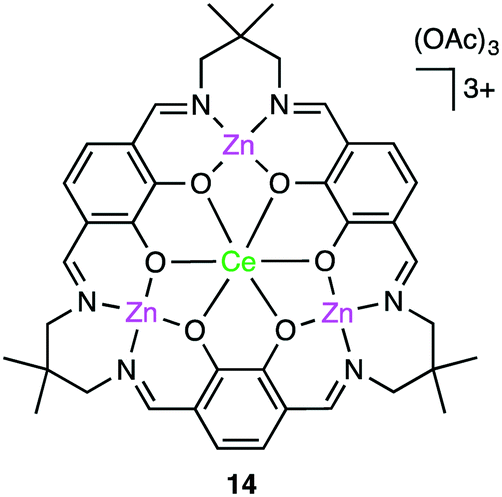
Okuda, Mashima, and co-workers synthesized several heterometallic tetranuclear complexes, among which CeZn3 complex 14 (Fig. 3) was the most effective for the copolymerization of CHO and CO2 at 100 °C, showing a TOF of 330 h−1 even at 0.6 MPa CO2 pressure with >99% carbonate-linkage selectivity.112 The molecular weight of the copolymer was controlled by adding tetrabutylammonium acetate as a chain-transfer agent. A rapid exchange of acetate anions is a key factor in the control of telomerization.
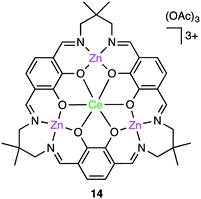 |
| | Fig. 3 Structure of CeZn3 complex 14. | |
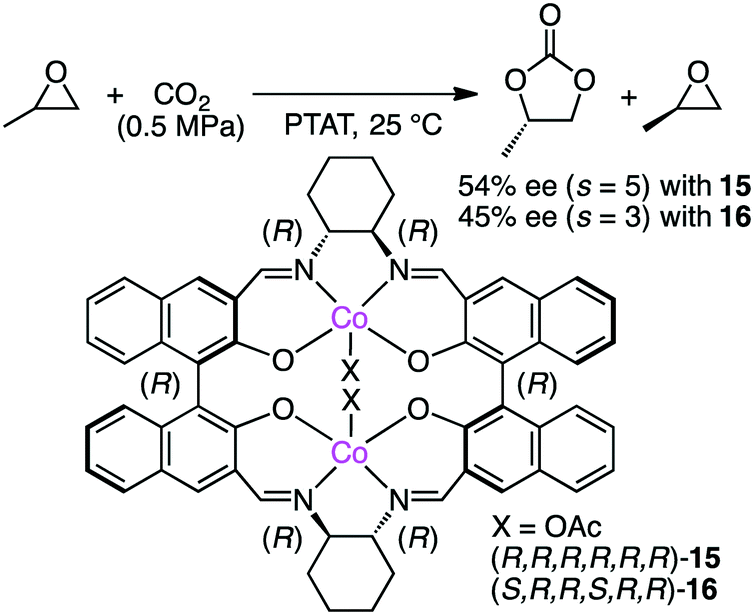
Jing and co-workers examined chiral dinuclear Co complexes 15 and 16 in the kinetic resolution of propylene oxide (PO) with CO2 using phenyltrimethylammonium tribromide (PTAT) as a nucleophilic co-catalyst (Scheme 3).113 (R,R,R,R,R,R)-15 showed higher enantioselectivity (s value 5) than (S,R,R,S,R,R)-16 (s value 3). The matched pair of two chiral moieties, BINOL and 1,2-cyclohexanediamine, is important for the enantioselectivity.
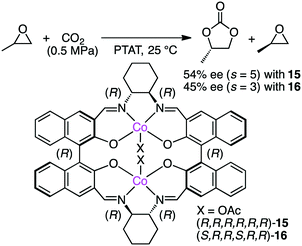 |
| | Scheme 3 Kinetic resolution of epoxides. | |
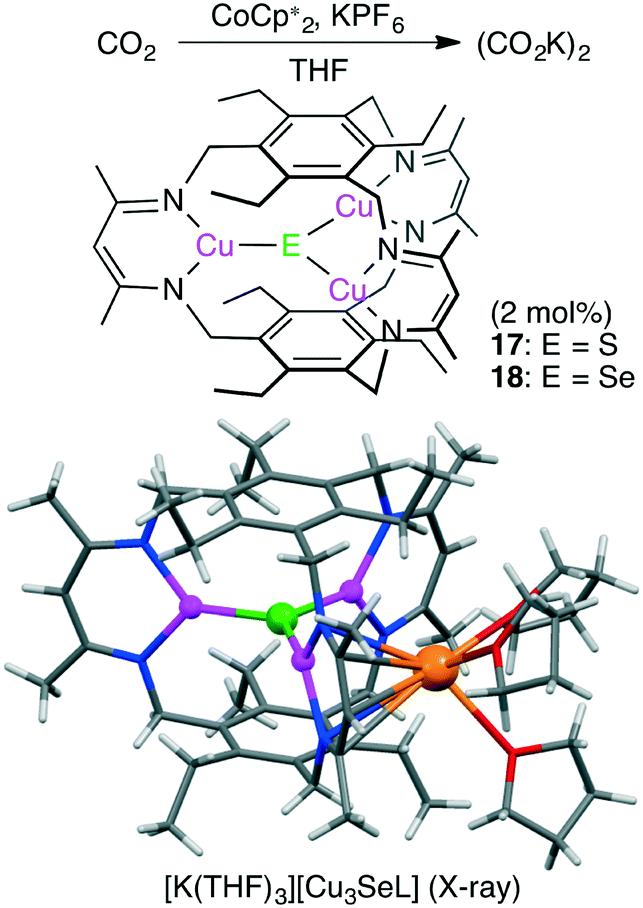
Murray and co-workers reported macrocyclic trinuclear Cu complexes, Cu3(μ3-S)L 17 and Cu3(μ3-Se)L 18, which showed catalytic activity for the selective reduction of CO2 to oxalate in the presence of a reductant (Scheme 4).114 To characterize the reactivity of 17 and 18, single-turnover reactions were carefully conducted. Anionic complex [Cu3EL]− was generated by the one-electron reduction of Cu3EL with various reductants such as [K(18-crown-6)][C10H8] in THF, and this anionic species successively reduced CO2 to (CO2K)2. Single crystals of [K(THF)3][Cu3EL] complexes were obtained by reduction of 17 or 18 with KC10H8 in THF, and X-ray analysis revealed that [K(THF)3]+ interacted with one β-diketiminate moiety of the [Cu3EL]− ion in an η5 fashion (Scheme 4). This crystal structure clearly illustrates how the solvent and cation stabilize the key intermediate. The pseudo-first-order rate constant for the reaction of [K(18-crown-6)][18] with CO2 was much greater than the corresponding value for [K(18-crown-6)][17]. These complexes and previously reported ones are promising catalysts for CO2 reduction to oxalate.115,116
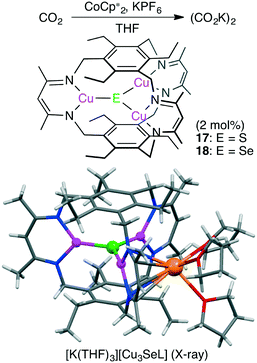 |
| | Scheme 4 Formation of oxalate from CO2. | |
2.2 Ring-opening copolymerization
Epoxide/anhydride copolymerization is an important area of research.93,117 Williams and co-workers prepared several copolymers by the ring-opening copolymerization (ROCOP) of phthalic anhydride (PA) and epoxides (Scheme 5).104,118,119 Although complex 1 was effective for PA/CHO copolymerization, it showed no activity for 1,4-cyclohexadiene oxide. On the other hand, complex 4 was almost equally effective for both epoxides.118,119 A synergistic effect between the two different metal centers in heterodinuclear metal complex 8 was observed; 8 was 20 times more active than 5.104
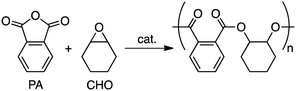 |
| | Scheme 5 Copolymerization of PA and CHO. | |
The mechanism of epoxide/anhydride copolymerization is proposed as follows: initially, a metal alkoxide is formed through epoxide-ring opening, which then rapidly reacts with the anhydride to form a metal carboxylate. The carboxylate reacts with the epoxide, producing a metal alkoxide.93,118,120
2.3 Olefin polymerization
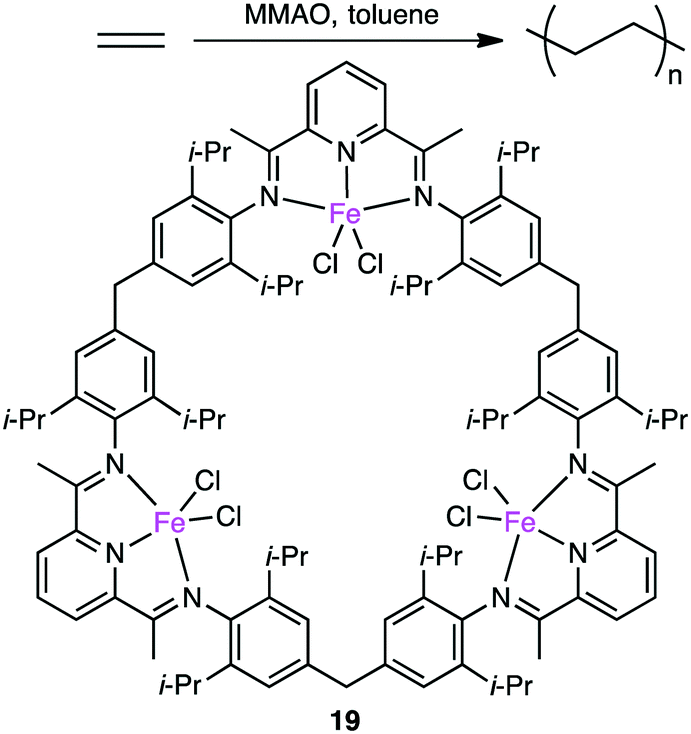
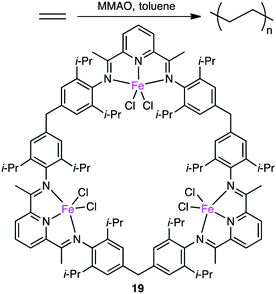
Since the discovery of Ziegler–Natta catalysts, numerous multinuclear metal complexes capable of catalyzing olefin polymerization have been reported so far.121 Several macrocyclic multinuclear metal complexes also worked as excellent catalysts.122–126 Li and co-workers reported that in the presence of a modified methylaluminoxane (MMAO) cocatalyst, trinuclear Fe(II) complex 19 showed excellent catalytic activity for the formation of polyethylene (PE) from ethylene as compared with a non-macrocyclic mononuclear counterpart. This fact clearly indicates the importance of the multiple metal centers in the macrocyclic skeleton (Scheme 6).122 The significance of the macrocyclic ligand was also demonstrated by a kinetic study. It was obvious that the macrocyclic ligand stabilized the catalytically active Fe center during polymerization and suppressed the chain transfer reaction to produce a high molecular-weight polymer. A maximum catalytic activity of 4300 kg molFe−1 h−1 bar−1 was achieved by 19 at 0 °C and 1 atm ethylene pressure. Both the catalytic activity of 19 and molecular weight of PE were temperature-dependent, decreasing with an increase in reaction temperature.
 |
| | Scheme 6 Ethylene polymerization. | |
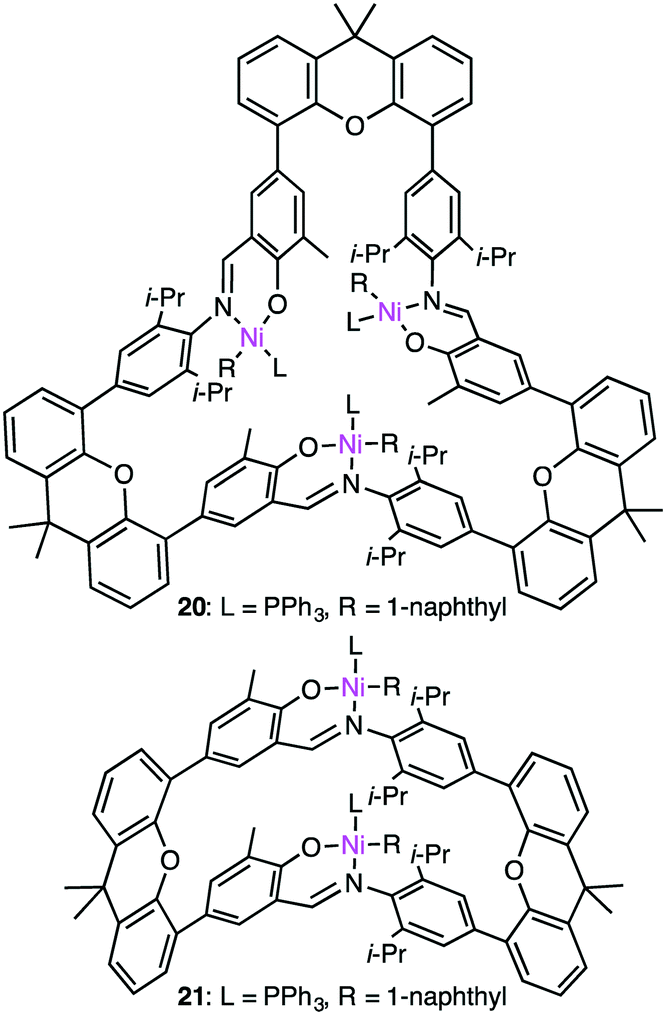
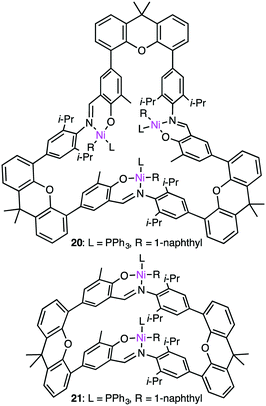
Ma and co-workers also reported the steric protection of the metal centers by the macrocyclic backbone in trinuclear Ni catalyst 20 during ethylene polymerization (Fig. 4).123 β-Hydrogen elimination and the chain-transfer process were suppressed by the steric protection of the metal centers to produce PE with a high molecular weight and a low branch density. Complex 20 showed a 4-fold greater polymerization activity than dinuclear analog 21. The same trend was observed for propylene polymerization. The most effective catalyst 20 provided the highest TOF at 15 °C although a polymer with the highest molecular weight was obtained at −10 °C. Similar to ethylene polymerization, the macrocyclic steric environment around the nickel centers in 20 was suggested to control the overall polymerization, which preferred sterically less hindered 1,2-insertion of propylene.
 |
| | Fig. 4 Structures of 20 and 21. | |
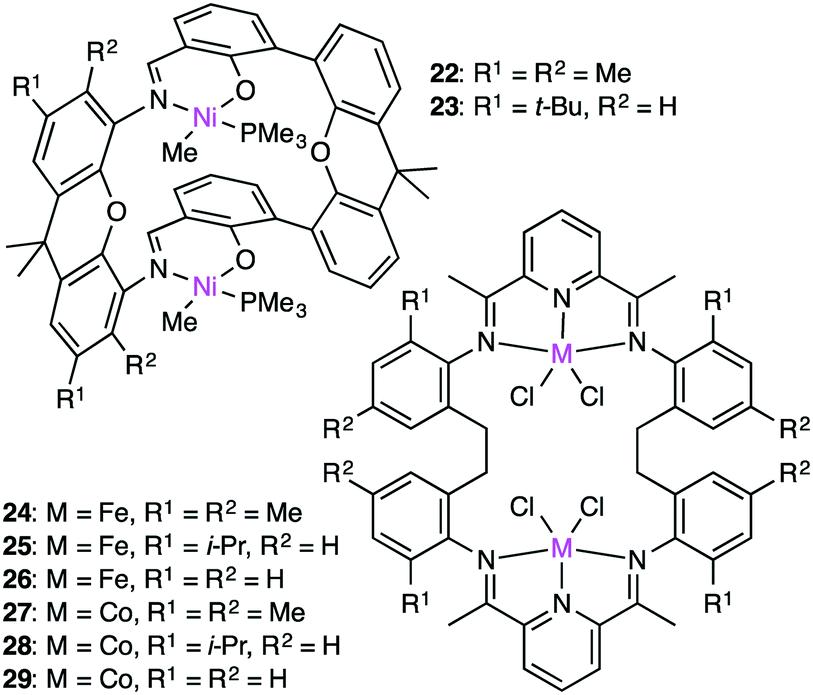
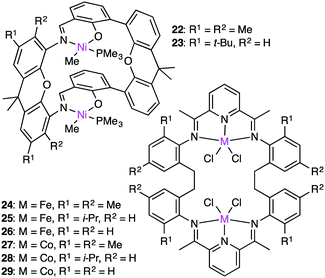
The cooperativity of two metal centers for olefin polymerization was demonstrated by Takeuchi, Osakada, and co-workers. Double-decker-type Ni catalyst 22 (Fig. 5) could copolymerize ethylene and bifunctional co-monomers such as dienes and unsaturated esters in the presence of a co-catalyst, Ni(cod)2, which served as a phosphine scavenger.124 The synergistic effect of the two Ni centers accelerated the polymerization. The growing polymer chain at one Ni center is stabilized by the coordination of the functional group to the other Ni center. Therefore, macrocyclic dinuclear Ni complex 22 was more effective than a monometallic counterpart.
 |
| | Fig. 5 Structures of 22–29. | |
The Takeuchi group studied the cooperative effect of the two Ni centers of 22 and 23 on ethylene polymerization in the presence of Ni(cod)2 (Fig. 5).125 The catalytic activity at room temperature decreased in the following order: 22 > 23 > a monometallic analog. The molecular weight of the polymers depended on the cooperativity of the two Ni centers, and higher-molecular-weight polymers were formed from di-activated catalysts, whereas lower-molecular-weight polymers were obtained by the mono-activated catalysts.
Dinuclear Fe and Co complexes 24–29 were synthesized to examine the polymerization activity at various temperatures and pressures in the presence of a MMAO co-catalyst.12624–29, which were thermodynamically more stable than the corresponding monometallic analogs, produced polymers with high molecular weights at room temperature and 1 atm ethylene pressure. Complex 24 showed the highest activity (975 g mmolFe−1 h−1 atm−1) at 100 °C and 5 atm ethylene pressure, while complex 27 showed the lowest activity (54.8 g mmolFe−1 h−1 atm−1) under the same reaction conditions. Complex 25 produced a higher molecular-weight polymer (Mn 62700) at room temperature than the corresponding monometallic complex (Mn 8930). The cooperative interaction between the growing polymer and the second metal center might retard the undesirable chain transfer and/or catalyst deactivation during the polymerization.
2.4 Aldol reaction
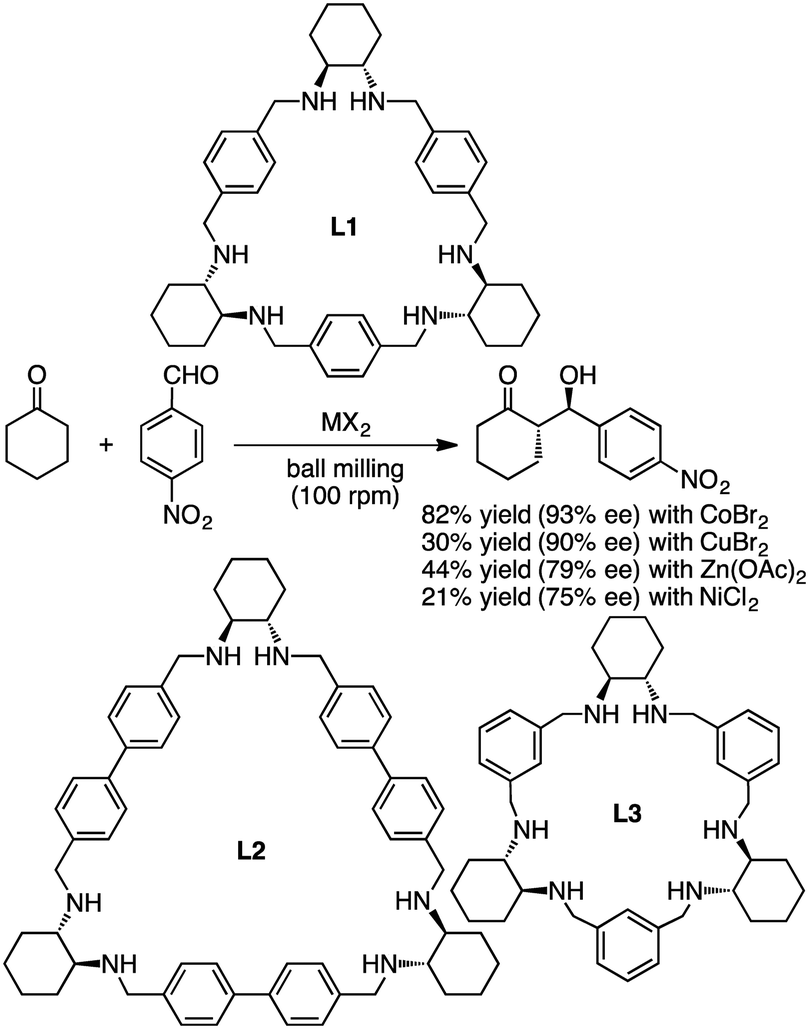
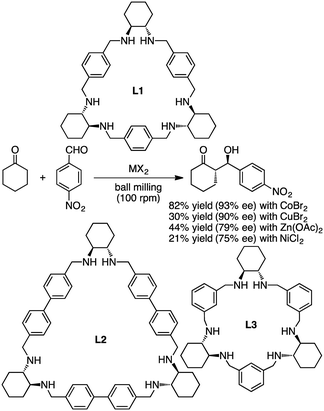
Tanaka and co-workers reported asymmetric aldol reactions catalyzed by metal complexes with chiral macrocyclic ligands L1, L2, and L3 under solvent-free conditions in a ball mill (Scheme 7).127 The most effective catalyst for the reaction of cyclohexanone with 4-nitrobenzaldehyde was obtained from a mixture of L1 and CoBr2 in a 1:2 ratio. The anti-aldol product was obtained as a major isomer in 82% yield and 93% ee. Other catalytic systems with CuBr2, Zn(OAc)2, and NiCl2 in combination with L1 gave 90, 79, and 75% ee, respectively.
 |
| | Scheme 7 Asymmetric aldol reaction. | |
2.5 Henry reaction
The Tanaka group employed similar catalytic systems in the enantioselective Cu-catalyzed Henry reaction of aldehydes with nitromethane under solvent-free conditions (Scheme 8).128 The reactions with aromatic aldehydes provided β-hydroxy nitroalkanes with up to 87% ee, and those with aliphatic aldehydes offered the products with up to 93% ee.
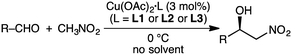 |
| | Scheme 8 Asymmetric Henry reaction. | |
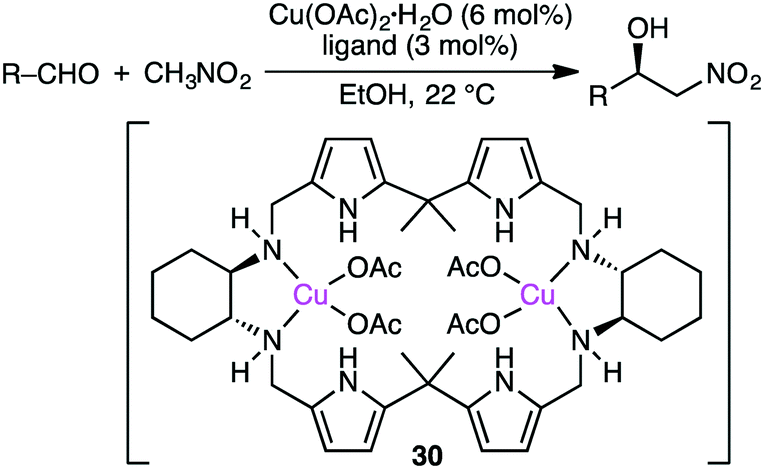
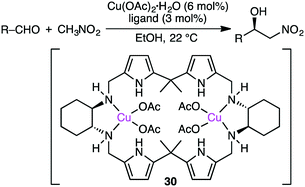
Savoia and co-workers developed an effective catalytic system for the asymmetric Cu-catalyzed Henry reaction of aldehydes with nitromethane (Scheme 9).129,130 The structure of complex 30, prepared from Cu(OAc)2·H2O and a C2-symmetric macrocyclic ligand, was confirmed by X-ray analysis. The reactions at room temperature gave a high enantioselectivity of up to 95% ee. The excellent performance of 30 as compared with a non-macrocyclic counterpart clearly demonstrates the essential role of the macrocyclic framework in the highly enantioselective Henry reaction.
 |
| | Scheme 9 Asymmetric Henry reaction. | |
2.6 Hydrolysis of phosphate esters
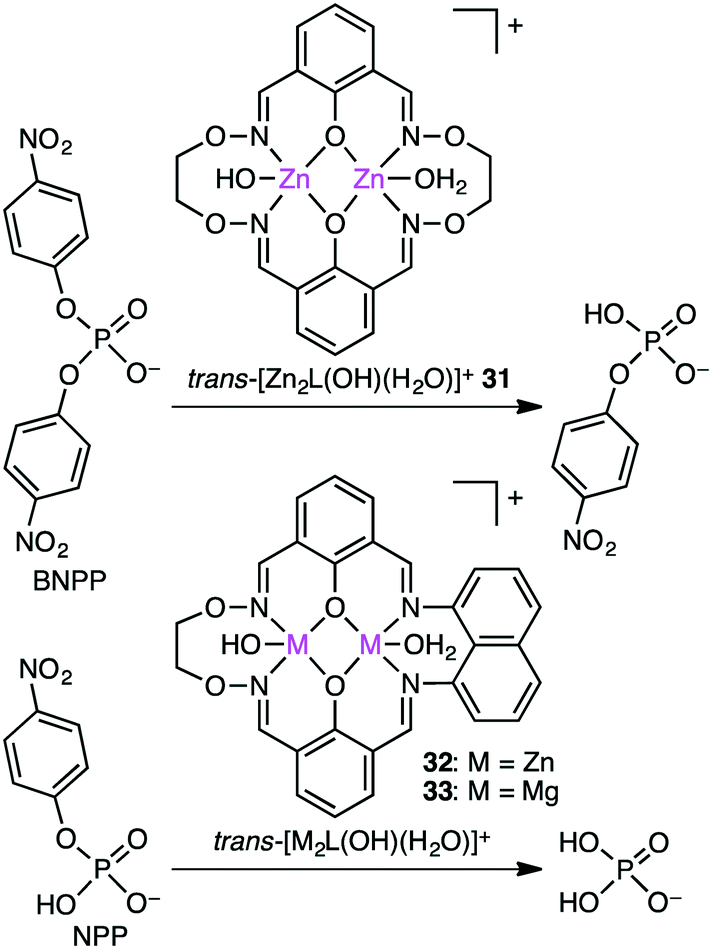
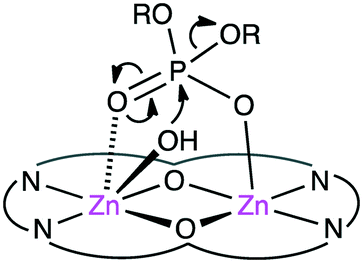
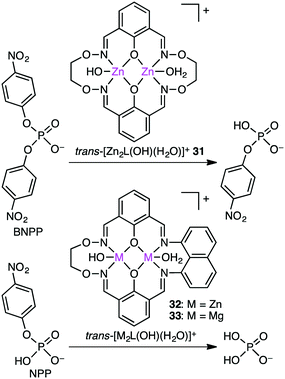
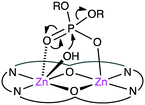
The biological roles of phosphate esters in living things are well known, and mechanistic studies on the hydrolytic cleavage of the P–O bond is an important area of research.131–134 Here, the hydrolysis of phosphate esters is briefly described in view of the potential significance in organic synthesis. Zhao and co-workers developed several symmetrical and unsymmetrical macrocyclic bimetallic systems to investigate the hydrolysis of phosphate diesters and monoesters.135,136 Among six possible forms of dinuclear Zn hydrates, trans-[Zn2L(OH)(H2O)]+ form 31 was considered to be the most active catalyst for the hydrolysis of phosphodiester BNPP (bis(4-nitrophenyl)phosphate) (Scheme 10).135 DFT calculations revealed that a ping-pong mechanism is most likely to occur, which involves a stepwise SN2-type nucleophilic addition–substitution reaction with inversion of the configuration at the P atom (Fig. 6). The two Zn(II) ions cooperatively facilitate the nucleophilic attack of the metal-bound OH−, stabilizing the anionic transition state. Mechanistic investigations on the hydrolytic cleavage of phosphate monoester NPP (4-nitrophenyl phosphate) were carried out by using unsymmetrical dinuclear catalysts 32 and 33.136 Zn catalyst 32 followed an energetically favorable pathway involving a concerted SN2-type addition–substitution reaction, while Mg catalyst 33 followed a stepwise SN2-type addition–substitution reaction.
 |
| | Scheme 10 Cleavage of phosphate esters. | |
 |
| | Fig. 6 Cooperative P–O bond cleavage. | |
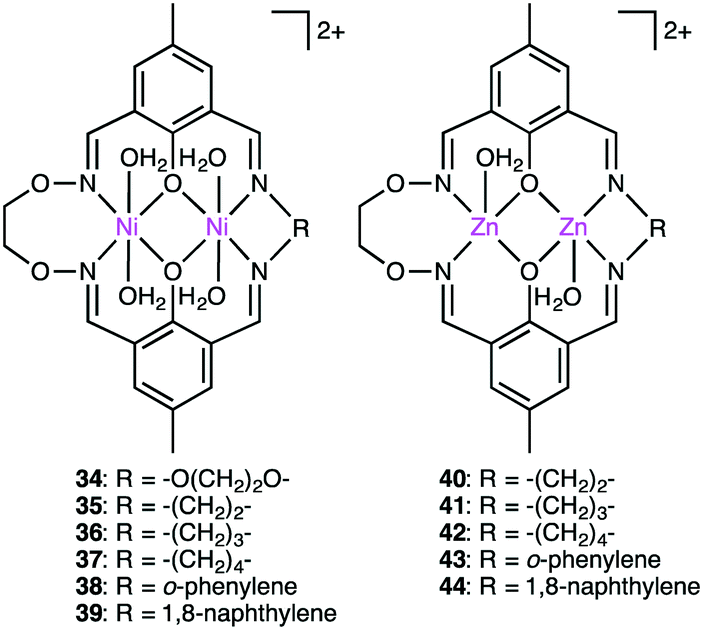
Kandaswamy and co-workers synthesized macrocyclic dinuclear Ni complexes 34–39 and Zn complexes 40–44 (Fig. 7) to study the hydrolytic cleavage of NPP and DNA.137,138 The pH-dependent formation of the nucleophilic OH− group by deprotonation of one of the metal-coordinated water molecules in the complexes was suggested to accelerate the hydrolysis of phosphate esters via nucleophilic attack on the P atom. Interestingly, symmetrical Ni complex 34 showed higher catalytic performance for the cleavage of NPP than unsymmetrical complexes 35–41. In the hydrolytic cleavage of DNA, macrocyclic Zn complexes exhibited higher catalytic performance than Ni analogs, which was attributed to the more Lewis acidic Zn(II) ion capable of tightly binding the phosphate group of DNA. The phosphate group of DNA is activated in a cooperative manner by the two Zn(II) ions, where the Zn(II)-bound OH− group attacks the P atom to break one of the P–O bonds of the DNA. Among the unsymmetrical Zn complexes, the best DNA cleavage activity was observed for 44.
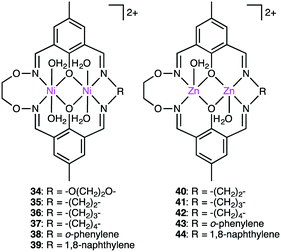 |
| | Fig. 7 Macrocyclic dinuclear complexes. | |
3. Multinuclear metal complexes forming a macrocyclic skeleton (category B)
3.1 CO2 fixation
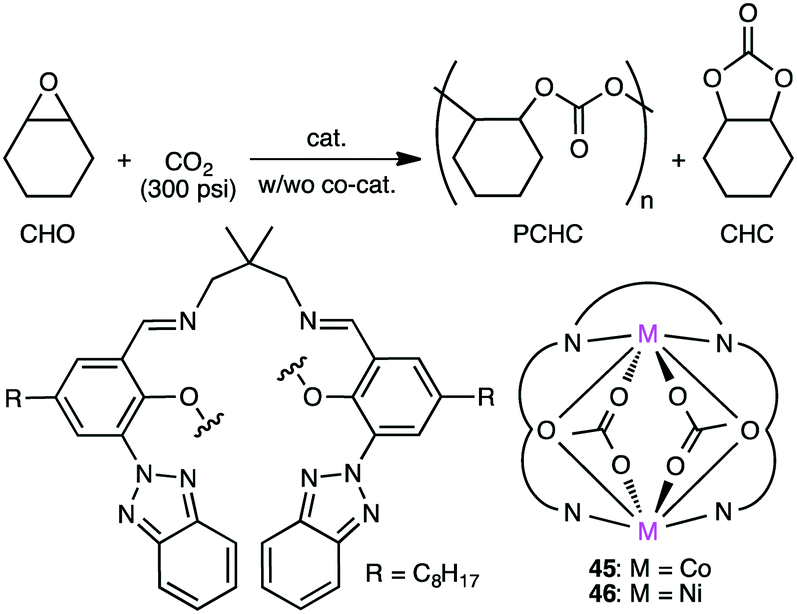
Ko and co-workers reported macrocyclic dinuclear Co complex 45 and Ni complex 46 for the reaction of epoxides with CO2 (Scheme 11).139 Ni complex 46 exhibited high polymerization activity (TOF 265 h−1) at 150 °C for the formation of PCHC (Mn 20800, carbonate linkage >99%) from CHO and CO2 in the absence of a co-catalyst. In the presence of a co-catalyst, Bu4NBr (TBAB), Co complex 45 produced cyclohexene carbonate (CHC) (cis-selectivity >99%) from CHO and CO2 at 120 °C with a TOF of 174 h−1.
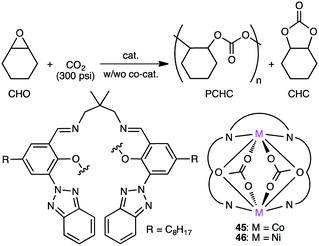 |
| | Scheme 11 Synthesis of PCHC and CHC. | |
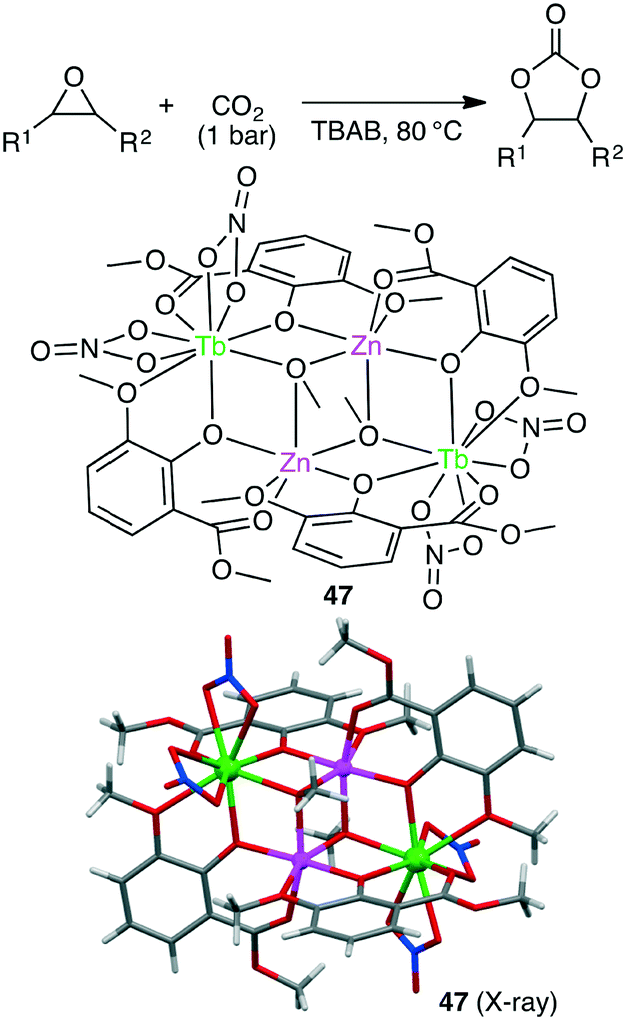
Liu and co-workers synthesized several anion-induced 3d–4f coordination clusters Zn2Ln2L4 (Ln = Nd, Eu, Tb, Er, Yb) and Zn4Ln2L4 (Ln = Nd, Tb).140 Zn2Tb2 complex 47 showed a TON of up to 9900 and a TOF of up to 700 h−1 (>99% selectivity) for the conversions of various epoxides and CO2 into cyclic carbonates under solvent-free conditions (Scheme 12).
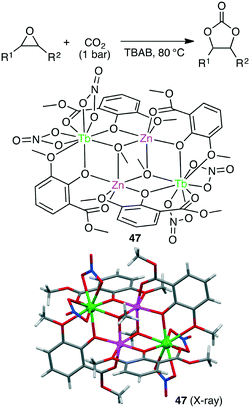 |
| | Scheme 12 Synthesis of cyclic carbonates. | |
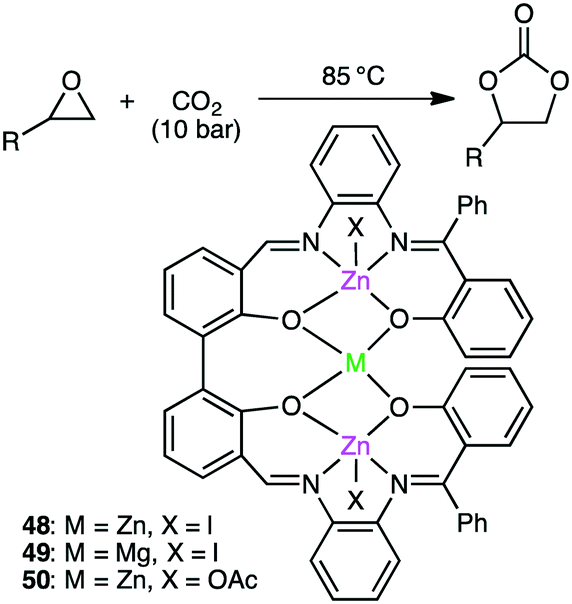
Kleij and co-workers reported trinuclear complexes 48–50 for the reaction of epoxides with CO2 (Scheme 13).141 Complex 48 gave 97% conversion even after 5 repeated uses of the recycled catalyst, and both the yield (>93%) and selectivity (>99%) remained high. The metal-bound I− is dissociated upon heating to become a nucleophile, which increases the Lewis acidity of the metal centers to facilitate the coordination and activation of the epoxide.
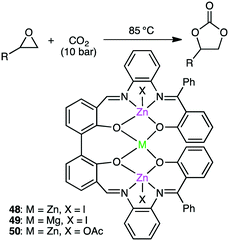 |
| | Scheme 13 Synthesis of cyclic carbonates. | |
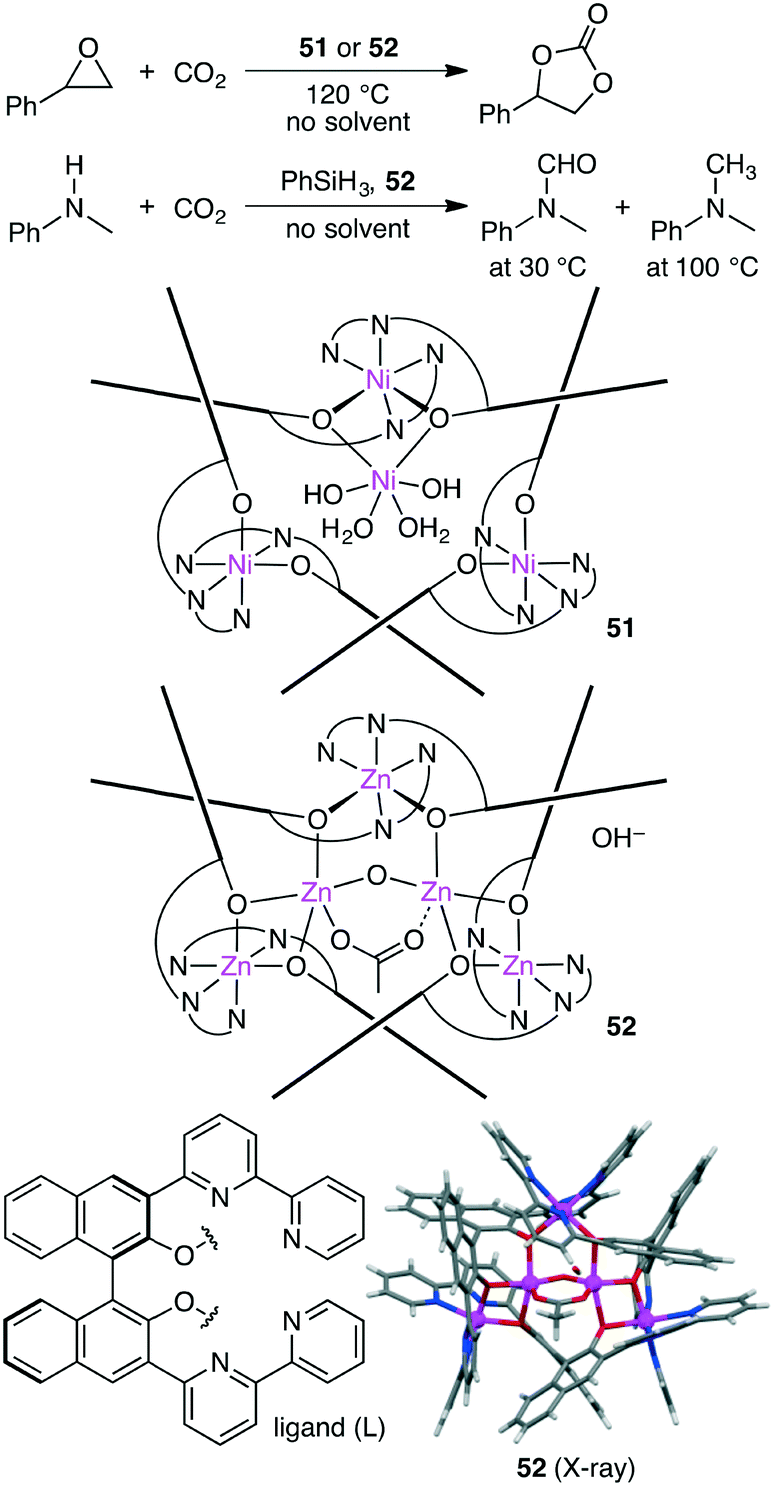
Takaishi, Ema, and co-workers synthesized macrocyclic multinuclear complexes Ni4L351 and Zn5L352 by the self-assembly of a chiral bipyridyl–binaphthyl ligand and metal salts (Scheme 14).142 X-ray analysis revealed unique complex@complex structures as represented by Ni@Ni3L3 and Zn2@Zn3L3. They exhibited dual catalytic activities for CO2 fixations under solvent-free conditions. The reaction of epoxides with CO2 at 120 °C gave cyclic carbonates, and 51 and 52 could be recycled at least 5 times without loss of catalytic activity. Zn5L3 complex 52 also acted as an excellent catalyst for the temperature-controlled N-functionalization reactions of amines with CO2 and PhSiH3; N-formylation products and N-methylation products were selectively obtained at 30 °C and 100 °C, respectively.
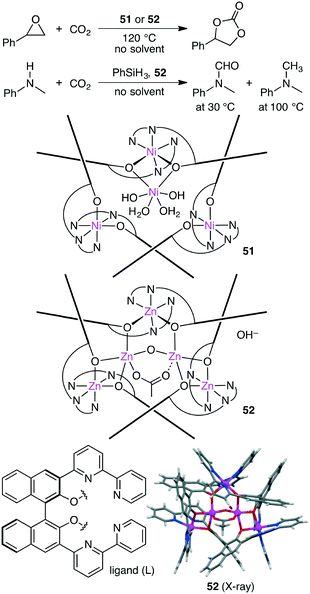 |
| | Scheme 14 Multitasking catalysts. | |
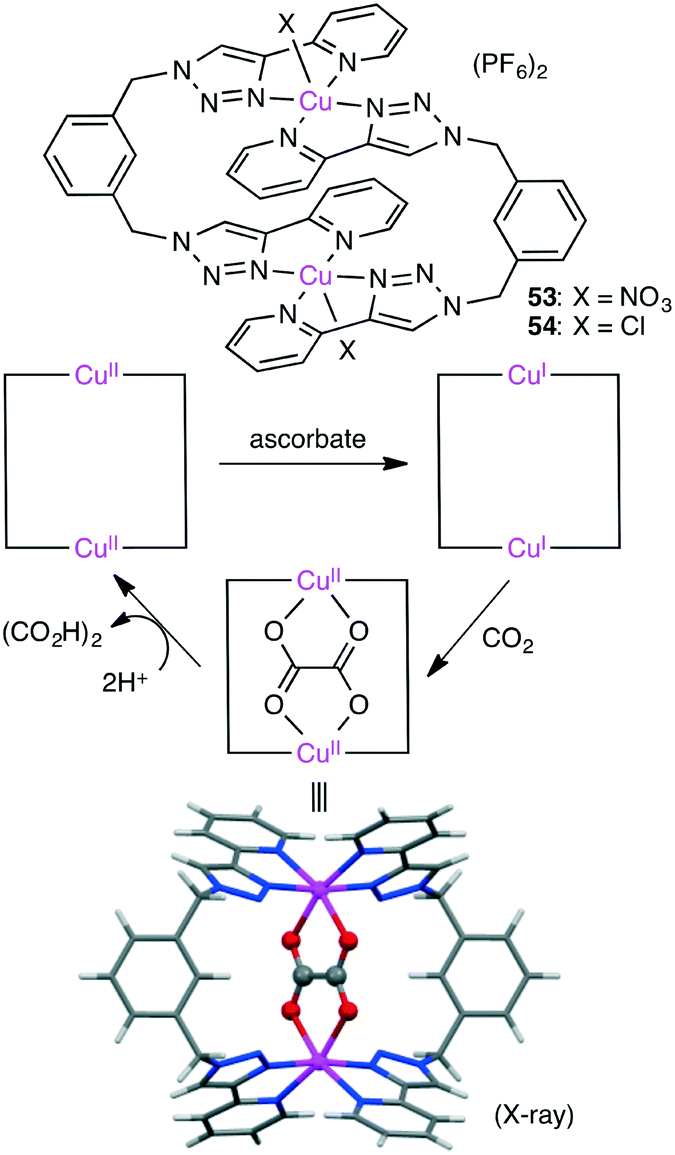
Maverick and co-workers synthesized well-designed macrocyclic dinuclear Cu complexes 53 and 54 capable of reducing CO2 to oxalate (Scheme 15).143 Reduction of the Cu(II) ions with a reductant (ascorbate) gave a Cu(I) species, which reduced CO2 to form the C–C bond. Acid treatment regenerated the starting Cu(II) species with the release of oxalic acid.
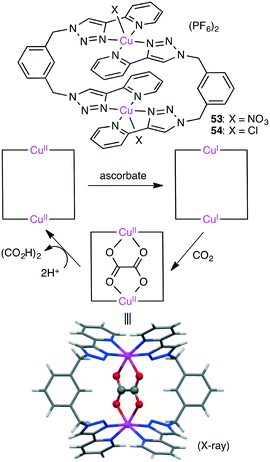 |
| | Scheme 15 Reduction of CO2 to oxalate. | |
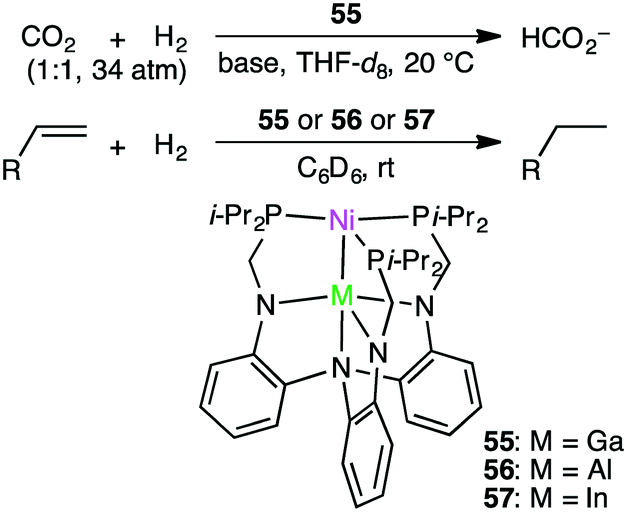
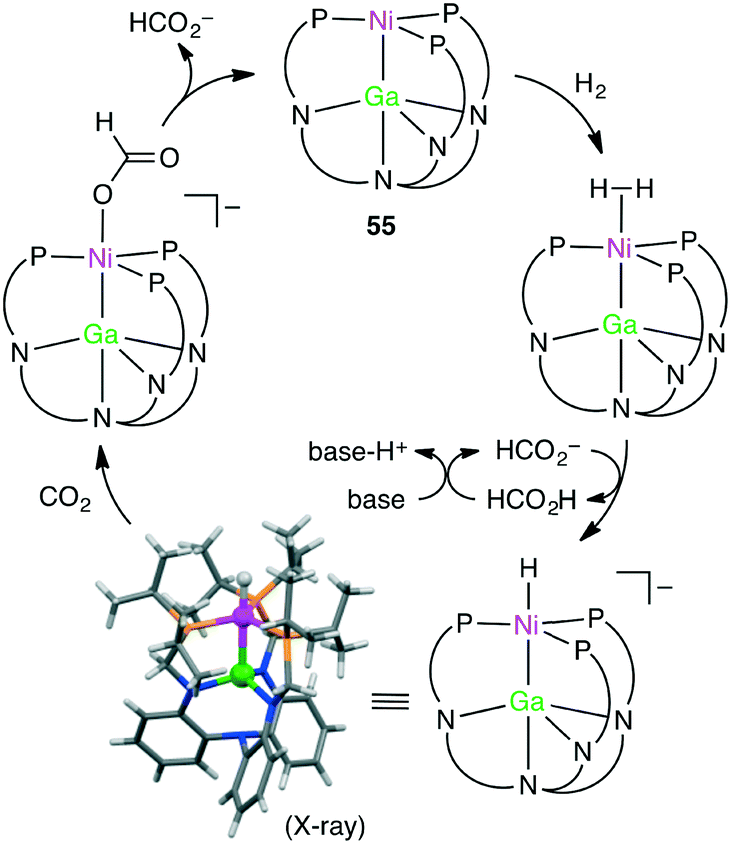
Homometallic or heterometallic metal–metal bonding produces unique catalytic activity and selectivity.45,144–146 Pairing of Ni0 with group 13 metal ions forms heterometallic complexes characterized by a strong Ni-to-M(III) dative bond. Because of the withdrawal of the electron density by the Lewis acidic M(III) ion, Ni becomes more electron-deficient, enabling the binding of small molecules such as H2. Lu and co-workers reported the hydrogenation of CO2 with NiGaL complex 55 to give formate at ambient temperature in the presence of a strong base (Scheme 16).147 The TON and TOF values reached 3150 and 6900 h−1, respectively. The essential role of the Ga(III) ion was clearly demonstrated by the fact that a Ni complex lacking the Ga(III) ion exhibited no catalytic activity. They performed an intensive mechanistic study to identify the most favorable reaction pathway: (i) binding of H2 to the Ni center to form adduct (η2-H2)NiGaL, (ii) deprotonation of the adduct with a base to generate a [HNiGaL]− species, (iii) production of formate adduct [(η1-HCO2)NiGaL]− through CO2 insertion, and (iv) the release of formate and regeneration of catalyst 55 (Scheme 17).148 On the other hand, Takaya and Iwasawa synthesized a macrocyclic Pd–Al complex from a different acyclic ligand, which acted as a catalyst for the hydrosilylation of CO2 (1 atm) to give silyl formate with a TOF of 19300 h−1 at 25 °C.149
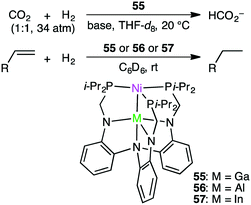 |
| | Scheme 16 Hydrogenation of CO2 and olefin. | |
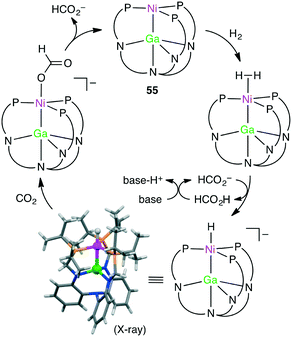 |
| | Scheme 17 Catalytic cycle. | |
3.2 Olefin hydrogenation
Lu and co-workers applied bimetallic complexes 55–57 to olefin hydrogenation (Scheme 16).150 Although Ni/In complex 57 showed modest activity for the hydrogenation of olefins, Ni/Al complex 56 was not effective at all. Ni/Ga complex 55 displayed the highest catalytic performance, delivering >99% yield under mild reaction conditions. The formation of a (H2)Ni–M adduct and then a HNi(μ-H)M species was likely to be key to the smooth olefin hydrogenation.
3.3 Hydrogenation of ketones
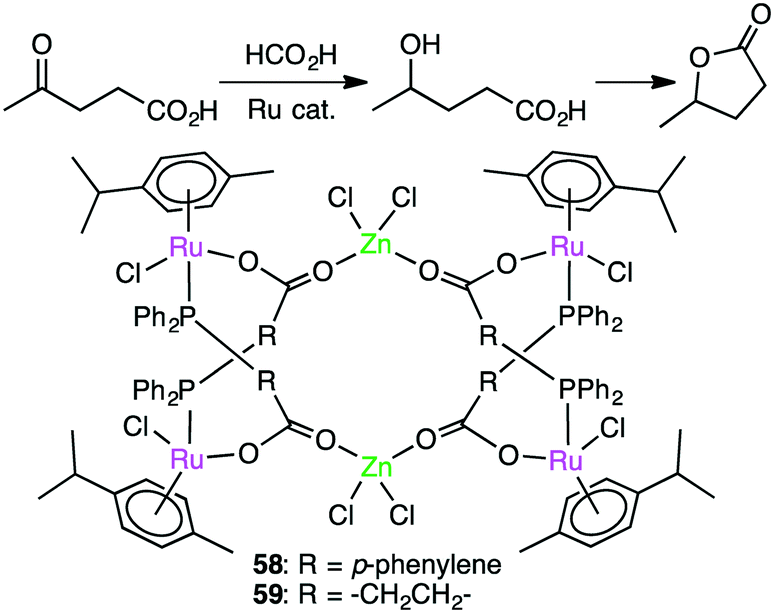
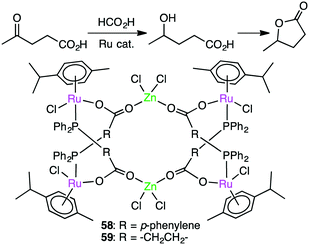
Darkwa, Makhubela, and co-worker developed hexanuclear Ru4Zn2 complexes 58 and 59 with phosphino–carboxylate bridges as precatalysts for the hydrogenation of levulinic acid to γ-valerolactone using formic acid as a hydrogen source (Scheme 18).151 These multimetallic complexes showed superior catalytic activities over the corresponding monometallic counterparts, one of which gave a maximum TOF of 540 h−1. Catalyst 58 was slightly more effective than 59. The high catalytic activity was ascribed to the presence of the Zn(II) ions, which are considered to decrease the electron density of the Ru centers to promote the H–H bond activation.
 |
| | Scheme 18 Hydrogenation of levulinic acid. | |
3.4 Ethylene polymerization
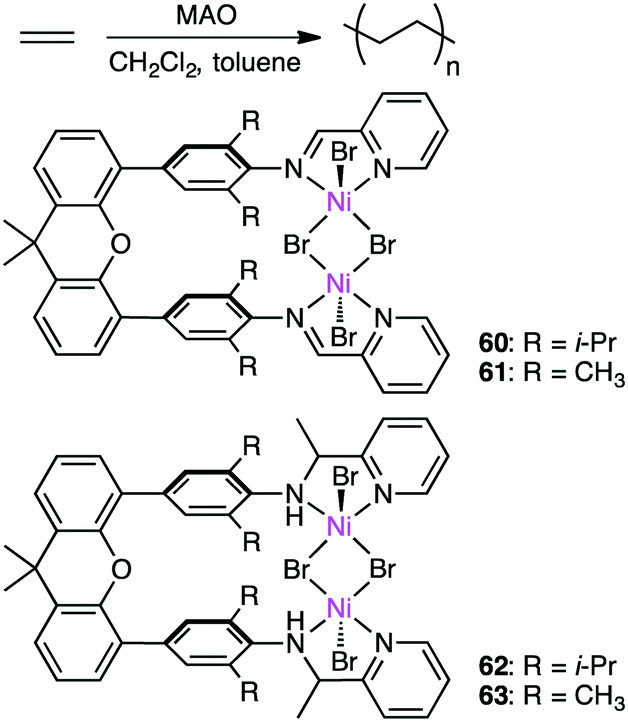
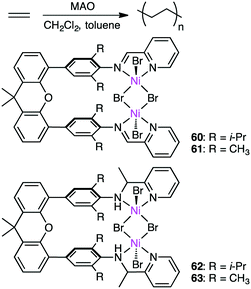
Wang, Chen, and co-workers prepared several dinuclear Ni complexes 60–63 for ethylene polymerization (Scheme 19).152 Iminopyridyl complexes 60 and 61 exhibited higher catalytic activity than aminopyridyl complexes 62 and 63, and the highest activity of up to 2200 kg molNi−1 h−1 was observed for 60 at 20 °C. Dinuclear complex 60 showed higher thermal stability and catalytic activity than the corresponding mononuclear analog, the former of which produced PE with a lower branching density than the latter. The cooperative effect of the two metal ions is considered to slow down β-hydride elimination and the chain-walking process. 60 and 62 with the isopropyl groups were catalytically more active than 61 and 63 with the methyl groups, respectively, and 60 and 62 produced PE with a higher molecular weight. It is suggested that the isopropyl groups on the aniline rings efficiently suppressed chain-transfer reactions. The steric bulkiness of the substituents on the aniline rings also affected the branching density of the polymers.
 |
| | Scheme 19 Ethylene polymerization. | |
3.5 Friedel–Crafts alkylation
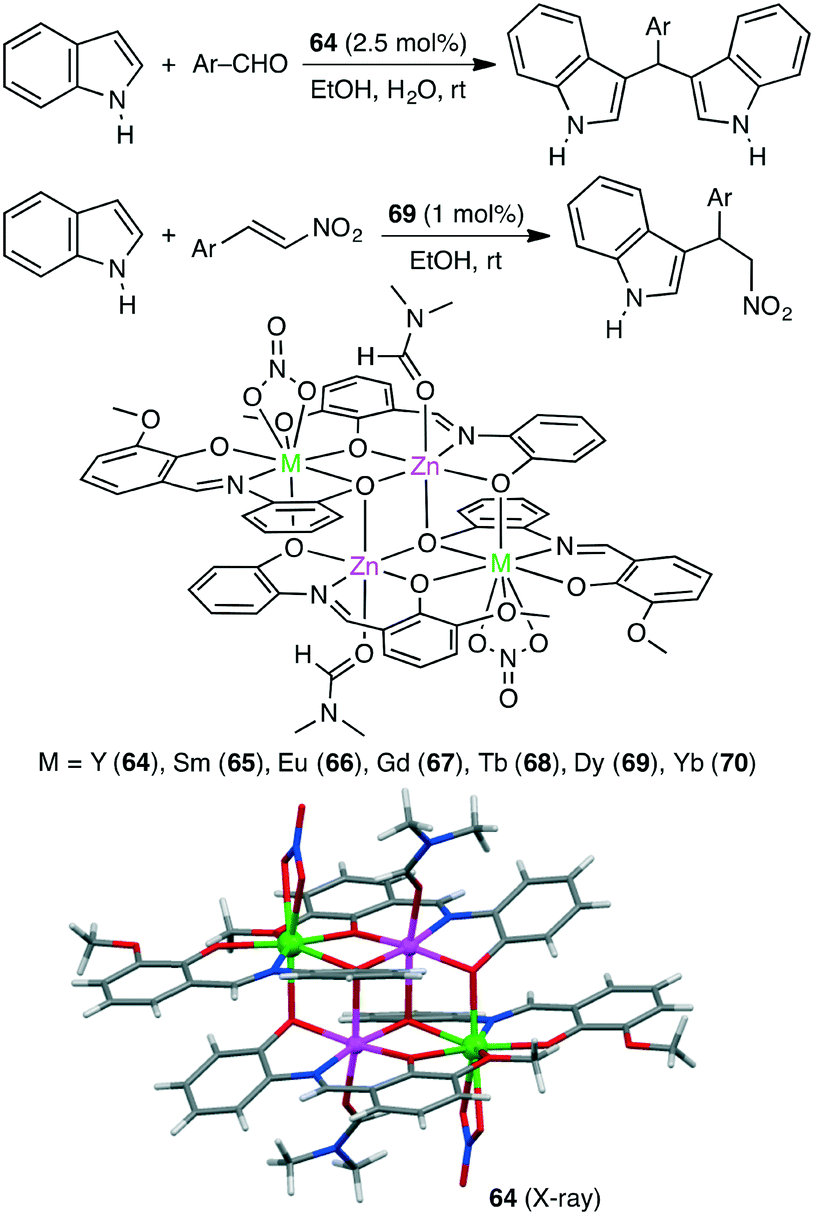
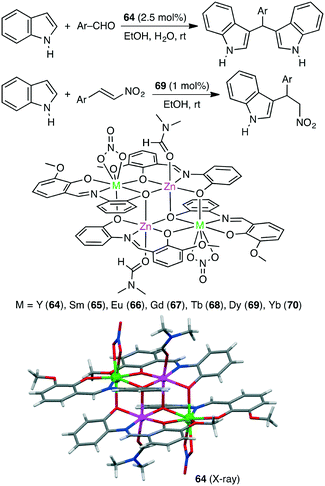
The Friedel–Crafts alkylation of aromatic compounds is extensively used in organic synthesis. Kostakis and co-workers synthesized a series of isoskeletal tetranuclear heterometallic clusters 64–70 (Scheme 20).153 X-ray analysis revealed the close proximity of the four metal ions in all cases. Each of them served as an excellent catalyst for the Friedel–Crafts alkylation of indoles with aldehydes, and Zn2Y2 complex 64 showed the highest catalytic activity. The Zn(II) and Y(III) ions are considered to activate indole and aldehyde molecules, respectively, in a cooperative manner. Tetranuclear clusters also catalyzed the Friedel–Crafts reaction of indoles with trans-β-nitrostyrenes at room temperature.154 The maximum yield of 99% was obtained by using Zn2Dy2 complex 69.
 |
| | Scheme 20 Friedel–Crafts alkylation. | |
3.6 Domino reaction
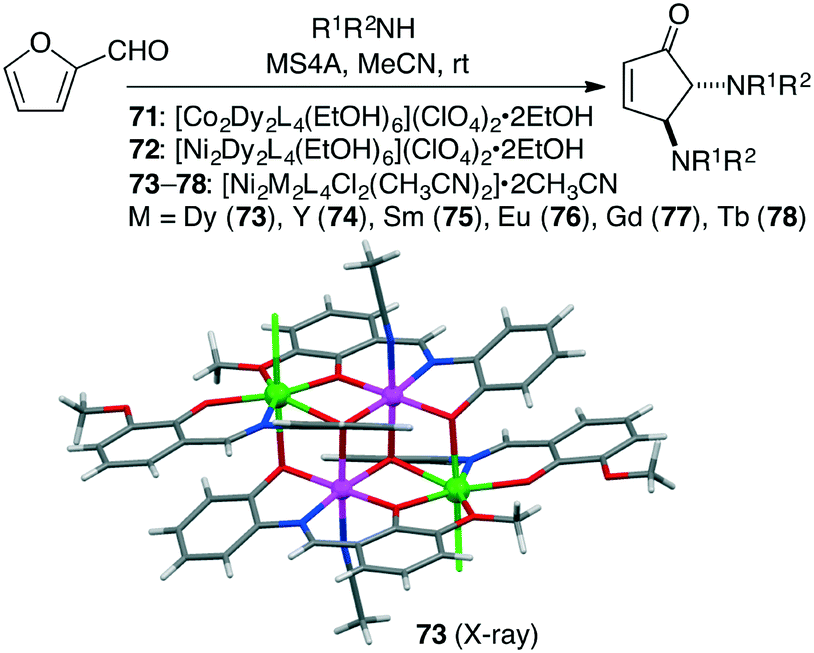
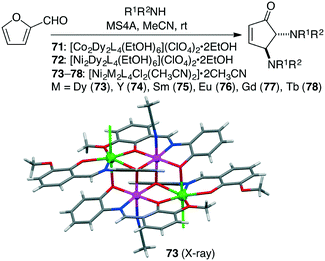
Kostakis and co-workers prepared three isoskeletal tetranuclear complexes 71–73, among which Ni2Dy2 complex 73 (1 mol%) was the most effective for the domino condensation/ring-opening/electrocyclization of amines and 2-furaldehyde at room temperature to produce trans-4,5-diaminocyclopent-2-enones (Scheme 21).155 A series of Ni2Ln2 complexes 74–78 analogous to 73 were further synthesized, and it was found that a lower amount of Ni2Y2 complex 74 (0.5 mol%) catalyzed the reaction to afford the products in a quantitative yield.156 The catalytic activity was mainly induced by the Y(III) or 4f ions rather than the Ni(II) ions.
 |
| | Scheme 21 Domino reaction. | |
3.7 Suzuki–Miyaura reaction
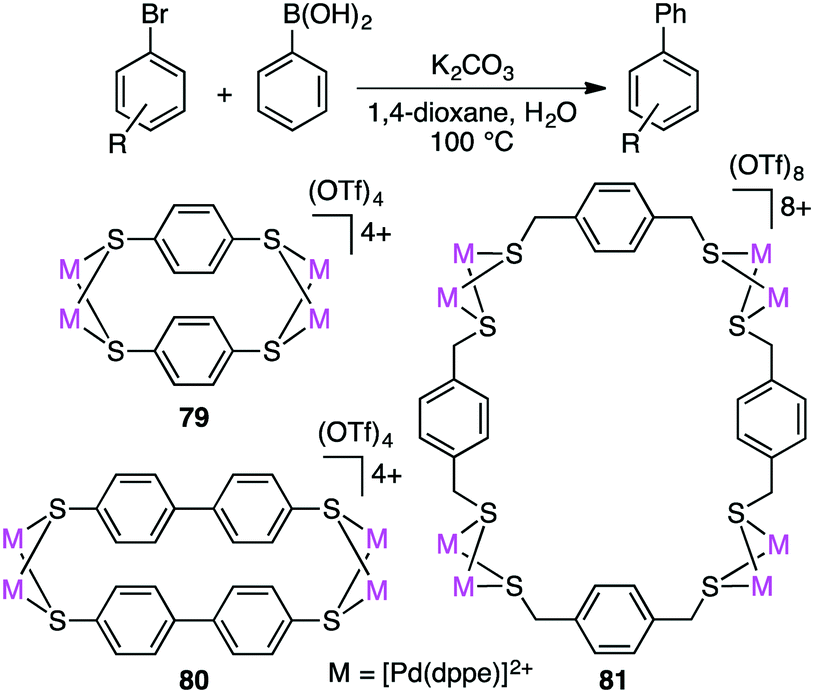
Dey and co-workers prepared self-assembled tetranuclear and octanuclear Pd-based macrocyclic dithiolates 79–81, which exhibited excellent catalytic activity for the Suzuki–Miyaura reaction of both electron-rich and electron-poor aryl bromides with phenylboronic acids (Scheme 22).157 Although all the complexes exhibited high catalytic efficiency, rigid macrocyclic dithiolates 79 and 80 were more effective than the octanuclear complex 81. With a trace amount of catalyst 79 (0.000004 mol%), the product was obtained in 85% yield (TON 21 million).
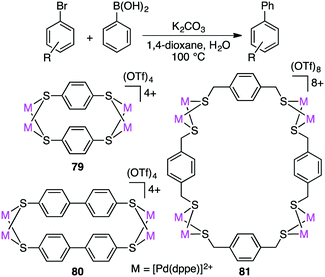 |
| | Scheme 22 Suzuki–Miyaura reaction. | |
3.8 Heck reaction
Macrocyclic dithiolate complexes 79–81 also showed excellent catalytic activity in the Heck reaction of aryl bromides with alkenes; the TON and TOF reached 410000 and 25625 h−1, respectively (Scheme 23).158 As observed for the Suzuki–Miyaura reaction, the catalytic activity of the three palladium complexes decreased in the following order: 79 > 80 > 81.
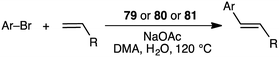 |
| | Scheme 23 Heck reaction. | |
3.9 Michael addition
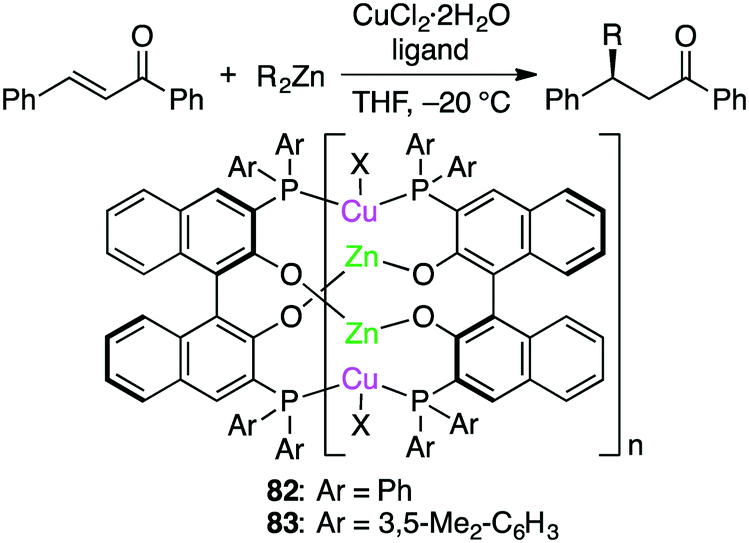
Endo, Shibata, and co-workers developed self-assembled oligomeric multinuclear Cu/Zn complexes 82 and 83 as asymmetric catalytic systems (Scheme 24).159 The specific ligand scaffold was essential for the generation of the metal cluster that could achieve highly enantioselective asymmetric conjugate addition of organozinc reagents to enones. A small amount (0.5 mol%) of 83 was enough to promote the reactions at −20 °C to furnish products in high yields with almost complete enantiomeric purities (up to >98% ee), and even 0.05 mol% of 83 could catalyze the same reactions with slightly lower enantioselectivity. A wide range of enones bearing various electron-donating or electron-withdrawing groups were tolerated.
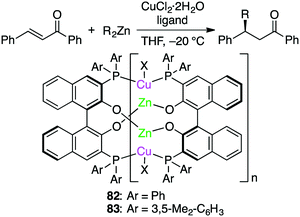 |
| | Scheme 24 Asymmetric conjugate addition. | |
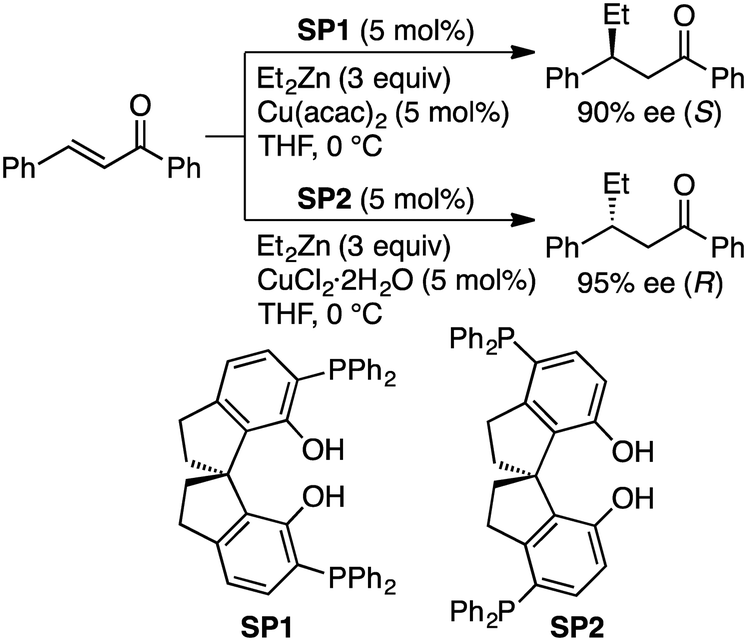
They also reported that multinuclear complexes prepared from a regioisomeric ligand (SP1 or SP2), Et2Zn, and a Cu salt showed opposite enantioselectivity in the asymmetric conjugate addition (Scheme 25).160 The oligomeric multinuclear Cu/Zn complex with SP1 provided the (S)-isomer with 90% ee, while that with SP2 afforded the (R)-isomer with 95% ee. A similar catalytic system with a self-assembled multinuclear Pd/Zn complex was successfully applied to the asymmetric alkylative ring-opening reaction of oxabicyclic alkenes.161
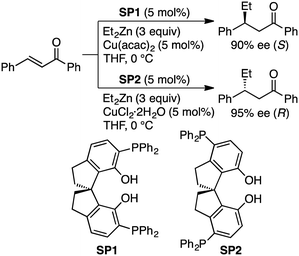 |
| | Scheme 25 Inversion of enantioselectivity. | |
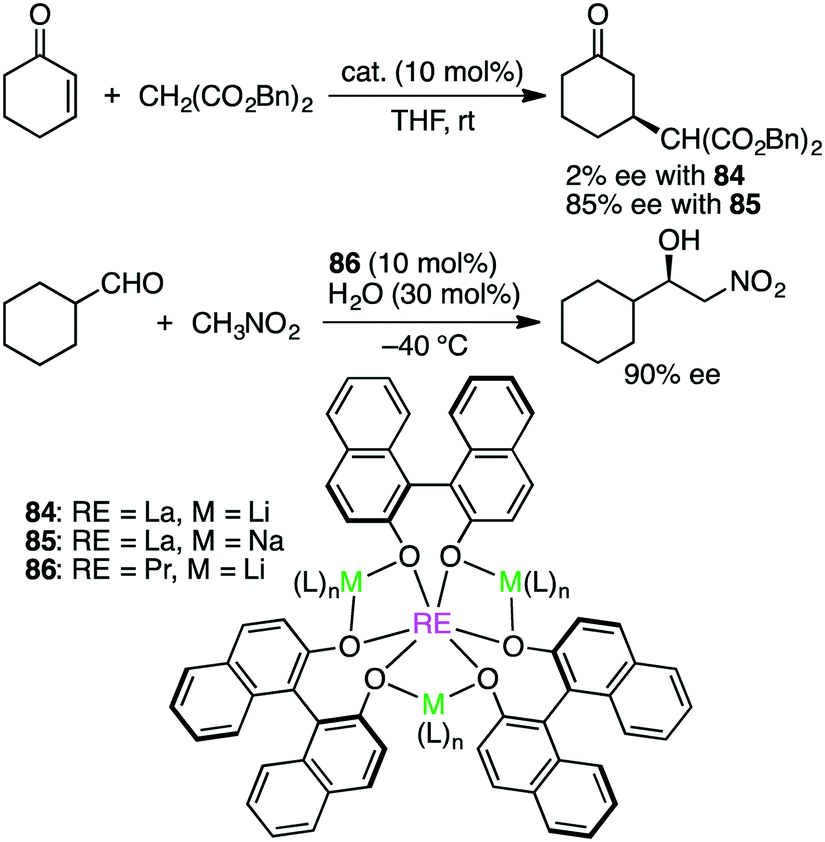
Shibasaki and co-workers made a great contribution to the development of asymmetric catalysis with multinuclear metal complexes.162 Further studies are continued to explore this exciting research area. Schelter, Walsh, and co-workers studied both Michael and Henry reactions catalyzed by Shibasaki's catalysts 84–86 containing rare earth (RE), alkali metal (M), and BINOLate (Scheme 26).163 Dynamic ligand exchange behaviors were clarified by 2D EXSY NMR measurements; both alkali metal cations and BINOLate ligands in the RE/M frameworks were rapidly exchanged under catalytically relevant conditions. In addition, the first X-ray crystal structure of a substrate-bound complex of 86 was determined, and using this crystal, stoichiometric and catalytic reactivity studies were done to observe that substrate deprotonation by the catalyst framework was necessary for the high enantioselectivity. Rather than a tris(BINOLate) species, a bis(BINOLate) or oligomeric heterobimetallic species may be the catalytically active species.
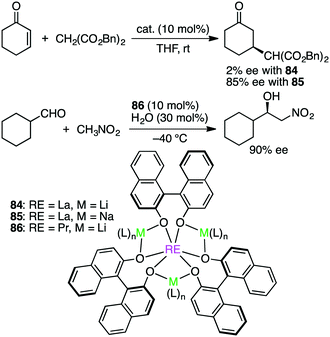 |
| | Scheme 26 Michael and Henry reactions. | |
3.10 Transesterification
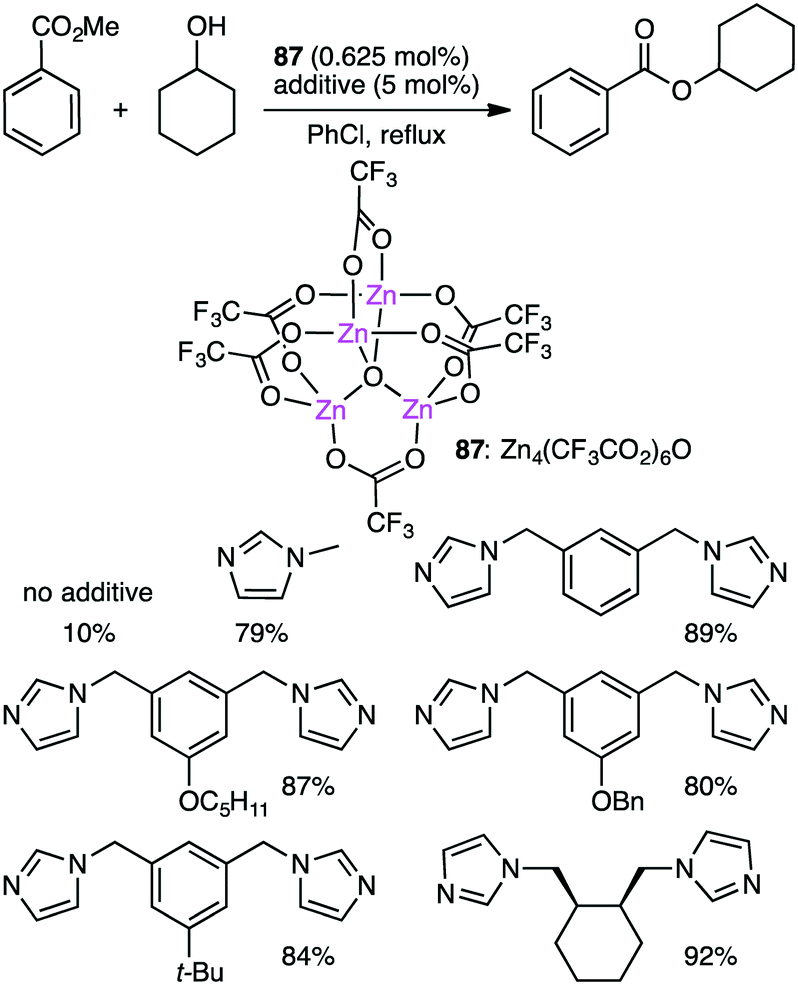
Ohshima and co-workers reported the transesterification between methyl benzoate and cyclohexanol using tetranuclear Zn cluster 87 in the presence of an N-heteroaromatic ligand as an additive (Scheme 27).164,165 The yield was dramatically improved by the addition of the N-heteroaromatic ligand. X-ray analysis of single crystals obtained from 87 and meta-bis(imidazolylmethyl)benzene revealed that an active species was a coordination polymer containing macrocyclic dinuclear complexes as a repeating unit.
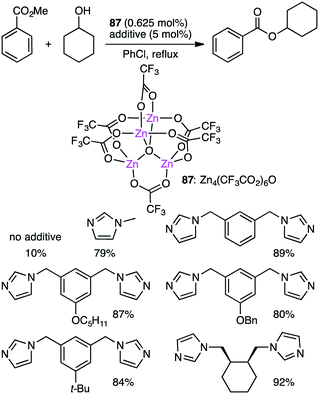 |
| | Scheme 27 Transesterification. | |
3.11 Ring-opening polymerization
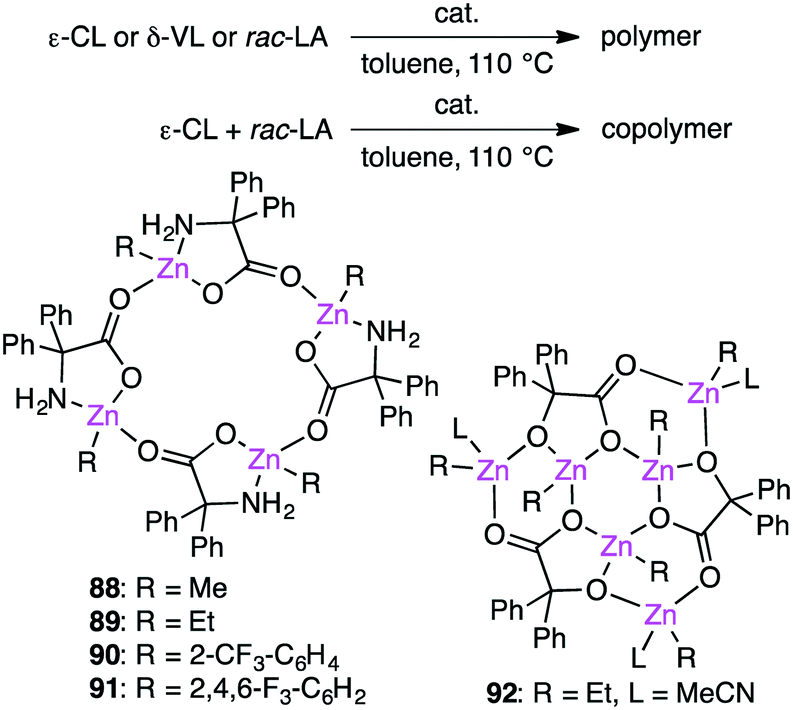
Redshaw and co-workers investigated the ring-opening polymerization of ε-caprolactone (ε-CL), δ-valerolactone (δ-VL), or rac-lactide (rac-LA) and the ring-opening copolymerization of ε-CL and rac-LA in the presence of macrocyclic Zn clusters 88–92 (Scheme 28).166,167 Fluorine-containing catalysts 90 and 91 were more active for the homopolymerizations, whereas all of them showed comparable activities for the copolymerization of ε-CL and rac-LA.
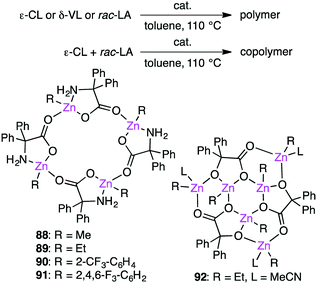 |
| | Scheme 28 Ring-opening polymerization. | |
3.12 Oxidation of alkanes
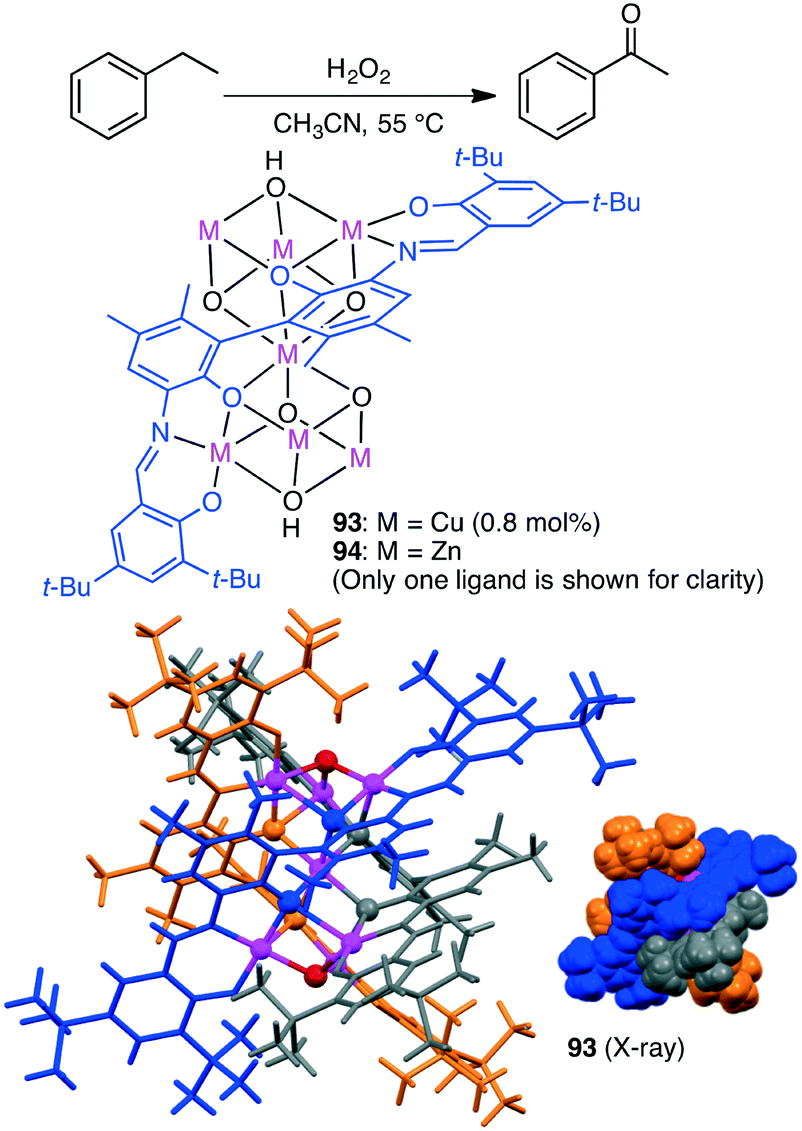
Selective oxidation of alkanes is a challenging subject. Liu, Cui and co-workers synthesized triple-stranded cluster helicates with a heptametallic dicubane core 93 and 94 from a hexadentate Schiff-base ligand (Scheme 29).168 Cu species 93 catalyzed the peroxidative oxidation of alkanes by a radical-type mechanism. Selective C–H oxidation of cyclopentane, cyclohexane, cyclooctane, toluene, and ethylbenzene proceeded to give the corresponding ketones along with a trace amount of alcohols.
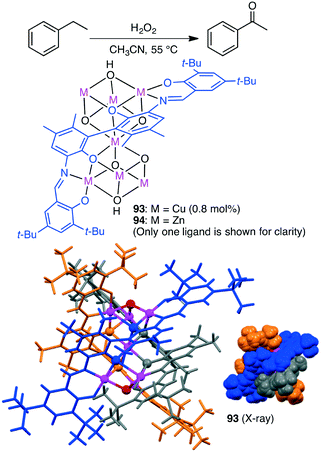 |
| | Scheme 29 Oxidation of alkane. | |
3.13 Oxidation of alcohols
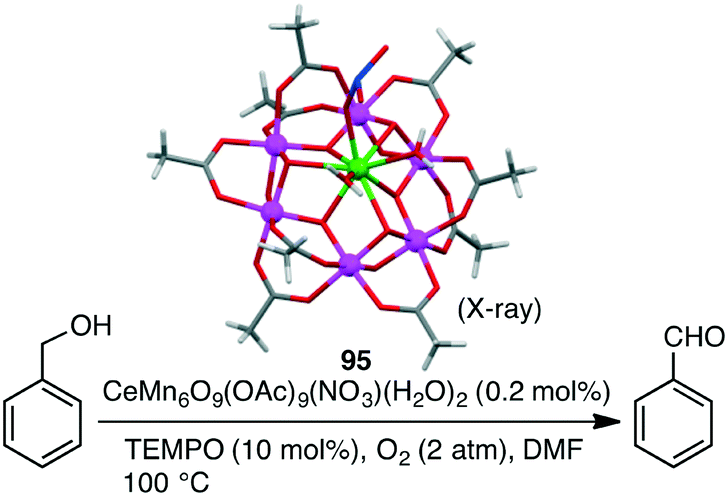
Christou and co-workers investigated the selective oxidation of benzyl alcohol to benzaldehyde with a series of macrocyclic Mn clusters, such as Mn12O12(OAc)16(H2O)4 and congeners, using O2 as an oxidant and TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) as a co-catalyst.169,170 The best catalytic activity (94% yield, TON 470) was observed for mixed-metal cluster CeMn6O9(OAc)9(NO3)(H2O)295 (Scheme 30). This performance must be due to the presence of the heterometallic Mn(IV)/Ce(IV) structure intimately connected via oxide bridges (CeMn6O9 core) in the cluster.
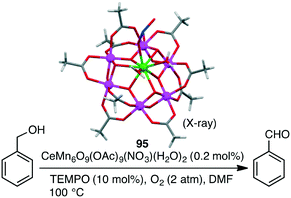 |
| | Scheme 30 Oxidation of alcohol. | |
Photosystem II (PSII) in higher plants contains a Mn4Ca cluster for water oxidation.9–12 Because water oxidation is an inorganic reaction, being outside the range of this perspective, we just briefly note that there are several excellent classes of macrocyclic multinuclear metal complexes for photocatalytic water oxidation.171–177 They are potential photocatalysts for organic reactions.
4. Self-assembled supramolecular coordination complexes (category C)
4.1 Diels–Alder reaction
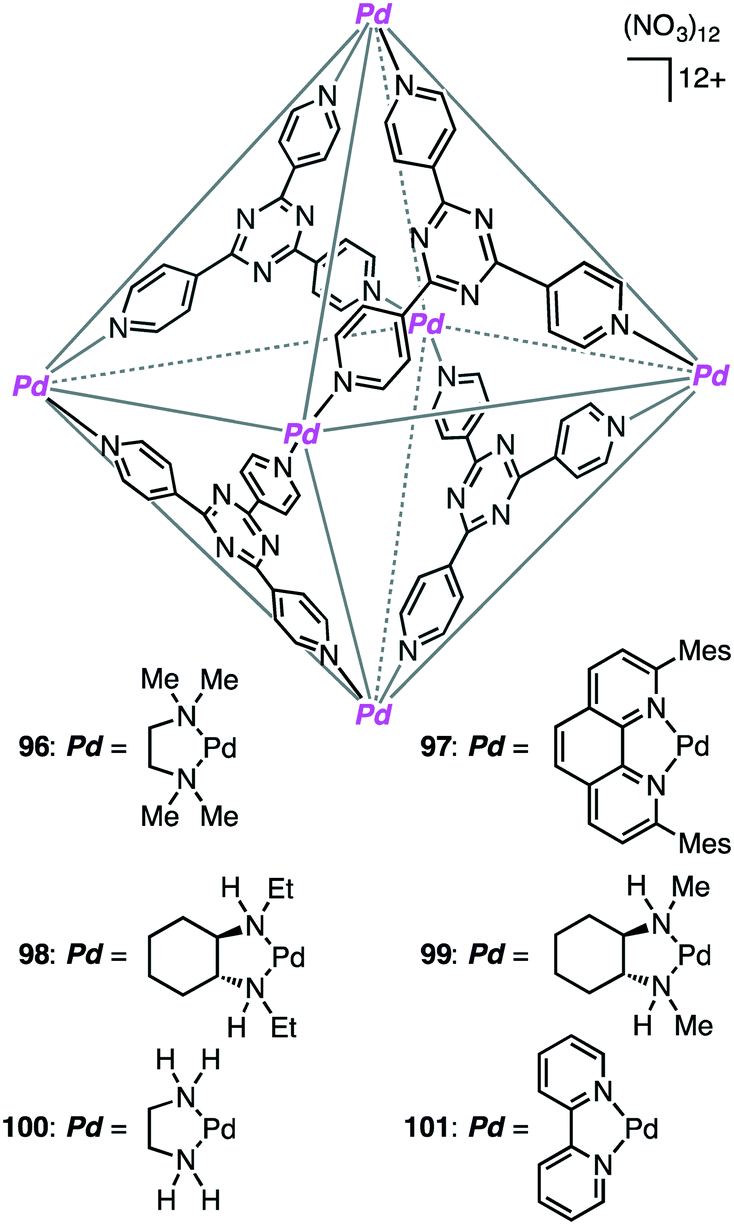
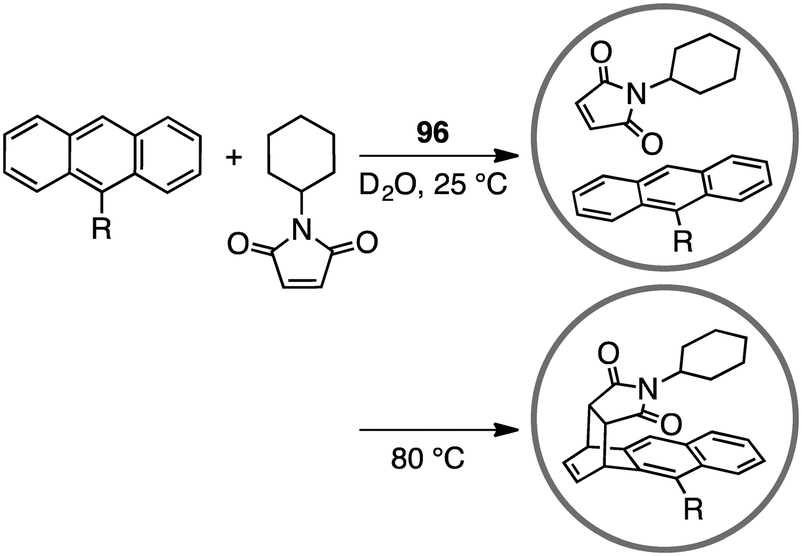
A variety of self-assembled supramolecular coordination complexes have been synthesized and used as catalysts for organic reactions, taking advantage of the vacant inner space, positive or negative charges, and peripheral catalytic functional groups.14–19,178,179 Fujita has done pioneering work, creating self-assembled supramolecular coordination complexes as exemplified in Fig. 8. Supramolecular complex 96 promoted the Diels–Alder reaction of anthracenes with N-cyclohexylmaleimide to furnish syn-adducts with unusual regioselectivity (Scheme 31).180 Although the formation of a 9,10-Diels–Alder adduct was favored in the absence of 96, the 1,4-regioselective Diels–Alder reaction was mediated by 96. This 1,4-regioselectivity results from steric restrictions inside 96. Unusual regioselectivity was also observed for the Diels–Alder reaction of alkyl-substituted naphthalenes with N-cyclohexylmaleimide in 96.181,182 No Diels–Alder reaction occurred in the absence of 96, clearly indicating the essential role of 96 as a molecular flask to bring the reactants into close proximity inside the cavity. The precise tuning of the cavity volume of the coordination cage as well as the substrate sizes can lead to the enhancement in reactivity. Indeed, 97 (cavity volume of 380 Å3) offered suitable space inside the cavity to promote the Diels–Alder reaction of unsubstituted naphthalene, which was inert in 96 (cavity volume of 462 Å3), with N-tert-butylmaleimide in D2O at 100 °C (46% yield).183
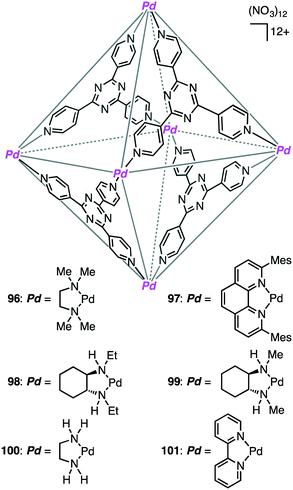 |
| | Fig. 8 Self-assembled supramolecular coordination complexes. | |
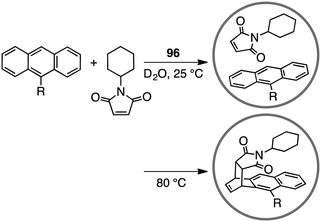 |
| | Scheme 31 Diels–Alder reaction. | |
4.2 [2 + 2] olefin cross photoaddition
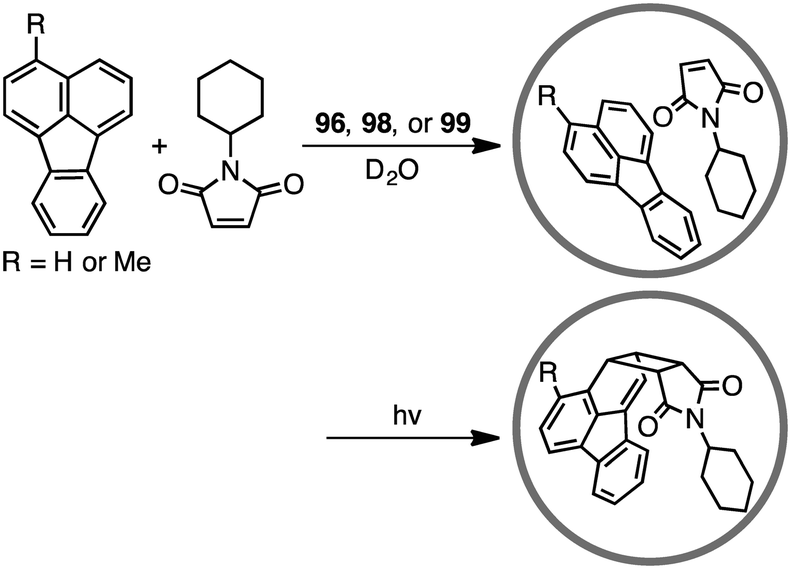
The first efficient [2 + 2] cross photoaddition reaction of fluoranthenes with N-cyclohexylmaleimide proceeded in the cavity of 96 to give the [2 + 2] adducts with high regioselectivity (Scheme 32).184,185 Interestingly, chiral cage 98 achieved an asymmetric induction of 40–50% ee, whereas chiral cage 99 resulted in an asymmetric induction of 20% ee. These results originate from the chiral deformation of the cavity induced by the different chiral diamino ligands located at the remote peripheral positions of the cage.
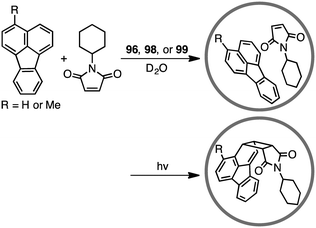 |
| | Scheme 32 [2 + 2] cross photoaddition. | |
4.3 Knoevenagel condensation
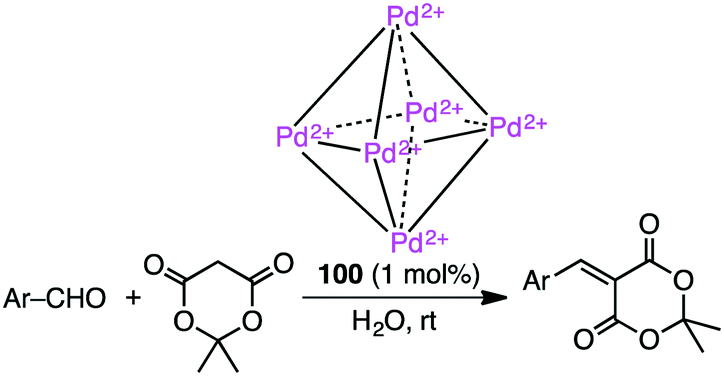
Knoevenagel condensation of aromatic aldehydes was accelerated in the hydrophobic cavity of cationic cage 100 in water under neutral conditions (Scheme 33).186 Anionic intermediates are effectively stabilized by the Pd2+ centers located at every corner of the cage. Owing to the host–guest size discrepancy, the product was spontaneously released from the cage, and the empty cavity allowed for further incorporation of a substrate molecule to continue the catalytic cycle. Only a small amount of 100 (1 mol%) was enough to produce the condensation product under mild conditions. Little or no condensation occurred in the absence of 100.
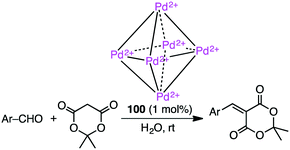 |
| | Scheme 33 Knoevenagel condensation. | |
Using different supramolecular coordination cages, Zhang, Lin, Wang, and co-workers investigated the substrate-dependent catalytic activity for the Knoevenagel condensation of malononitrile with various aldehydes.187 The reaction was switched on by the size/shape-selective substrate recognition and the electrostatic modulation of the binding pocket via cationic regulators.
4.4 Alkyne hydration
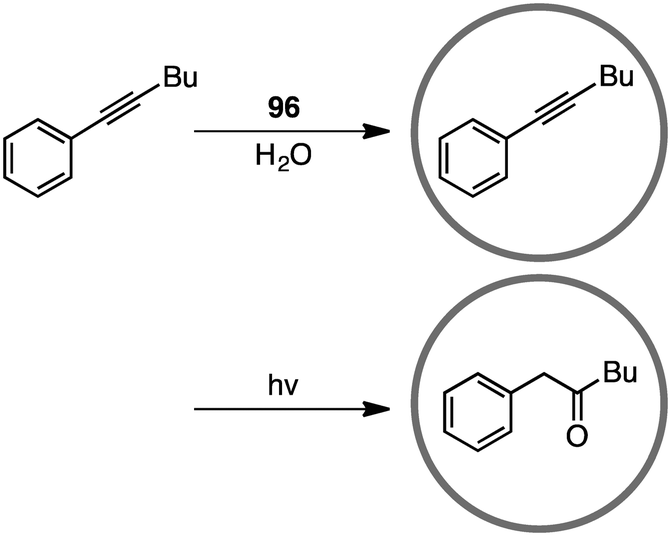
Coordination cage 96, which contains an electron-deficient triazine core and metal-coordinated pyridyl moieties, can act as a good electron acceptor as well as a molecular flask. Indeed, this cage served as a photosensitizing molecular flask, facilitating a cage-mediated guest-to-host electron transfer leading to new reactivity and selectivity (Scheme 34).188 Under UV-light irradiation, the photo-hydration of internal aryl alkynes occurred in the hydrophobic cavity of 96 to give benzyl ketones with anti-Markovnikov selectivity. After the formation of benzyl ketones, 96 acted as a protection chamber to suppress further photochemical reaction or degradation of the ketone.
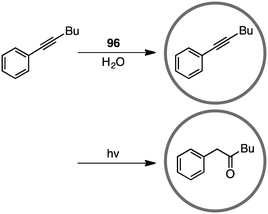 |
| | Scheme 34 Anti-Markovnikov hydration of alkyne. | |
4.5 Nucleophilic substitution
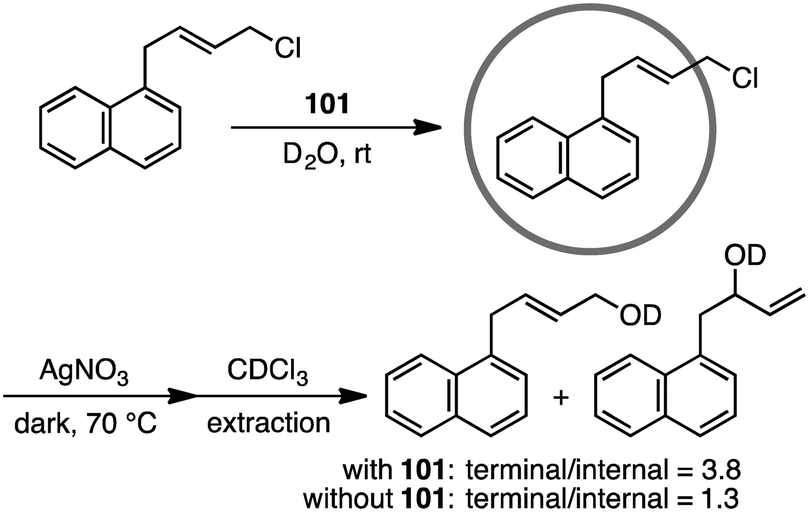
Coordination cage 101 non-covalently protected the internal reaction site of aryl-substituted allylic chlorides through encapsulation to control the regioselectivity of the nucleophilic substitution reaction (Scheme 35).189 The incoming nucleophile preferentially attacked the less protected terminal site of the allylic chlorides.
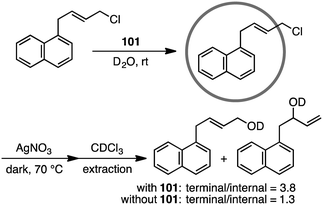 |
| | Scheme 35 Nucleophilic substitution. | |
4.6 Organometallic transformation
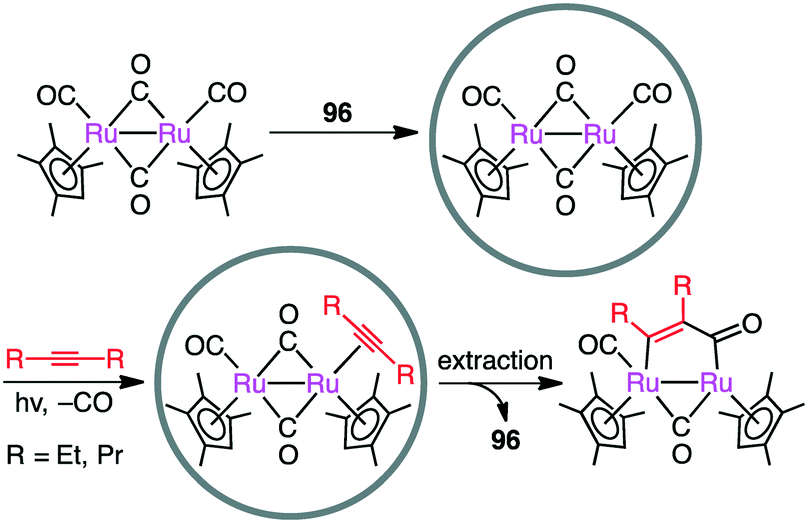
Metal–metal bonds are generally photolabile, which imposes some restriction in photochemical transformations. Fujita and co-workers demonstrated that the photocleavage of the Ru–Ru bond of a dinuclear Ru complex could be successfully suppressed by the tight encapsulation of the complex in 96 (Scheme 36).190 The remarkable photostability of [(Me4Cp)Ru(CO)2]2 by the cage effect enabled CO/alkyne photosubstitution to generate a Ru–alkyne π complex. This π complex was a metastable species, which was rearranged into diruthenacyclopentenone by intramolecular CO insertion upon extraction from the cage with CH2Cl2. Interestingly, sterically less demanding alkyne, 2-butyne, was directly converted into the corresponding diruthenacyclopentenone product in 96 probably because of the loose packing of the Ru–alkyne π complex intermediate in the cavity.
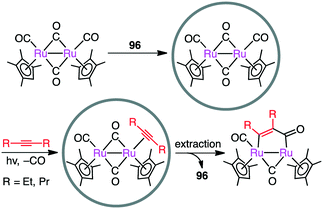 |
| | Scheme 36 Photosubstitution. | |
4.7 Oxidation/Diels–Alder cascade reaction
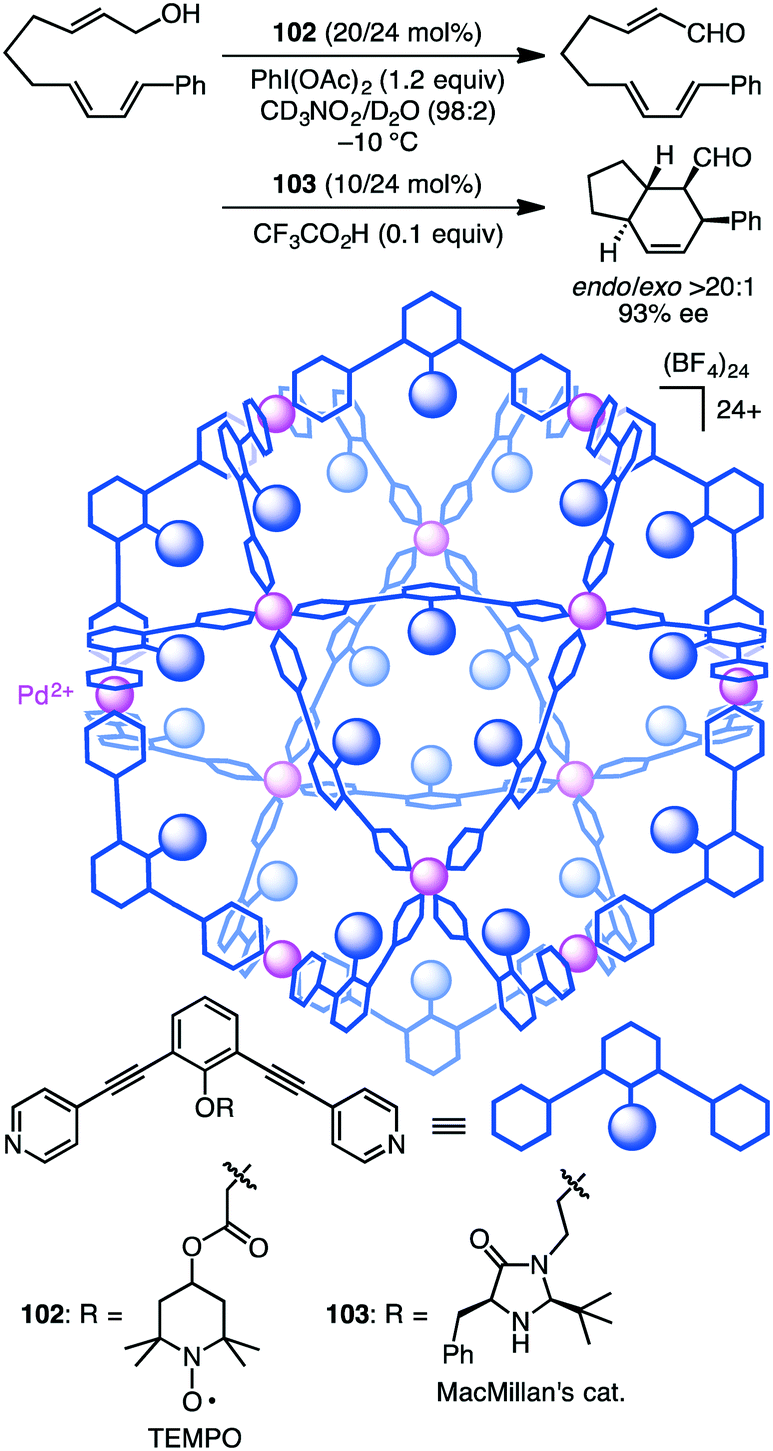
Fujita and co-workers developed supramolecular capsules [Pd12L24]24+102 and 103 bearing TEMPO and MacMillan's catalyst, respectively (Scheme 37).191 A solution of the substrate, 102, 103, PhI(OAc)2, and CF3CO2H in CD3NO2/D2O (98:2) was kept at −10 °C for 72 h. The two-step cascade reaction, allylic oxidation followed by the intramolecular asymmetric Diels–Alder reaction, proceeded successfully, providing a bicyclic product with high enantio- and diastereoselectivity. Control experiments indicated that only the combined use of 102 and 103 accelerated the cascade reaction, clearly demonstrating cooperative catalysis. It should be noted that the M12L24 spherical framework protected both of the active sites, TEMPO and MacMillan's catalyst, from undesirable deactivation, realizing the smooth one-pot cascade reaction (site-isolation strategy).
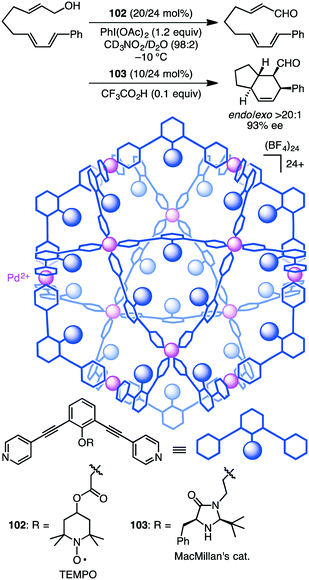 |
| | Scheme 37 Cascade reaction. | |
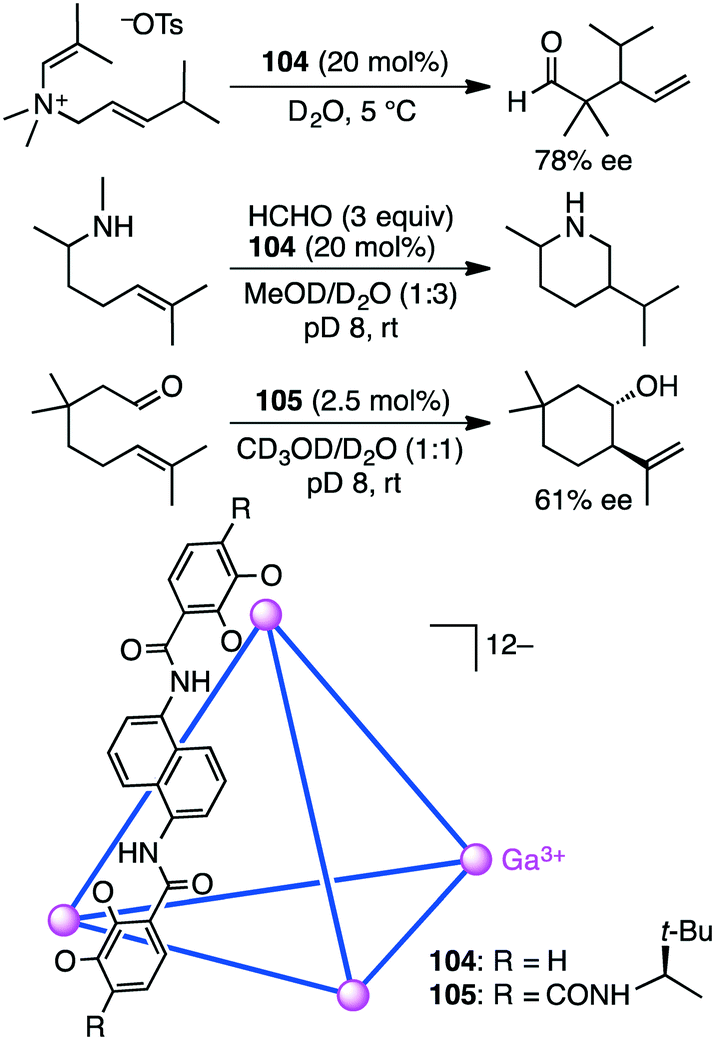
4.8 Aza-Cope rearrangement and aza-Prins and Prins cyclizations
Bergman, Raymond, and co-workers synthesized and evaluated supramolecular tetrahedral cage [Ga4L6]12−104 as a chiral catalyst for the enantioselective aza-Cope rearrangement (Scheme 38).192 A catalytic amount of enantiopure ΔΔΔΔ-104 was used to induce the reactivity of a series of enammonium tosylates. The aza-Cope rearrangement product, the iminium ion, was hydrolyzed to neutral aldehydes with up to 78% ee. The enammonium substrate is considered to be tightly encapsulated in the cage to adopt a chiral reactive conformation. The enantioselectivities were found to depend on the size and shape of the substrate molecules as well as on the reaction temperature.
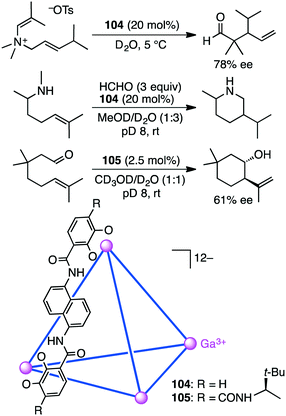 |
| | Scheme 38 Aza-Cope rearrangement and aza-Prins and Prins cyclizations. | |
They also reported the bimolecular aza-Prins cyclization of amines with formaldehyde via the formation of an iminium intermediate in the cavity of 104, which was followed by an unexpected 1,5-hydride transfer (Scheme 38).193 The piperidine products with no methyl group on the nitrogen atom were obtained; they are unusual as compared to the products in bulk solution without 104. Deuterium labeling experiments strongly suggested that the aza-Prins cyclization product underwent 1,5-hydride transfer from the N-methyl group to the tertiary carbocation. This reactivity is the result of the constrictive binding of the intermediate in 104 that favors a more compact transition state. The carbocation of the intermediate is preorganized in close proximity to the N-methyl C–H bonds, which facilitates a 1,5-hydride transfer.
Synthesis of enantiopure supramolecular tetrahedral cage [Ga4L6]12−104 is challenging because a racemic mixture of 104 is usually formed. Toste, Bergman, Raymond, and co-workers solved this problem by using a ligand with additional chiral amide groups; enantiopure cluster K12Ga4L6105 was obtained as a single diastereomer, which was strongly controlled by the chiral terephthalamide-based ligand (Scheme 38).194105 was stable in both the solid and solution states at elevated temperature and in aerobic D2O, in contrast to 104 that was sensitive to temperature and oxidation. The additional amide group of 105 stabilized the assembly via intramolecular hydrogen bonding with the catecholate oxygen atom, preventing the catecholate ligand from being oxidized and decomposed. ΔΔΔΔ-105 presented a rare example of the chiral host-catalyzed enantioselective Prins cyclization of carbonyl–ene compounds, and 105 displayed better catalytic performance (catalyst loading of 2.5 mol%) than 104 (10 mol%).195
4.9 Cross coupling
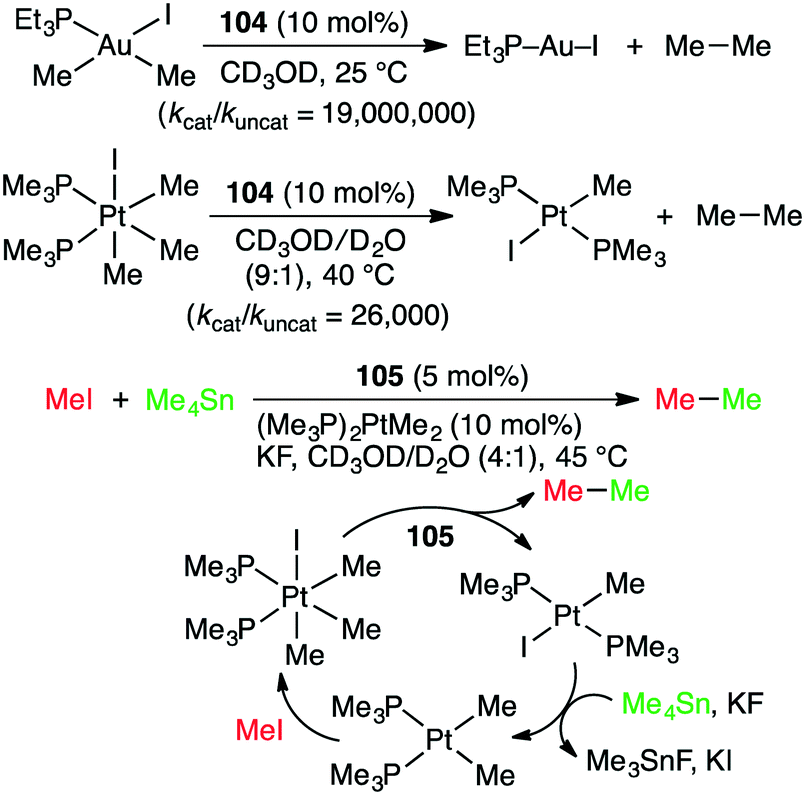
Alkyl–alkyl cross coupling processes often suffer from poor turnover and undesired side reactions because of sluggish reductive elimination of sp3 fragments from transition metals. Bergman, Raymond, Toste, and co-workers reported that the alkyl–alkyl reductive elimination from high-valent transition metal complexes was greatly accelerated by the supramolecular microenvironment strategy (Scheme 39).196 The reductive elimination of ethane from Au(III) and Pt(IV) complexes in the presence of 104 (10 mol%) was apparently 80000-fold and 2300-fold faster, respectively, than the uncatalyzed reactions. The kcat/kuncat values determined by kinetic measurements were 19000000 and 26000 for Au(III) and Pt(IV) complexes, respectively. Furthermore, when supramolecular capsule 105 and (Me3P)2PtMe2 were used as dual catalysts for the reaction of MeI with Me4Sn, the alkyl–alkyl cross coupling reaction proceeded to give ethane (Scheme 39). Both catalysts were essential for this reaction, but the role of 105 in the acceleration of the reductive elimination is quite important for the catalytic turnover. The same group also studied the oxidative addition of aryl halides that was facilitated by 104.197 The microenvironment inside the cage of 104 was useful for the encapsulation of guest molecules, increasing the local substrate concentration, and for the stabilization of cationic organometallic intermediate.
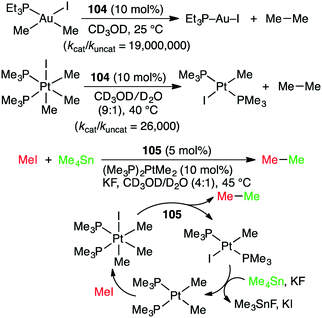 |
| | Scheme 39 Acceleration of alkyl–alkyl reductive elimination. | |
4.10 Epoxidation
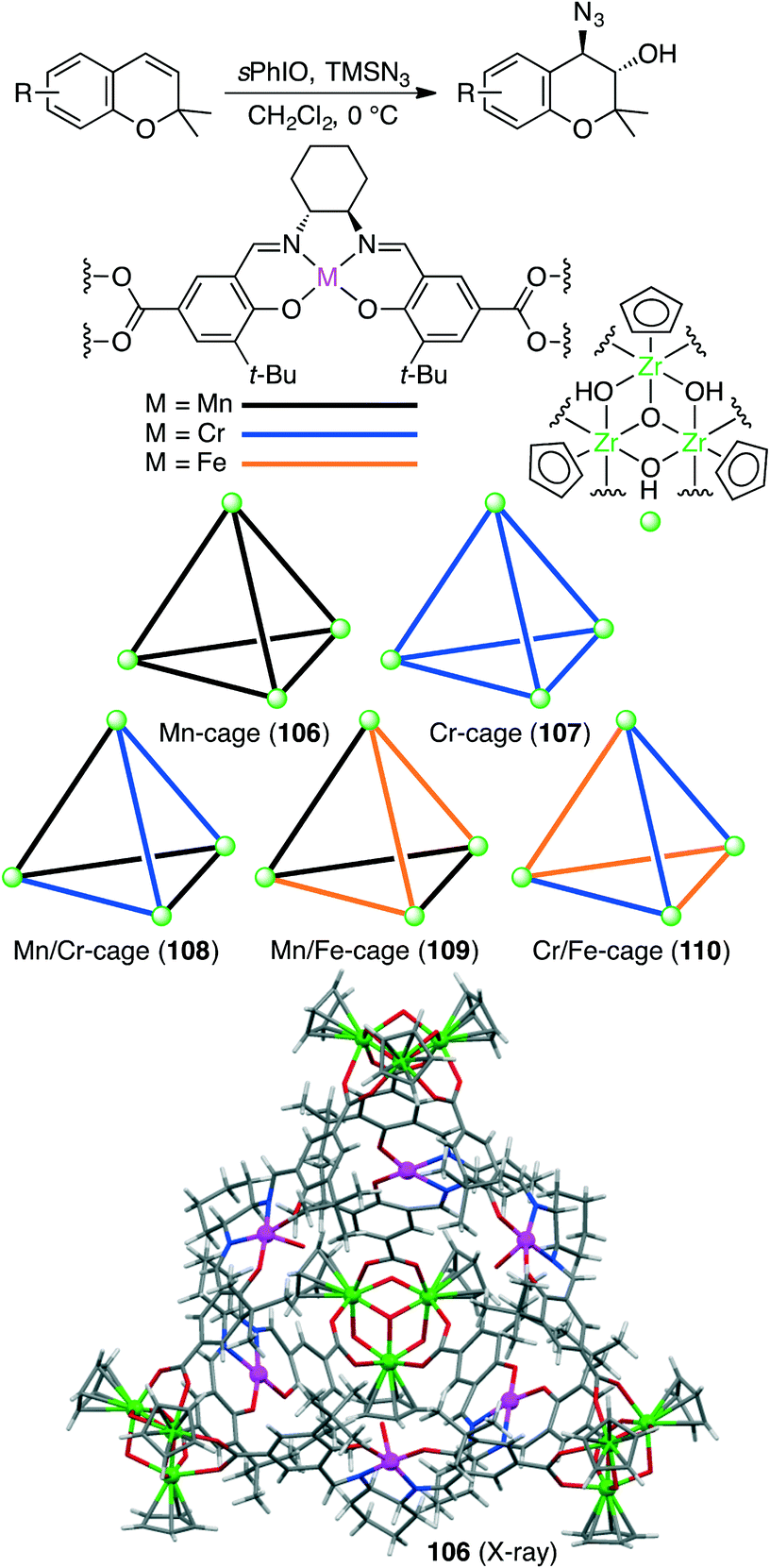
Liu, Cui, and co-workers constructed five chiral single- or mixed-linker tetrahedral coordination cages 106–110 to achieve the sequential asymmetric epoxidation/epoxide ring-opening reaction (Scheme 40).198 Four bowl-like Cp3Zr3(μ3-O)(μ2-OH)3 clusters located in the vertices are linked by six dicarboxylate ligands of Mn(salen), Cr(salen), and/or Fe(salen) to form a cationic cage {[Cp3Zr3(μ3-O)(μ2-OH)3]4(ML)6}Cl6. Mn/Cr mixed-linker cage 108 (0.2 mol%) catalyzed the sequential reaction in the presence of 2-(tert-butylsulfonyl)iodosylbenzene (sPhIO) and TMSN3 in CH2Cl2 at 0 °C; Mn(salen) and Cr(salen) moieties acted as catalysts for alkene epoxidation and epoxide-ring opening, respectively. In contrast, each of the single-linker cages 106 and 107 exhibited no catalytic activity. Interestingly, Mn/Cr cage 108 showed higher activity and enantioselectivity than a mixture of two catalytic components. The increased stability of the metallosalen units, the increased reactant concentration, and the efficient sequential reaction in the single cavity led to the high catalytic efficacy of 108.
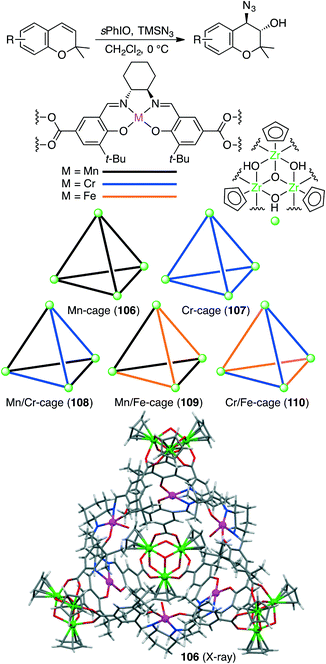 |
| | Scheme 40 Cascade reaction. | |
Bruin and co-workers synthesized a self-assembled cubic molecular cage with an encapsulated manganese porphyrin catalyst and conducted the epoxidation of styrene derivatives in 50% acetonitrile aqueous media. The epoxide products were obtained with a TON of up to 319, and substrate size-selectivity was observed.199 The cage not only acted as a phase-transfer catalyst but also improved the stability of the encapsulated metalloporphyrin catalyst.
4.11 Kemp elimination
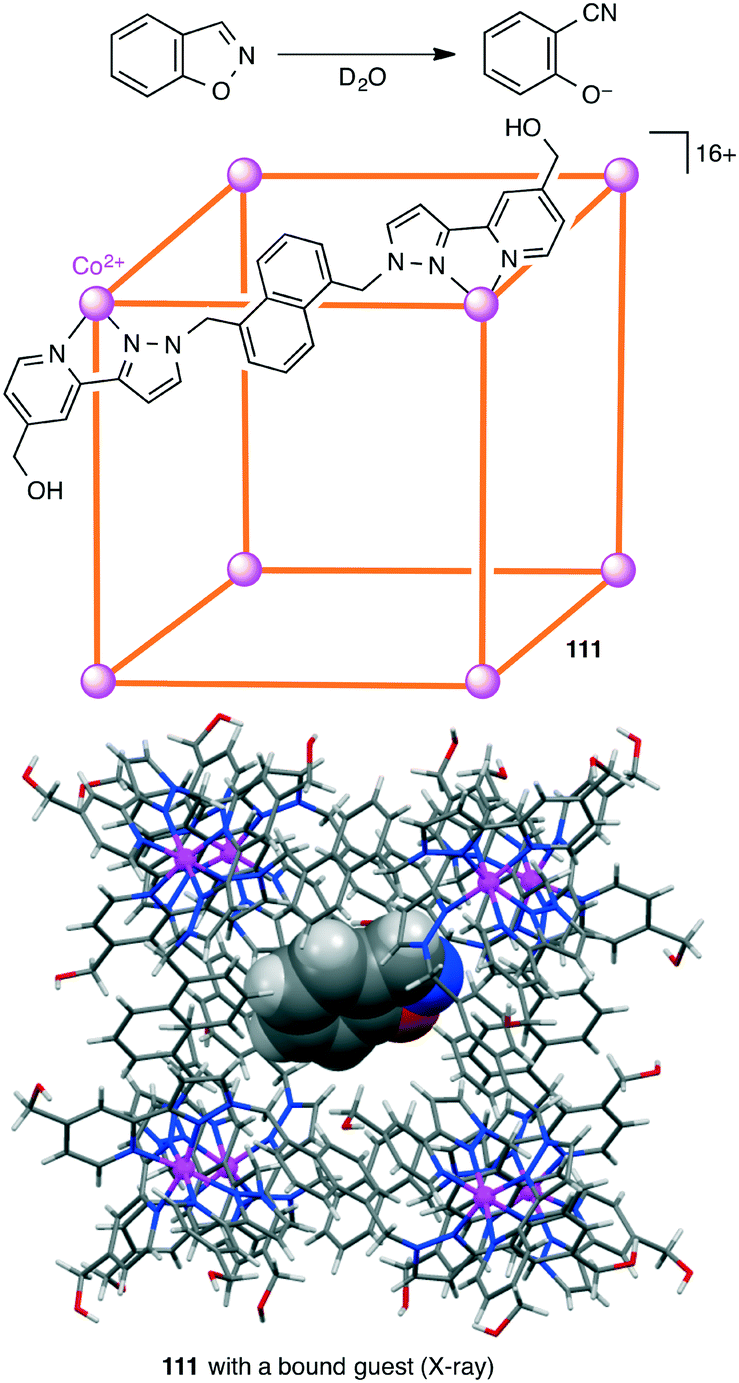
The catalytic efficiency of a supramolecular cage catalyst depends on the ability to recognize and bind substrate molecules and to accelerate the reaction by increasing the local reactant concentrations and/or stabilizing the transition state. In addition, the ability to expel the product is also important for a high catalytic turnover. Hunter, Williams, Ward, and co-workers studied the Kemp elimination catalyzed by cubic coordination cage [Co8L12]16+111 (Scheme 41).200 Deprotonation of benzisoxazole at the 3-position with hydroxide results in ring-opening to generate a 2-cyanophenolate anion. Hydrophobic binding of benzisoxazole in the cavity and polar binding of hydroxide ions at the cage surface accelerated the reaction with a rate enhancement (kcat/kuncat) of 200000 at pD 8.5. The poor binding of the anionic product in the hydrophobic cage allowed easy ejection from the cage, providing a smooth catalytic turnover. The effect of different surface-bound anions on the reactivity of the cavity-bound benzisoxazole was also reported.201
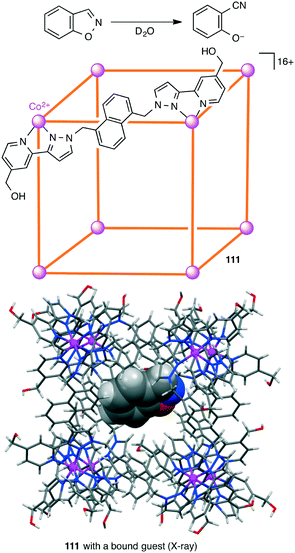 |
| | Scheme 41 Kemp elimination reaction. | |
4.12 Hydroformylation reaction
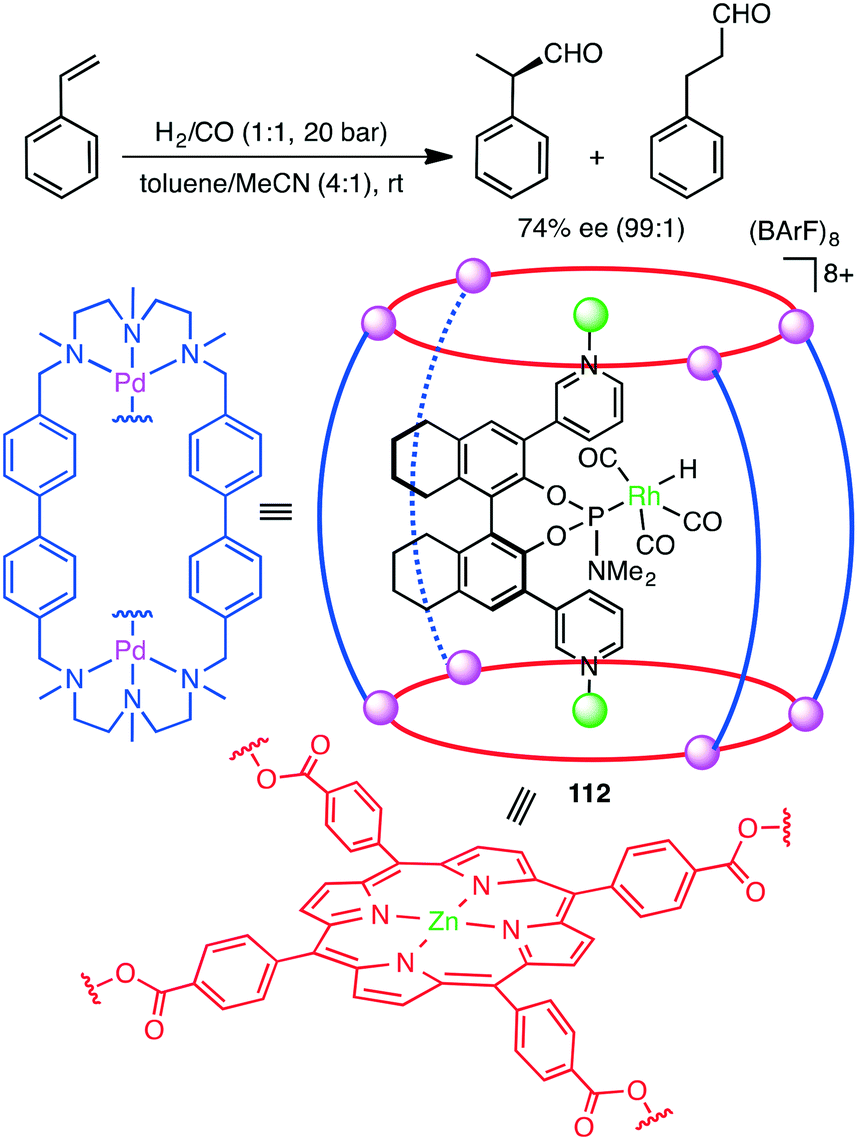
Encapsulation of a transition metal complex in a self-assembled coordination cage may form an efficient supramolecular catalyst. Costas, Ribas, Reek, and co-workers constructed elegant supramolecular multi-component catalyst 112in situ (Scheme 42).202 The chiral Rh complex was encapsulated in a supramolecular metallocage, the latter of which was constructed by the self-assembly of the macrocyclic Pd complex and Zn porphyrin complex. The Rh catalyst entrapped in the cage showed much higher regio- and enantioselectivity in the hydroformylation of styrenes than the non-encapsulated one. The supramolecular cage acted as a second coordination sphere, enabling a through-space chirality control around the catalyst center. The Reek group also achieved the size-selective hydroformylation of terminal alkenes with a Rh catalyst encapsulated in the cavity of a supramolecular tetrahedral cage.203
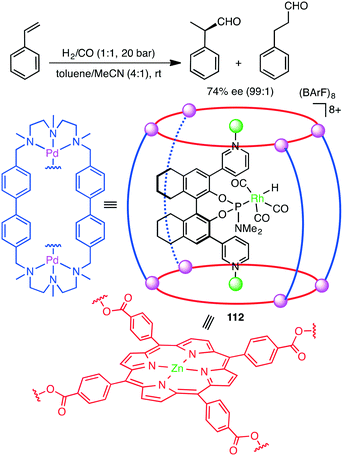 |
| | Scheme 42 Hydroformylation. | |
4.13 Michael addition
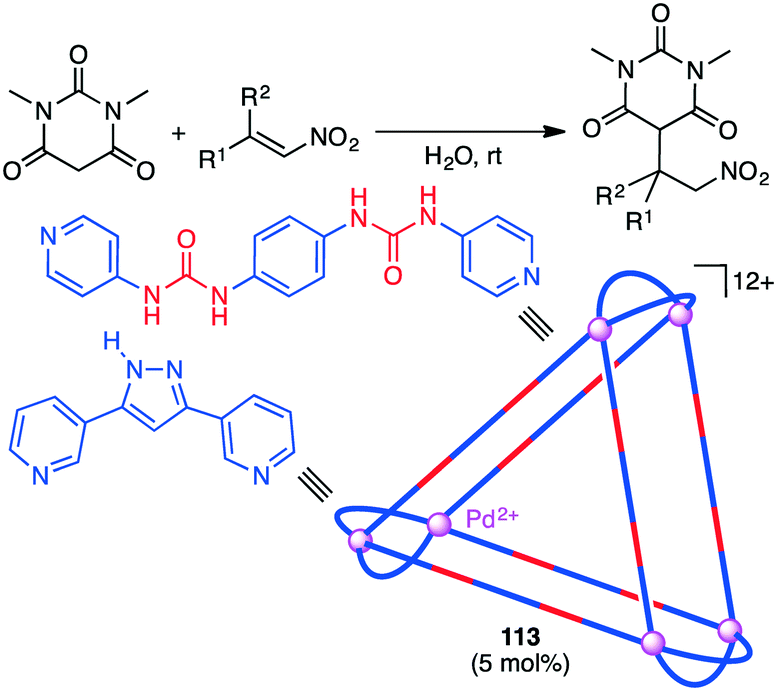
A substrate molecule can be bound with a supramolecular catalyst via hydrogen bonding. Mukherjee and co-workers reported that supramolecular trigonal prism 113 with multiple urea groups acted as a catalyst for the Michael addition reaction of nitro-olefins with dimethylbarbituric acid in aqueous medium (Scheme 43).204 Water-insoluble nitro-olefins that are hydrogen-bonded with the urea moiety of 113 undergoes the conjugate addition of the enolate form of dimethylbarbituric acid to generate a water-soluble nitronate intermediate, which is spontaneously released from 113 upon protonation because of the large size of the product.
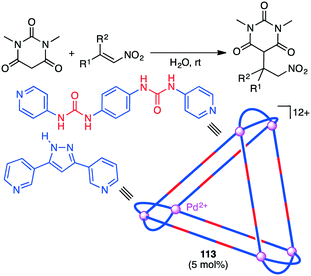 |
| | Scheme 43 Michael addition. | |
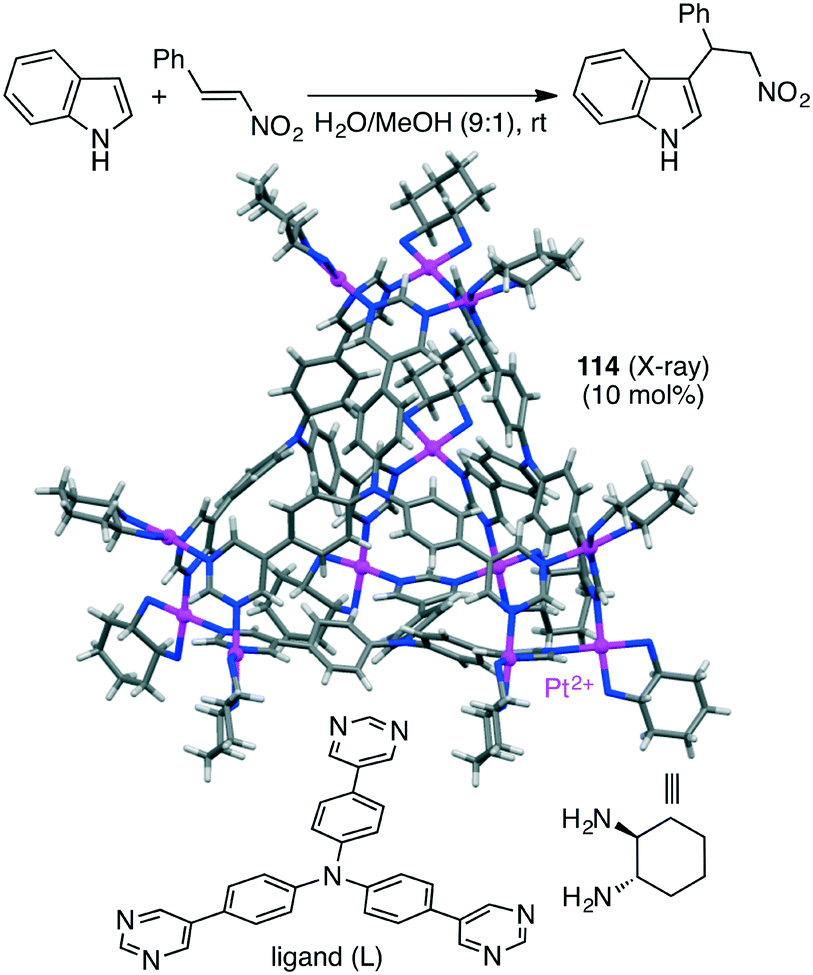
The Mukherjee group also reported self-assembled tetrahedral cage Pt12L4114 capable of accelerating the Michael addition of indole with nitrostyrenes (Scheme 44).205 The substrates can be stabilized by π–π interactions with the cage walls, and β-nitrostyrene can be activated by electrostatic interactions with the cationic charge at the cage vertices, both of which accelerated the electrophilic substitution on indole to give the Michael adduct.
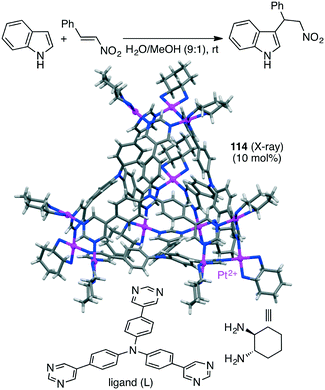 |
| | Scheme 44 Michael addition. | |
5. Conclusions and outlook
In this perspective, we provide an overview of the recent progress in homogeneous catalysis with macrocyclic multinuclear metal complexes: (i) multinuclear metal complexes with a covalently-linked macrocyclic ligand (category A), (ii) multinuclear metal complexes forming a macrocyclic skeleton (category B), and (iii) self-assembled supramolecular coordination complexes (category C). Useful organic reactions are catalyzed by these beautiful coordination systems. A variety of metal clusters can be formed in well-designed macrocyclic polydentate ligands, while metal coordinations can be used to construct macrocyclic frameworks and metal clusters at the same time. It has been demonstrated that the macrocyclic skeletons are useful for the construction, protection, and activation of metal clusters. Both homometallic and heterometallic clusters inside the macrocycles show excellent catalytic activity originating from the synergetic effect. On the other hand, self-assembled supramolecular coordination complexes with a hollow cavity can be constructed by the self-assembly of metal ions and polyvalent ligands. Some of the cage complexes are positively or negatively charged by an unbalanced combination of metal cations and anionic or neutral ligands. The net charge and the hydrophobic cavity can attract substrates effectively, and the specific cavity is useful for reaction acceleration and selectivity. The design flexibility of all these types of complexes will enable further success in catalytic applications. In many cases, the macrocyclic multinuclear metal complexes featured herein exerted remarkable and/or unusual catalytic performances that the corresponding non-macrocyclic/monometallic counterparts could not achieve, which is well rationalized by the proposed mechanisms; for example, see Schemes 2, 15, 17, 31, 36 and 39 and Fig. 6. Creation of artificial catalysts that are superior to enzymes containing metal clusters in terms of catalytic efficiency, reaction diversity, selectivity, substrate scope, and robustness will be one of the most challenging and fascinating goals. There are many enzymes and proteins containing an unstable multiple-metal cluster, for which even model systems are difficult to synthesize. A collection of the macrocyclic multinuclear metal complex systems shown here will be useful for a broad range of scientific studies, giving hints or clues to the creation of a high level of coordination systems for innovative breakthroughs.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- C.-Y. Lin and P. P. Power, Chem. Soc. Rev., 2017, 46, 5347–5399 RSC.
- G. Tseberlidis, D. Intrieri and A. Caselli, Eur. J. Inorg. Chem., 2017, 3589–3603 CrossRef CAS.
- D. S. Nesterov, O. V. Nesterova and A. J. L. Pombeiro, Coord. Chem. Rev., 2018, 355, 199–222 CrossRef CAS.
- E. Y. Tsui, J. S. Kanady and T. Agapie, Inorg. Chem., 2013, 52, 13833–13848 CrossRef CAS PubMed.
- T. Cadenbach, J. R. Pankhurst, T. A. Hofmann, M. Curcio, P. L. Arnold and J. B. Love, Organometallics, 2015, 34, 2608–2613 CrossRef CAS.
- K. Omoto, S. Tashiro, M. Kuritani and M. Shionoya, J. Am. Chem. Soc., 2014, 136, 17946–17949 CrossRef CAS.
- T. Nakamura, Y. Kaneko, E. Nishibori and T. Nabeshima, Nat. Commun., 2017, 8, 129 CrossRef.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS.
- C. Chen, C. Zhang, H. Dong and J. Zhao, Chem. Commun., 2014, 50, 9263–9265 RSC.
- C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS.
- C. Chen, Y. Chen, R. Yao, Y. Li and C. Zhang, Angew. Chem., Int. Ed., 2019, 58, 3939–3942 CrossRef CAS.
- M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS.
- M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS.
- C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Acc. Chem. Res., 2018, 51, 2447–2455 CrossRef CAS.
- C. Tan, D. Chu, X. Tang, Y. Liu, W. Xuan and Y. Cui, Chem. – Eur. J., 2019, 25, 662–672 CrossRef CAS.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef.
- S. Kitagawa, Acc. Chem. Res., 2017, 50, 514–516 CrossRef CAS.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- R. Gaillac, P. Pullumbi, K. A. Beyer, K. W. Chapman, D. A. Keen, T. D. Bennett and F.-X. Coudert, Nat. Mater., 2017, 16, 1149–1154 CrossRef CAS.
- H.-C. Hu and B. Zhao, Chem. – Eur. J., 2018, 24, 16702–16707 CrossRef CAS.
- M. J. Kalmutzki, C. S. Diercks and O. M. Yaghi, Adv. Mater., 2018, 30, 1704304 CrossRef PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- A. Schoedel, Z. Ji and O. M. Yaghi, Nat. Energy, 2016, 1, 16034 CrossRef CAS.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ACS Catal., 2017, 7, 2896–2919 CrossRef CAS.
- Y.-B. Huang, J. Liang, X.-S. Wang and R. Cao, Chem. Soc. Rev., 2017, 46, 126–157 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- F. N. Al-Rowaili, A. Jamal, M. S. Ba Shammakh and A. Rana, ACS Sustainable Chem. Eng., 2018, 6, 15895–15914 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri, M. Alvaro and H. Garcia, Green Chem., 2018, 20, 86–107 RSC.
- A. Dhakshinamoorthy, Z. Li and H. Garcia, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ACS Catal., 2019, 9, 1081–1102 CrossRef CAS.
- M. Liu, J. Wu and H. Hou, Chem. – Eur. J., 2019, 25, 2935–2948 CAS.
- S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166–11194 CrossRef CAS PubMed.
- J.-F. Ayme, J. E. Beves, C. J. Campbell and D. A. Leigh, Chem. Soc. Rev., 2013, 42, 1700–1712 RSC.
- M. Marenda, E. Orlandini and C. Micheletti, Nat. Commun., 2018, 9, 3051 CrossRef PubMed.
- F. B. L. Cougnon, K. Caprice, M. Pupier, A. Bauzá and A. Frontera, J. Am. Chem. Soc., 2018, 140, 12442–12450 CrossRef CAS PubMed.
- N. C. H. Lim and S. E. Jackson, J. Phys.: Condens. Matter, 2015, 27, 354101 CrossRef.
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS.
- T. Shima, Y. Luo, T. Stewart, R. Bau, G. J. McIntyre, S. A. Mason and Z. Hou, Nat. Chem., 2011, 3, 814–820 CrossRef CAS.
- Y. Li, Y. Li, B. Wang, Y. Luo, D. Yang, P. Tong, J. Zhao, L. Luo, Y. Zhou, S. Chen, F. Cheng and J. Qu, Nat. Chem., 2013, 5, 320–326 CrossRef CAS.
- T. Shima, S. Hu, G. Luo, X. Kang, Y. Luo and Z. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS.
- S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS.
- K. Wang, G. Luo, J. Hong, X. Zhou, L. Weng, Y. Luo and L. Zhang, Angew. Chem., Int. Ed., 2014, 53, 1053–1056 CrossRef CAS.
- G. Luo, Y. Luo, Z. Hou and J. Qu, Organometallics, 2016, 35, 778–784 CrossRef CAS.
- E. J. L. McInnes, G. A. Timco, G. F. S. Whitehead and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2015, 54, 14244–14269 CrossRef CAS.
- S. Castellanos, F. Kapteijn and J. Gascon, CrystEngComm, 2016, 18, 4006–4012 RSC.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275–11288 RSC.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, Chem. Soc. Rev., 2015, 44, 1922–1947 RSC.
- S. Subudhi, D. Rath and K. M. Parida, Catal. Sci. Technol., 2018, 8, 679–696 RSC.
- T. Joshi, B. Graham and L. Spiccia, Acc. Chem. Res., 2015, 48, 2366–2379 CrossRef CAS PubMed.
- B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916–11919 CrossRef CAS PubMed.
- Z. Sun, M. Yang, Y. Ma and L. Li, Cryst. Growth Des., 2017, 17, 4326–4335 CrossRef CAS.
- E. A. Dolgopolova, A. M. Rice, C. R. Martin and N. B. Shustova, Chem. Soc. Rev., 2018, 47, 4710–4728 RSC.
- C. García-Simón, M. Garcia-Borràs, L. Gómez, T. Parella, S. Osuna, J. Juanhuix, I. Imaz, D. Maspoch, M. Costas and X. Ribas, Nat. Commun., 2014, 5, 5557 CrossRef.
- X. Li, B. Wang, Y. Cao, S. Zhao, H. Wang, X. Feng, J. Zhou and X. Ma, ACS Sustainable Chem. Eng., 2019, 7, 4548–4563 CrossRef CAS.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167–170 CrossRef CAS PubMed.
- K. S. Walton, Nat. Chem., 2014, 6, 277–278 CrossRef CAS PubMed.
- A. Cadiau, K. Adil, P. M. Bhatt, Y. Belmabkhout and M. Eddaoudi, Science, 2016, 353, 137–140 CrossRef CAS PubMed.
- H. Li, L. Li, R.-B. Lin, G. Ramirez, W. Zhou, R. Krishna, Z. Zhang, S. Xiang and B. Chen, ACS Sustainable Chem. Eng., 2019, 7, 4897–4902 CrossRef CAS.
- Y. Ye, Z. Ma, R.-B. Lin, R. Krishna, W. Zhou, Q. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2019, 141, 4130–4136 CrossRef CAS PubMed.
- W.-Y. Zhang, Y.-J. Lin, Y.-F. Han and G.-X. Jin, J. Am. Chem. Soc., 2016, 138, 10700–10707 CrossRef CAS.
- K. Fujie, K. Otsubo, R. Ikeda, T. Yamada and H. Kitagawa, Chem. Sci., 2015, 6, 4306–4310 RSC.
- M. Sadakiyo, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2014, 136, 13166–13169 CrossRef CAS PubMed.
- M. Sadakiyo, T. Yamada and H. Kitagawa, Inorg. Chem. Commun., 2016, 72, 138–140 CrossRef CAS.
- A. Casini, B. Woods and M. Wenzel, Inorg. Chem., 2017, 56, 14715–14729 CrossRef CAS PubMed.
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9–25 RSC.
- S. K. Samanta, D. Moncelet, V. Briken and L. Isaacs, J. Am. Chem. Soc., 2016, 138, 14488–14496 CrossRef CAS.
- J. Ren, H. W. Langmi, B. C. North and M. Mathe, Int. J. Energy Res., 2015, 39, 607–620 CrossRef CAS.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557–580 CAS.
- G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 2014, 13, 802–806 CrossRef CAS.
- M. Inukai, M. Tamura, S. Horike, M. Higuchi, S. Kitagawa and K. Nakamura, Angew. Chem., Int. Ed., 2018, 57, 8687–8690 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395–3403 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef.
- G. Fiorani, W. Guo and A. W. Kleij, Green Chem., 2015, 17, 1375–1389 RSC.
- B. Yu and L.-N. He, ChemSusChem, 2015, 8, 52–62 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- D. J. Darensbourg and S. J. Wilson, Green Chem., 2012, 14, 2665–2671 RSC.
- X.-B. Lu, W.-M. Ren and G.-P. Wu, Acc. Chem. Res., 2012, 45, 1721–1735 CrossRef CAS.
- N. Ikpo, J. C. Flogeras and F. M. Kerton, Dalton Trans., 2013, 42, 8998–9006 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- A. Buchard, F. Jutz, M. R. Kember, A. J. P. White, H. S. Rzepa and C. K. Williams, Macromolecules, 2012, 45, 6781–6795 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291–2298 CrossRef CAS.
- M. R. Kember, F. Jutz, A. Buchard, A. J. P. White and C. K. Williams, Chem. Sci., 2012, 3, 1245–1255 RSC.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- P. K. Saini, C. Romain and C. K. Williams, Chem. Commun., 2014, 50, 4164–4167 RSC.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- A. C. Deacy, C. B. Durr, J. A. Garden, A. J. P. White and C. K. Williams, Inorg. Chem., 2018, 57, 15575–15583 CrossRef CAS PubMed.
- M. W. Lehenmeier, C. Bruckmeier, S. Klaus, J. E. Dengler, P. Deglmann, A.-K. Ott and B. Rieger, Chem. – Eur. J., 2011, 17, 8858–8869 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem. – Eur. J., 2015, 21, 8148–8157 CrossRef CAS PubMed.
- S. Kissling, M. W. Lehenmeier, P. T. Altenbuchner, A. Kronast, M. Reiter, P. Deglmann, U. B. Seemann and B. Rieger, Chem. Commun., 2015, 51, 4579–4582 RSC.
- H. Nagae, R. Aoki, S. Akutagawa, J. Kleemann, R. Tagawa, T. Schindler, G. Choi, T. P. Spaniol, H. Tsurugi, J. Okuda and K. Mashima, Angew. Chem., Int. Ed., 2018, 57, 2492–2496 CrossRef CAS PubMed.
- L. Jin, Y. Huang, H. Jing, T. Chang and P. Yan, Tetrahedron: Asymmetry, 2008, 19, 1947–1953 CrossRef CAS.
- B. J. Cook, G. N. Di Francesco, K. A. Abboud and L. J. Murray, J. Am. Chem. Soc., 2018, 140, 5696–5700 CrossRef CAS PubMed.
- M. Rudolph, S. Dautz and E.-G. Jäger, J. Am. Chem. Soc., 2000, 122, 10821–10830 CrossRef CAS.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Science, 2010, 327, 313–315 CrossRef CAS PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- M. Winkler, C. Romain, M. A. R. Meier and C. K. Williams, Green Chem., 2015, 17, 300–306 RSC.
- R. C. Jeske, J. M. Rowley and G. W. Coates, Angew. Chem., Int. Ed., 2008, 47, 6041–6044 CrossRef CAS PubMed.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- J. Liu, Y. Li, J. Liu and Z. Li, Macromolecules, 2005, 38, 2559–2563 CrossRef CAS.
- Z. Chen, X. Zhao, X. Gong, D. Xu and Y. Ma, Macromolecules, 2017, 50, 6561–6568 CrossRef CAS.
- D. Takeuchi, Y. Chiba, S. Takano and K. Osakada, Angew. Chem., Int. Ed., 2013, 52, 12536–12540 CrossRef CAS PubMed.
- D. Takeuchi, Y. Chiba, S. Takano, H. Kurihara, M. Kobayashi and K. Osakada, Polym. Chem., 2017, 8, 5112–5119 RSC.
- D. Takeuchi, S. Takano, Y. Takeuchi and K. Osakada, Organometallics, 2014, 33, 5316–5323 CrossRef CAS.
- K. Tanaka, A. Asakura, T. Muraoka, P. Kalicki and Z. Urbanczyk-Lipkowska, New J. Chem., 2013, 37, 2851–2855 RSC.
- K. Tanaka and S. Hachiken, Tetrahedron Lett., 2008, 49, 2533–2536 CrossRef CAS.
- A. Gualandi, L. Cerisoli, H. Stoeckli-Evans and D. Savoia, J. Org. Chem., 2011, 76, 3399–3408 CrossRef CAS PubMed.
- D. Savoia, A. Gualandi and H. Stoeckli-Evans, Org. Biomol. Chem., 2010, 8, 3992–3996 RSC.
- H. Lönnberg, Org. Biomol. Chem., 2011, 9, 1687–1703 RSC.
- H. Korhonen, T. Koivusalo, S. Toivola and S. Mikkola, Org. Biomol. Chem., 2013, 11, 8324–8339 RSC.
- M. Zhao, H.-B. Wang, L.-N. Ji and Z.-W. Mao, Chem. Soc. Rev., 2013, 42, 8360–8375 RSC.
- C. I. Maxwell, N. J. Mosey and R. S. Brown, J. Am. Chem. Soc., 2013, 135, 17209–17222 CrossRef CAS PubMed.
- X. Zhang, X. Zheng, D. L. Phillips and C. Zhao, Dalton Trans., 2014, 43, 16289–16299 RSC.
- X. Zhang, Y. Zhu, X. Zheng, D. L. Phillips and C. Zhao, Inorg. Chem., 2014, 53, 3354–3361 CrossRef CAS PubMed.
- S. Anbu, M. Kandaswamy and B. Varghese, Dalton Trans., 2010, 39, 3823–3832 RSC.
- S. Anbu, S. Kamalraj, B. Varghese, J. Muthumary and M. Kandaswamy, Inorg. Chem., 2012, 51, 5580–5592 CrossRef CAS PubMed.
- C.-Y. Yu, H.-J. Chuang and B.-T. Ko, Catal. Sci. Technol., 2016, 6, 1779–1791 RSC.
- R. Zhang, L. Wang, C. Xu, H. Yang, W. Chen, G. Gao and W. Liu, Dalton Trans., 2018, 47, 7159–7165 RSC.
- M. V. Escárcega-Bobadilla, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2013, 19, 2641–2648 CrossRef PubMed.
- K. Takaishi, B. D. Nath, Y. Yamada, H. Kosugi and T. Ema, Angew. Chem., Int. Ed., 2019, 58, 9984–9988 CrossRef CAS PubMed.
- U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Nat. Commun., 2014, 5, 5883 CrossRef CAS PubMed.
- R. J. Eisenhart, L. J. Clouston and C. C. Lu, Acc. Chem. Res., 2015, 48, 2885–2894 CrossRef CAS PubMed.
- N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed.
- J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- G. Amenuvor, J. Darkwa and B. C. E. Makhubela, Catal. Sci. Technol., 2018, 8, 2370–2380 RSC.
- C. Rong, F. Wang, W. Li and M. Chen, Organometallics, 2017, 36, 4458–4464 CrossRef CAS.
- K. Griffiths, P. Kumar, G. R. Akien, N. F. Chilton, A. Abdul-Sada, G. J. Tizzard, S. J. Coles and G. E. Kostakis, Chem. Commun., 2016, 52, 7866–7869 RSC.
- P. Kumar, S. Lymperopoulou, K. Griffiths, S. I. Sampani and G. E. Kostakis, Catalysts, 2016, 6, 140 CrossRef.
- K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed.
- K. Griffiths, P. Kumar, J. D. Mattock, A. Abdul-Sada, M. B. Pitak, S. J. Coles, O. Navarro, A. Vargas and G. E. Kostakis, Inorg. Chem., 2016, 55, 6988–6994 CrossRef CAS PubMed.
- K. V. Vivekananda, S. Dey, D. K. Maity, N. Bhuvanesh and V. K. Jain, Inorg. Chem., 2015, 54, 10153–10162 CrossRef CAS PubMed.
- P. A. Mane, S. Dey and K. V. Vivekananda, Tetrahedron Lett., 2017, 58, 25–29 CrossRef CAS.
- K. Endo, M. Ogawa and T. Shibata, Angew. Chem., Int. Ed., 2010, 49, 2410–2413 CrossRef CAS PubMed.
- K. Endo, D. Hamada, S. Yakeishi, M. Ogawa and T. Shibata, Org. Lett., 2012, 14, 2342–2345 CrossRef CAS PubMed.
- K. Endo, K. Tanaka, M. Ogawa and T. Shibata, Org. Lett., 2011, 13, 868–871 CrossRef CAS PubMed.
- N. Kumagai, M. Kanai and H. Sasai, ACS Catal., 2016, 6, 4699–4709 CrossRef CAS.
- J. R. Robinson, J. Gu, P. J. Carroll, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2015, 137, 7135–7144 CrossRef CAS PubMed.
- T. Ohshima, Chem. Pharm. Bull., 2016, 64, 523–539 CrossRef CAS.
- D. Nakatake, Y. Yokote, Y. Matsushima, R. Yazaki and T. Ohshima, Green Chem., 2016, 18, 1524–1530 RSC.
- C. Redshaw and M. R. J. Elsegood, Angew. Chem., Int. Ed., 2007, 46, 7453–7457 CrossRef CAS PubMed.
- Y. F. Al-Khafaji, M. R. J. Elsegood, J. W. A. Frese and C. Redshaw, RSC Adv., 2017, 7, 4510–4517 RSC.
- Y. Fang, W. Gong, L. Liu, Y. Liu and Y. Cui, Inorg. Chem., 2016, 55, 10102–10105 CrossRef CAS PubMed.
- A. J. Tasiopoulos, P. L. Milligan, Jr., K. A. Abboud, T. A. O'Brien and G. Christou, Inorg. Chem., 2007, 46, 9678–9691 CrossRef CAS PubMed.
- G. Maayan and G. Christou, Inorg. Chem., 2011, 50, 7015–7021 CrossRef CAS PubMed.
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC.
- J.-H. Xu, L.-Y. Guo, H.-F. Su, X. Gao, X.-F. Wu, W.-G. Wang, C.-H. Tung and D. Sun, Inorg. Chem., 2017, 56, 1591–1598 CrossRef CAS PubMed.
- F. Evangelisti, R. Moré, F. Hodel, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2015, 137, 11076–11084 CrossRef CAS PubMed.
- F. Evangelisti, R. Güttinger, R. Moré, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2013, 135, 18734–18737 CrossRef CAS.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS.
- P. F. Smith, C. Kaplan, J. E. Sheats, D. M. Robinson, N. S. McCool, N. Mezle and G. C. Dismukes, Inorg. Chem., 2014, 53, 2113–2121 CrossRef CAS.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS PubMed.
- Y. Inokuma, M. Kawano and M. Fujita, Nat. Chem., 2011, 3, 349–358 CrossRef CAS PubMed.
- K. Harris, D. Fujita and M. Fujita, Chem. Commun., 2013, 49, 6703–6712 RSC.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- T. Murase, S. Horiuchi and M. Fujita, J. Am. Chem. Soc., 2010, 132, 2866–2867 CrossRef CAS PubMed.
- S. Horiuchi, T. Murase and M. Fujita, Chem. – Asian J., 2011, 6, 1839–1847 CrossRef CAS.
- Y. Fang, T. Murase and M. Fujita, Chem. Lett., 2015, 44, 1095–1097 CrossRef CAS.
- Y. Nishioka, T. Yamaguchi, M. Kawano and M. Fujita, J. Am. Chem. Soc., 2008, 130, 8160–8161 CrossRef CAS.
- S. Horiuchi, Y. Nishioka, T. Murase and M. Fujita, Chem. Commun., 2010, 46, 3460–3462 RSC.
- T. Murase, Y. Nishijima and M. Fujita, J. Am. Chem. Soc., 2012, 134, 162–164 CrossRef CAS PubMed.
- Y. Qiao, L. Zhang, J. Li, W. Lin and Z. Wang, Angew. Chem., Int. Ed., 2016, 55, 12778–12782 CrossRef CAS PubMed.
- T. Murase, H. Takezawa and M. Fujita, Chem. Commun., 2011, 47, 10960–10962 RSC.
- Y. Kohyama, T. Murase and M. Fujita, Chem. Commun., 2012, 48, 7811–7813 RSC.
- S. Horiuchi, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12029–12031 CrossRef CAS PubMed.
- Y. Ueda, H. Ito, D. Fujita and M. Fujita, J. Am. Chem. Soc., 2017, 139, 6090–6093 CrossRef CAS PubMed.
- C. J. Brown, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2009, 131, 17530–17531 CrossRef CAS.
- D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2015, 137, 9202–9205 CrossRef CAS PubMed.
- C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed.
- D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235–1238 CrossRef CAS.
- T. A. Bender, M. Morimoto, R. G. Bergman, K. N. Raymond and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 1701–1706 CrossRef CAS.
- J. Jiao, C. Tan, Z. Li, Y. Liu, X. Han and Y. Cui, J. Am. Chem. Soc., 2018, 140, 2251–2259 CrossRef CAS PubMed.
- P. F. Kuijpers, M. Otte, M. Dürr, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, ACS Catal., 2016, 6, 3106–3112 CrossRef CAS.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- W. Cullen, A. J. Metherell, A. B. Wragg, C. G. P. Taylor, N. H. Williams and M. D. Ward, J. Am. Chem. Soc., 2018, 140, 2821–2828 CrossRef CAS PubMed.
- C. García-Simón, R. Gramage-Doria, S. Raoufmoghaddam, T. Parella, M. Costas, X. Ribas and J. N. H. Reek, J. Am. Chem. Soc., 2015, 137, 2680–2687 CrossRef PubMed.
- S. S. Nurttila, W. Brenner, J. Mosquera, K. M. van Vliet, J. R. Nitschke and J. N. H. Reek, Chem. – Eur. J., 2019, 25, 609–620 CAS.
- P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS.
- I. A. Bhat, A. Devaraj, P. Howlader, K.-W. Chi and P. S. Mukherjee, Chem. Commun., 2018, 54, 4814–4817 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Tadashi
Ema
and
Tadashi
Ema