
Understanding the role of surface basic sites of catalysts in CO2 activation in dry reforming of methane: a short review
M. A. A.
Aziz
 *ab,
A. A.
Jalil
ab,
S.
Wongsakulphasatch
c and
Dai-Viet N.
Vo
d
*ab,
A. A.
Jalil
ab,
S.
Wongsakulphasatch
c and
Dai-Viet N.
Vo
d
aSchool of Chemical and Energy Engineering, Faculty of Engineering, Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor, Malaysia. E-mail: m.arif@utm.my
bCenter of Hydrogen Energy, Institute of Future Energy, Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor, Malaysia
cDepartment of Chemical Engineering, Faculty of Engineering, King Mongkut's University of Technology North Bangkok, 10800 Bangkok, Thailand
dCenter of Excellence for Green Energy and Environmental Nanomaterials (CE@GrEEN), Nguyen Tat Thanh University, 300A Nguyen Tat Thanh, District 4, Ho Chi Minh City 755414, Vietnam
First published on 13th November 2019
Abstract
The surface of basic sites in catalysts plays an important role in many catalytic applications, such as dry reforming of methane for generating renewable hydrogen energy. The reaction concept of methane dry reforming is initiated by an acid–base interaction, followed by catalytic cycles. For instance, the reactants of CO2 act as acids toward catalysts, which act as bases. The basic sites of catalysts are generated from a severe pre-treatment temperature by desorbing the surface species. The current consensus on the role of basic sites is to enhance the activation of acidic CO2 on the catalyst support's surface and to inhibit carbon deposition on the catalyst, thus enhancing the catalytic stability. This review elucidated the behaviour of surface basic sites towards CO2 activation in dry reforming of methane. The method for characterising the basicity of catalysts was also reviewed to strategically design catalysts, which could increase the catalytic activity.
 M. A. A. Aziz | Dr. Muhammad Arif Ab Aziz is a senior lecturer of Chemical Engineering at Universiti Teknologi Malaysia (UTM) in Skudai, Johor, Malaysia. He obtained his undergraduate degree in Chemical Engineering (2009) from UTM. He received his PhD in 2015 working on the valorization of CO2 over solid acid/base heterogeneous catalysts. His current research interests focus on the design and synthesis of advanced materials and heterogeneous catalysis relevant to clean environment and energy. |
 A. A. Jalil | Prof. Dr Aishah Abdul Jalil received Ph.D. degree in Molecular Chemistry from Hokkaido University, Japan. Since 1996, she has been serving as lecturer in the School of Chemical and Energy Engineering at Universiti Teknologi Malaysia. Her research interests are in the area of electrochemistry, organic chemistry, zeolitic and advanced materials, catalysis, photocatalysis and also adsorption. Her research has been published in more than 60 peer-reviewed articles in international/national journals. Currently, as one of the Center of Hydrogen Energy member, she is very active earnestly involved in teaching as well as in research with the sole intent to guide the upcoming generation towards a higher level of education. |
 S. Wongsakulphasatch | Dr. Suwimol Wongsakulphasatch earned her Ph.D. in Chemical Engineering & Analytical Science from the University of Manchester, UK. She is currently Assistant Professor at the Department of Chemical Engineering, King Mongkut’s University of Technology North Bangkok (KMUTNB), Thailand. Interest research areas are on gas adsorption by porous solids, H2 production technology, molecular thermodynamics, and colloid and interface science. |
 Dai-Viet N. Vo | Dr. Dai-Viet N. Vo is the Director of Center of Excellence for Green Energy and Environmental Nanomaterials, Nguyen Tat Thanh University, Vietnam. He received his Ph.D. in Chemical Engineering from The University of New South Wales (UNSW), Australia in 2011. He worked as a postdoctoral fellow at UNSW and Texas A&M University in Doha, Qatar before joining Universiti Malaysia Pahang, Malaysia as a senior lecturer in 2013. His research areas are synthetic fuel generation via reforming processes and Fischer–Tropsch synthesis. He is the assistant subject editor for the International Journal of Hydrogen Energy and Editorial Board Member of PLOS One and Scientific Reports. |
1. Introduction
Catalysis depends significantly on the active acid–base centre found in oxide catalysts.1–3 Acid and base-pairing concepts have been applied to a number of chemical reactions. The hydroxyl group on the metal oxide surface is amphoteric in nature (i.e. concomitant occurrence of both acidic and basic sites) and determines how basic and acidic the surface is. The overall OH content and amount of acidic and basic OH groups can be measured through a number of reactions. The OH groups are equally divided into acidic and primarily basic groups accordingly, with the potential for exchange with other anions according to empirical studies conducted by using anatase, rutile, η-alumina, α-Fe2O3, CeO2, and SnO2. The way in which the hydroxylated surface is structured elucidates this behaviour.4 It is possible for two centres to function either together or separately. For example, dehydration and dehydrogenation are respectively prioritised on acidic and basic sites in the context of alcohol transformation.5,6A solid base catalyst is defined as a material that acts as a base, which is favourable for an interaction with an acid reactant. A solid base catalyst is generally characterised by the exposure of a basic group on the catalyst surface. Mostly, basic sites can be activated by the removal of water or CO2 from the catalyst surface. Basicity is exhibited by numerous materials, such as single-metal oxides (MgO, TiO2, CaO, ZnO, etc.), supported alkali metals (Na/MgO, K/K2CO3, etc.), mixed-metal oxides (MgO–A12O3, ZnO–SiO2, MgO–TiO2, etc.), zeolites (X, USY, etc.), hydrotalcite-type anionic clay, asbestos-based materials, carbon-based materials, and anion exchange resin. Hydrogenation,7,8 transesterification,9,10 CO2 reforming11 and aldol condensation12 are among the wide-ranging catalysis applications in which base catalysts are employed. The reaction of CO2 reforming requires an interplay between an acid and base, which is why base catalysts have been slowly considered as suitable for this reaction. The reaction occurs between the acid CO2 and the basic surface sites within the catalyst. The significant contribution of weak and medium basic sites to the activation of CO2 on the catalyst is common knowledge. For example, as reported in ref. 13, the addition of La to a catalyst has made weak and medium basic sites stronger, leading to increased catalyst activity. In a different study, the integration of a Ba promoter having a greater basicity into a La/MnO3 catalyst for CO2 reforming of methane has been achieved via an appealing approach. The basic sites proliferate in number and become stronger as a result of Ba2+ introduction, as revealed by a CO2 temperature programmed desorption (TPD) study.14
The improvement of CO2 dissociative adsorption in the context of dry reforming of methane (DRM), enhancement of CO2 conversion, and reduction of coke deposition on the surface of catalysts is one of the contributions of basic supports. The basic character of Al2O3 and MgO increases the adsorption of CO2, which is probably the reason for their intensified activity. CO2-TPD has indicated that CO2 desorption for Al2O3 is 33.7 mol g−1, followed by MgO (12.8 mol g−1), ZrO2 (7.6 mol g−1), TiO2 (1.2 mol g−1), and SiO2 (2.1 mol g−1).15 CO2 adsorption is greater in Al2O3 overlayers than in other metal oxides. Furthermore, more potent CO2 adsorption has been suggested by the CO2-TPD results as the CO2 desorption peak is associated with MgO, which occurred at higher temperatures. The activity of ZrO2 and SiO2 has been found to be comparable. Although it does not seem to improve the catalytic activity, ZrO2 is perceived to facilitate the elimination of deposited carbon species as it possesses high oxygen mobility.
At present, basic sites are considered to contribute to the improvement of acidic CO2 chemisorption via a catalyst support and towards preventing carbon deposition on the catalyst, thus making the catalyst more stable. However, the knowledge remains limited with regard to the character of surface basic sites, especially the amount and strength they possess, as well as their involvement in dry reforming of methane. Hence, it can be helpful to establish the behaviour in which surface basic sites and the activity of dry reforming of methane are correlated.
CO2-TPD and in situ Fourier-transform infrared (FTIR) spectroscopy employing appropriate probe molecules are widely implemented for characterising the basic sites of solid catalysts in terms of qualitative and quantitative analyses. However, the present and main challenges in determining the basic sites on the catalyst surface are complex experimental procedures and the accurate interpretation of acquired spectra for various types, strengths, and concentrations of basic sites. Besides, other obstacles include the differentiated roles of supports and active metals in basic sites, and the minimised interference of gaseous environments in FTIR measurement. The establishment of a correlation between the basic sites of catalysts and their performance (i.e. reactant conversion, selectivity, and product yield) in the dry reforming of methane is also crucial to generate an advanced catalyst design and understand the intrinsic reaction mechanism and kinetic modelling.
The current work draws from earlier research to investigate the impact of surface basic sites on dry reforming of methane. To the best of our knowledge, the significance and/or contribution of basic sites to dry reforming of methane have not been thoroughly reviewed so far. Therefore, it is vital to review the recent study about the character of basic sites and their role in improving the reaction activity. In addition, to enable the creation of a catalyst that can improve the catalytic activity, this review encompasses the approach for customising catalyst basicity as well.
2. The nature of basic sites and their characterisation techniques
The process whereby the rate of a particular chemical reaction is improved by an electron-pair donor through an interplay with an acceptor donor in a reagent or substrate is known as Lewis base catalysis. Any substance (e.g. OH− ion) capable of donating a pair of electrons without bonding represents a Lewis base, which can thus be considered as an electron-pair donor. Essentially constituting solid base catalysts, metal oxides uncover O atoms on the surfaces; all O atoms are capable of H+ abstraction from molecules with high acidity.16Pre-treatment, where pure H2 is used for in situ reduction of the catalyst bed at a high temperature, can facilitate the activation of the basic character for solid base catalysts. The elimination of water and CO2 from the catalyst surface is the goal of reduction pre-treatment. This suggests that a catalyst will become more basic as the temperature of treatment increases. Pre-treatment results in the reduction of metal oxide to metal, which enhances the rate of methane decomposition. Moreover, the creation of oxygen vacancies due to reduction facilitates the activation of CO2 and C–O bond cleavage. Such vacancies can induce the oxygen to move from the lattice to the surface and oxidise carbon deposits towards forming CO and carbonate intermediate substances. In contrast, the vacant sites created are reserved for CO2 activation.17,18 At present, the correlation between basic sites and oxygen vacancies is still unclear although the basic oxygen in the catalyst's framework may exhibit the oxygen vacancy's character.19,20 A study of TiO2 rutile (110) with ceria nanoclusters by density functional theory (DFT) has shown that CO2 interaction is stronger at oxygen vacancy sites. That was evidenced by higher formation of energy which indicates the most stable oxygen vacancy for CO2 adsorption at that site.21 In contrast, a study of CO2 adsorption on an In2O3(110) system has shown that CO2 is preferably adsorbed on CO2− configuration on hydroxylated In2O3(110) rather than in oxygen vacancy sites, indicating that the oxygen vacancy sites does not facilitate CO2 adsorption.20 Albeit, the role of oxygen vacancies is still under debate, and the role of surface basic sites in CO2 adsorption and/or activation is well documented in the literature.22,23
In Fig. 1A, the character and strength of the surface basic sites are determined by how severe the pre-treatment conditions are.24 The production of basic sites of CaO catalysts through the elimination of surface CO2 and water at high temperatures is in line with 1-butene isomerisation activity. Pre-treatment also involves the reorganisation of surface and bulk atoms, which elevates the temperature required for the process and thus alters the count and character of basic sites.24,25 Meanwhile, catalysts can be rendered more basic by microwave heating.26 The impact of pre-treatment temperature on the reaction has been examined in many studies in the context of dry reforming of methane.27 The study by Perez-Lopez has shown that the CO2 conversion in dry reforming of methane is significantly affected by the reduction temperature (i.e. treatment at 500 °C, 600 °C and 700 °C) on Ni–Mg–Al catalysts.27 As depicted in Fig. 1B, the result reveals that a higher reduction temperature is associated with better catalytic activity. Recently, Zhang et al. have observed different distributions of oxygen species over the surface of a ZrO2 support.28 The experiment is conducted by treatment under H2 (denoted as ZRH), N2 (denoted as ZRN), and O2 (denoted as ZRO) atmospheres; the treatment under a H2 flow has been able to promote the formation of most adsorbed oxygen species over the ZrO2 surface, followed by the treatment under a N2 flow. In contrast, the O2 atmosphere yields the most unfavourable outcome. The adsorbed oxygen species are efficient in removing the deposited carbon and subsequently enhancing the performance for the dry reforming of methane. The observation of basic sites may be explained by CO2 adsorption on the treated samples of ZrO2 catalysts, as shown in Fig. 1B and C. It can be observed that the band intensities for the H2 treated ZrO2 catalysts are higher than that for the N2 and O2-treated ZrO2 catalysts.
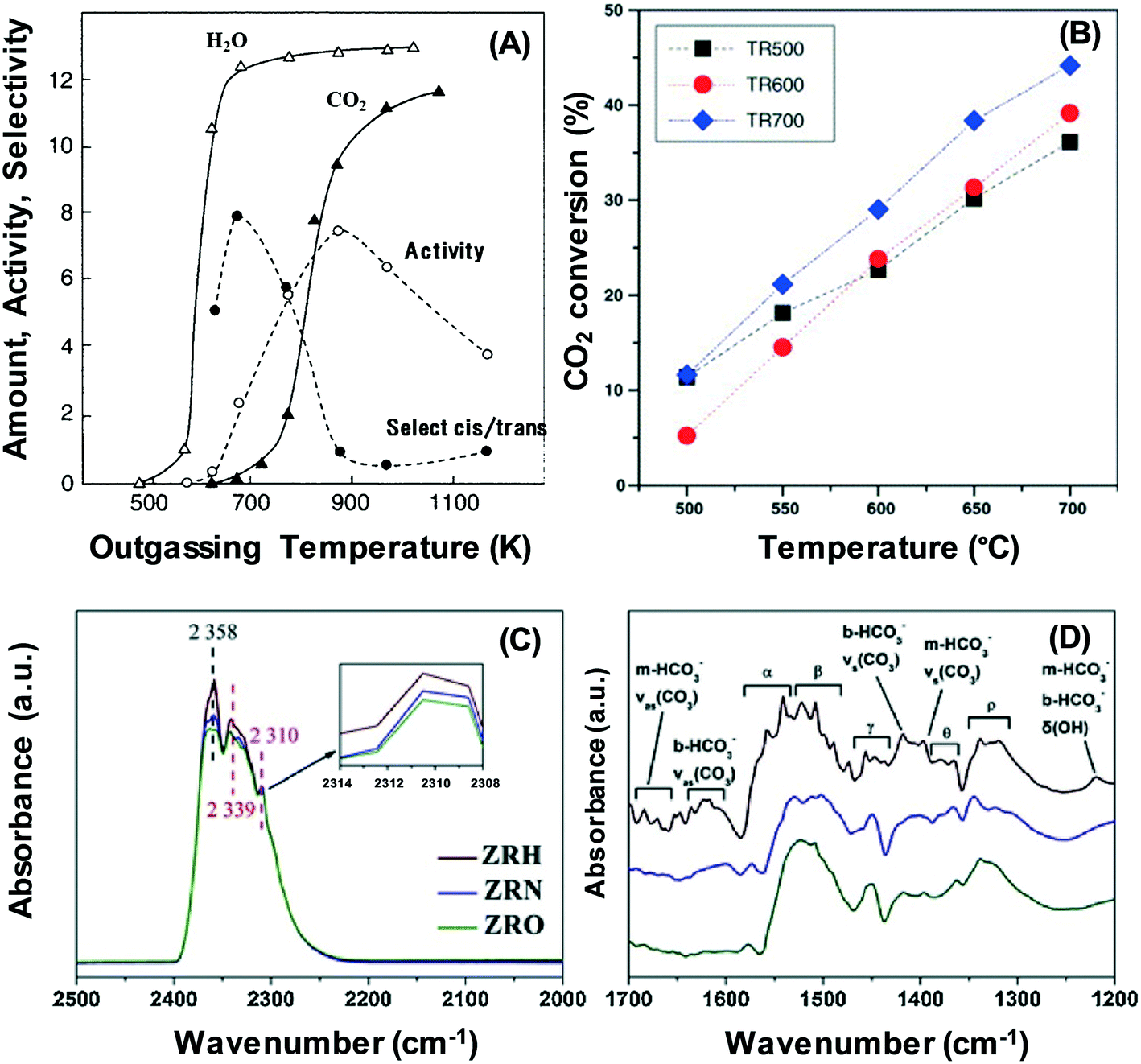 | ||
| Fig. 1 (A) Generation of H2O and CO2 and the activity of 1-butene isomerisation over CaO derived from Ca(OH)2 at different outgassing temperatures.24 (B) CO2 conversion on Ni–Mg–Al catalysts at different reduction temperatures: (■) 500 °C; (●) 600 °C and (♦) 700 °C.27 (C) and (D) FTIR spectra of CO2 adsorption on ZrO2 samples treated under different atmospheres (i.e. H2, N2 and O2).28 Reproduced from ref. 24, 27 and 28, and with permission from Elsevier. | ||
The acidity or basicity strength of a catalyst surface is dependent upon various properties of metal, namely charge status, coordination number, size and electronegativity in the oxidation state. The acidity is characterised by coordinating the unsaturated metal surface sites, whereas the basicity of the catalyst surface is determined by surface oxygen sites. A surface defect, especially defective energy levels thus correlated to electronic transitions, is a factor that is used to determine the defect behaviour into acting as a Lewis acid or Lewis base, as well as its acid or basic strength. When a defect is formed, the local bonding structure and lattice polarisation are transformed. As metal oxides have high ionicity, the electrostatic potential generated is thus able to localise the electrons at vacant sites; this serves as a key role in catalytic activities.29 The basic strength of lattice oxygen ions depends on the pre-treatment temperature. The strongest basic sites can be revealed at high pre-treatment temperature.25 For example, one superior solid base catalyst for a range of catalytic activities (e.g. methane dry reforming, isomerisation, and transesterification) is nanocrystalline MgO.30–32 Its surfaces are known to present various anomalies.29 Point defects of the unsaturated O2− species (e.g. cationic vacancies) and low-coordinated anionic sites can emerge from the surface reconstruction in the context of annealing.33 The major types of basic sites incorporated in MgO are the surface hydroxy groups, while the three types of O2− sites that can be found on the material surface consist of: (ii) edges, (iii) vertex, and (iv) pits. These kinds of surface defects are produced by Mg2+ vacancies or the occurrence of lower coordination (110) and (111) facets, which are some of the possible reasons for the basic character depicted by the oxides. In addition, (111) facets of the nanocrystalline MgO have a higher basicity, causing them to display the greatest activity for the reaction, whereas the thermal treatment renders the strong base sites denser.34,35
CO2-TPD facilitates the identification of the character, number, and strength of basic sites, resulting in its wide use to chemically characterise surfaces. The approach is particularly popular for determining the number and strength of base sites present on solid base catalysts. The standard CO2-TPD desorption profiles consist of three contributions equivalent to distinct classes of basic groups. Weak Brønsted basic sites (e.g. surface OH groups), Lewis acid–base sites of medium strength, and low-coordination oxygen anions manifested as strong basic sites are henceforth associated with low temperatures (100–250 °C), medium temperatures (250–400 °C), and high temperatures (400–600 °C), respectively.13,36 For instance, Fig. 2 shows three major peaks exhibited by the CO2-TPD of a clay-based catalyst, which are equivalent to weak, medium, and strong basic sites. According to the results, the strong basic sites are restricted, while the weak and medium basic sites proliferated when Fe is added to the Ni/clay catalyst. Furthermore, the addition of Fe and Cu has diminished the overall basicity, particularly in the case of clay modified with Cu. Reduction at 900 °C has diminished the basicity particularly more and is most likely to be caused by Ni re-dispersion (i.e. Ni crystals of smaller dimensions).37
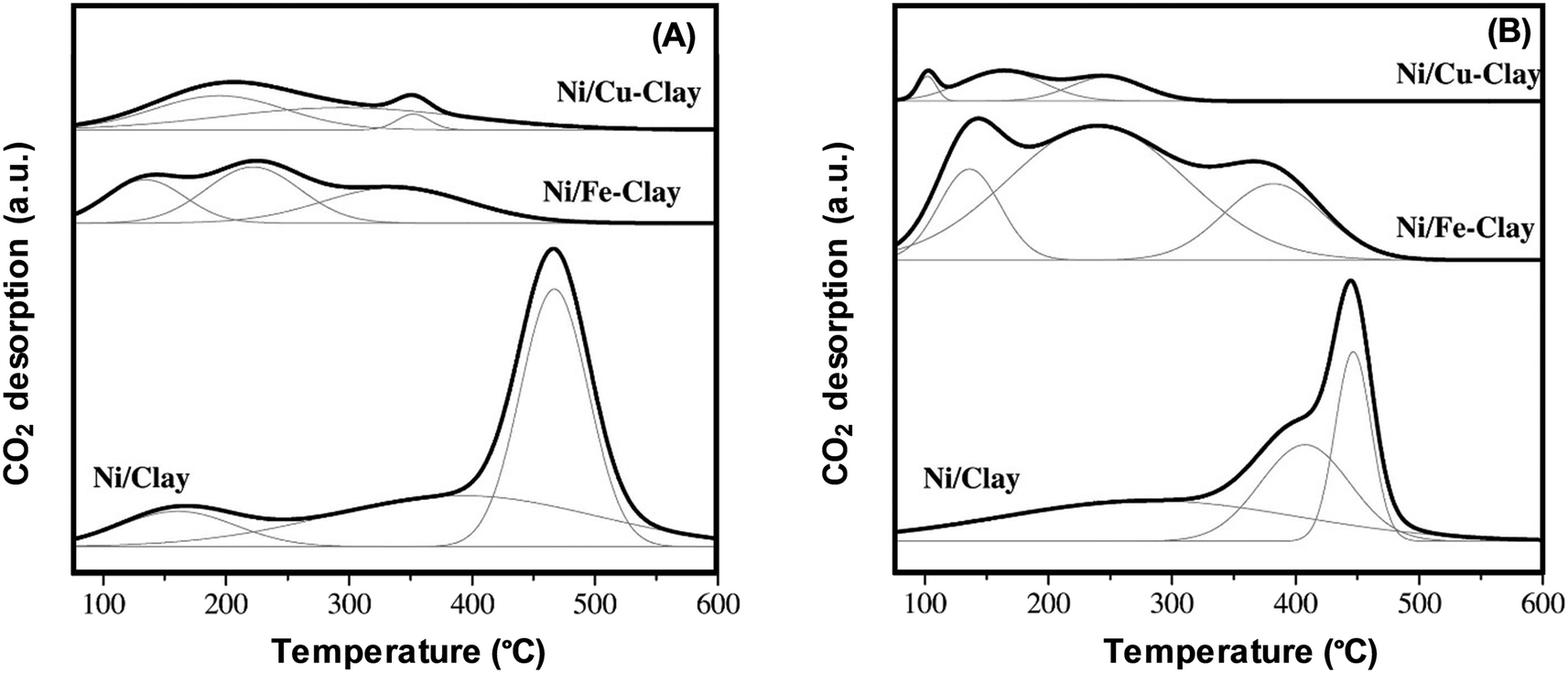 | ||
| Fig. 2 CO2-TPD profiles generated for Ni catalysts based on natural clay with reduction at (A) 800 °C and (B) 900 °C.37 reproduced from ref. 37 with permission from Elsevier. | ||
The catalyst basic strength can be investigated via probe molecule adsorption by employing FTIR spectroscopy methods. Pyrrole, carbon dioxide, carbon monoxide and methane are some of the probe molecules employed for this purpose.19,38–41 Pyrrole is applied to explore the basic qualities of mesoporous ZSM5 (m-ZSM5) via FTIR spectroscopy. The mesoporous ZSM5s were tailored by varying the aging time during preparation at 0.5 day (m-ZSM5-0.5D), 1 day (m-ZSM5-1D) and 3 day (m-ZSM5-3D) as depicted in Fig. 3.38 Pyrrole adsorption at ambient temperature (a) has been conducted before outgassing within a range of temperatures (spectra b–f). The interactions of N–H stretching vibrations of chemisorbed pyrrole (C4H4NH) with the framework oxygen atoms via hydrogen bonding (C4H4NH–Ozeolite) and with the non-framework cations accordingly via the aromatic system are found to be responsible for the chief broadband around 3700–3270 cm−1 for every catalyst. The two interactions have occurred at the same time and affected one another. The interaction between the disrupted N–H stretch of pyrrole molecules and basic site surface is thus considered as the reason for the band at 3478 cm−1. The catalyst has been concluded to have numerous basic sites due to the high intensity observed.
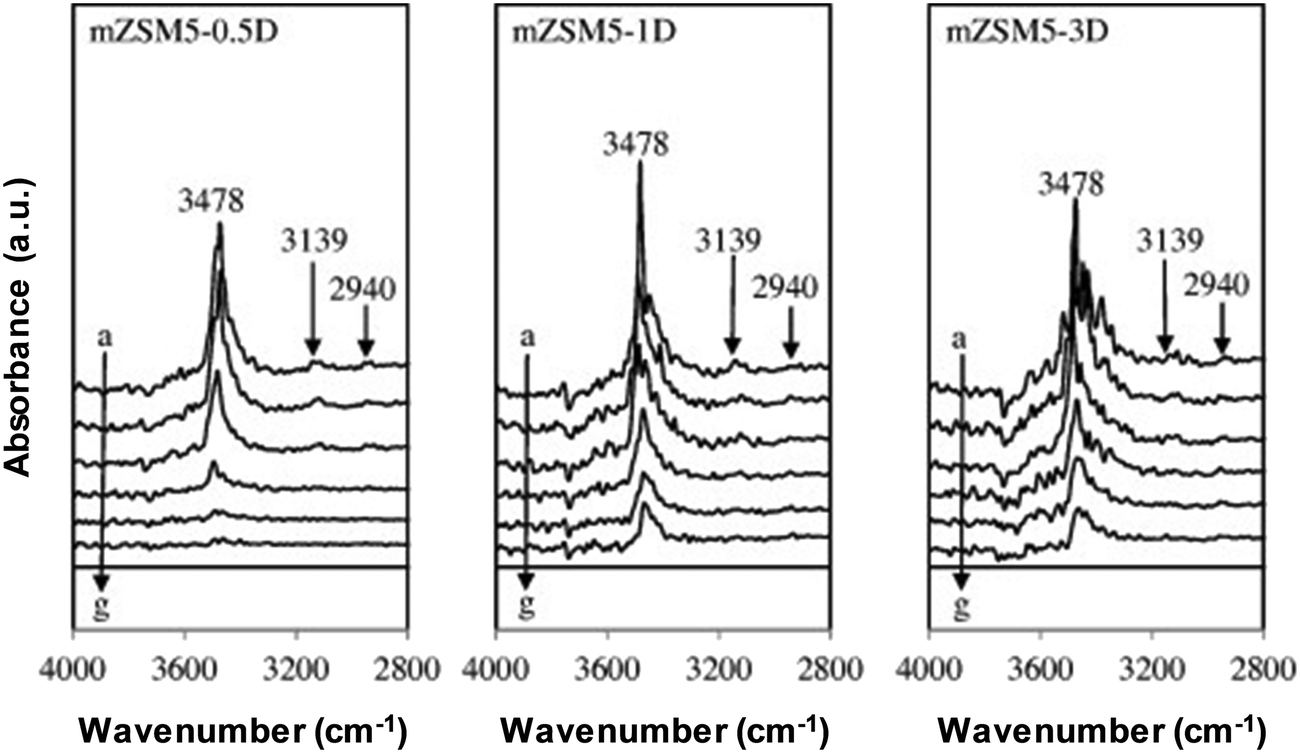 | ||
| Fig. 3 FTIR spectra of pyrrole adsorbed on activated mZSM5s at (a) 298 K, followed by outgassing at (b) 298 K, (c) 323 K, (d) 373 K, (e) 423 K, and (f) 473 K (g) before exposure to pyrrole.38 Reproduced from ref. 38 with permission from Elsevier. | ||
Furthermore, CO2 adsorbed FTIR spectroscopy has been used to study the basic properties of catalysts.42,43 Sidik et al. have explored the functional basicity of mesoporous silica nanoparticles (MSNs) and nickel loaded MSNs (NiM) through in situ observations of CO2 adsorption on the catalyst by using FTIR spectroscopy. The NiM catalysts have been varied by different preparation methods, namely in situ method (NiM_IS), physical mixing method (NiM_PM), and impregnation method (NiM_IM). The carbonate-like species with basic oxygen atoms are observed on the MSN and Ni/M catalysts within the region of 1800–1400 cm−1, as depicted in Fig. 4.
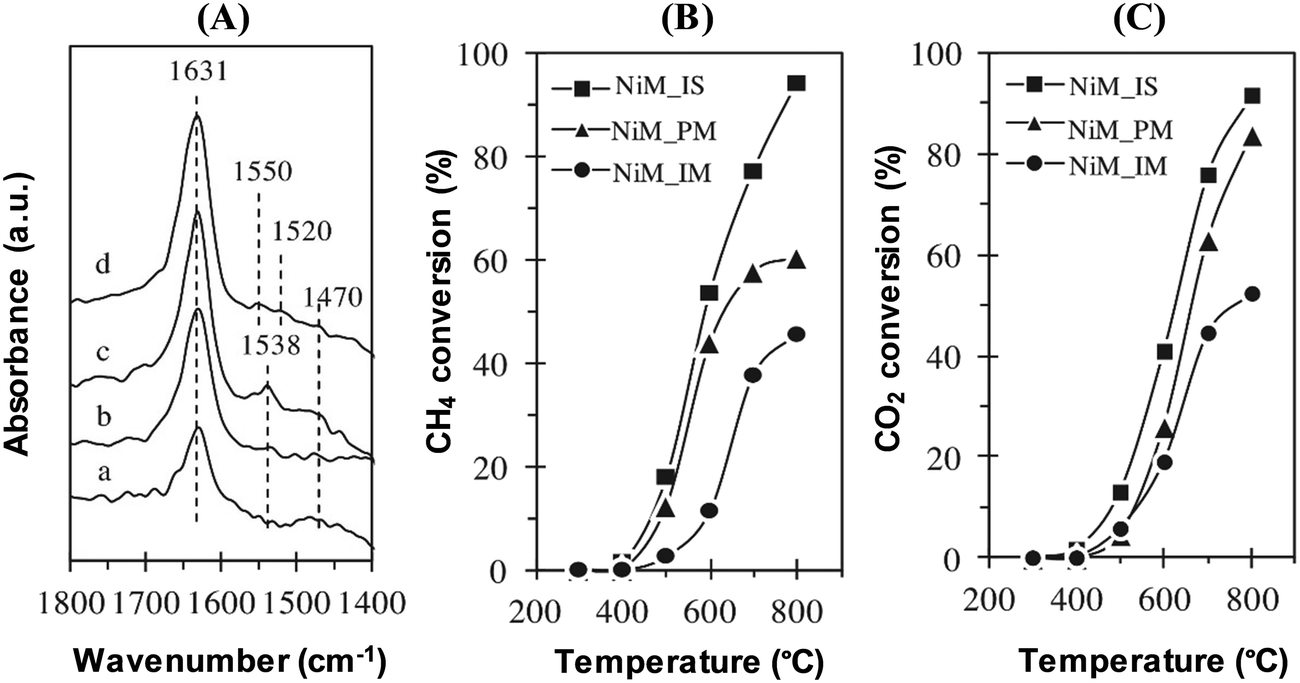 | ||
| Fig. 4 (A) CO2 adsorbed IR of (a) MSNs, (b) NiM_IM, (c) NiM_PM, and (d) NiM_IS catalysts. The catalytic activity of CH4 conversion (B) and CO2 conversion (C) of NiM_IS, NiM_PM, and NiM_IM catalysts in CO2 reforming of CH4.42 Reproduced from ref. 42 with permission from Elsevier. | ||
For MSNs, two prominent peaks have been observed at 1631 and 1470 cm−1, which are attributed to the symmetric and asymmetric O–C–O stretching modes of bidentate carbonates, respectively. Compared to the MSNs, the intensity of these peaks is increased for the NiM catalysts, whereby the highest intensity is observed for the NiM_IS catalyst. In addition, a new peak at around 1538 cm−1 has appeared for the NiM_IM and NiM_PM catalysts both, and at 1550 and 1520 cm−1 for NiM_IS, which may be attributed to unidentate carbonates associated with stronger basic sites. It is known that the formation of unidentate and bidentate carbonates requires surface basic oxygen atoms. In particular, unidentate carbonates are formed by CO2 dissociation with strong basic sites, while bidentate carbonates are formed by CO2 adsorption at medium-strength basic sites. Therefore, the addition of Ni into the MSN catalysts, irrespective of the preparation methods, has increased the concentration and strength of the basic sites found on the catalyst surface. Among the three NiM catalysts, the highest concentration of bidentate carbonate has been observed on NiM_IS, followed by NiM_PM and NiM_IM. Moreover, NiM_IS has shown the highest concentration of total basicity, which correlates to the catalytic activity in CH4 and CO2 conversions. Therefore, it can be deduced that high basic sites are vital for a high level of activity in the dry reforming of methane.
3. Correlation of catalyst surface basic sites with dry reforming of methane
Basic catalysts are inclined towards CO2 adsorption and activation with high resistance towards coke deposition. CO is generated from the reaction between the adsorbed CO2 species and adsorbed carbon (coke) on the surface. This is due to the interaction between coke deposits on the catalyst surface and the mobile oxygen of the catalyst, leading to better catalytic activity and stability.44–46 As a generally known information, one of the causes of coke deposition on the catalyst surface is due to methane decomposition, which is favourable at the acid sites. As a consequence, having basic properties is yet another option to limit coke formation. Increasing the basicity, which fundamentally means increasing the oxygen mobility, can accelerate CO2 activation and subsequently inhibit the formation of carbon from CH4 decomposition. Besides, the mobile oxygen can interact with coke deposits on the catalyst surface, resulting in the formation of CO. In particular, the reverse Boudouard reaction (CO2 + C → 2CO) is a well-known endothermic side reaction in the dry reforming of methane. It is favoured at a temperature greater than 673 °C. However, apart from increasing the reaction temperature, the extent of the reverse Boudouard reaction rate depends strongly on the basic nature of a catalyst. This is attributable to its involvement with the surface reaction between acidic CO2 adsorbed species and surface CHx fragments from dissociative CH4 adsorption. The increasing basic character of a catalyst can induce a greater degree of CO2 adsorption, and thus leads to higher coke gasification from the catalyst surface.The improvement of a catalyst's basic nature depends greatly on promoters. Fig. 5 shows the mechanistic study of dry reforming of methane based on CO2 and coke formation reduction by a La2O3 promoter in the case of Ni/La2O3/γ-Al2O3 catalysts.
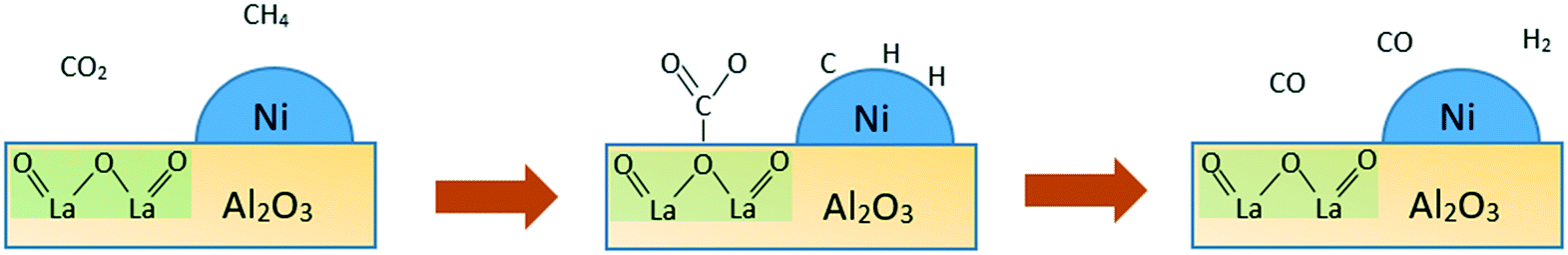 | ||
| Fig. 5 The probable mechanism of CO2 and CH4 adsorbed on Ni/La2O3/γ-Al2O3 catalysts. Adopted from ref. 47. | ||
The provision of more oxygen basic sites of the catalyst surface is the function of La2O3 as a promoter.13 Oxygen basic sites facilitate the adsorption of CO2 in the form of carbonate species on La2O3. While the adsorption of CH4 occurs on metal Ni sites, with separation of molecules from adsorbed C, which subsequently yields hydrogen gas and adsorbed H. The adsorbed C may also form a coke which aggregates on metal sites. In contrast, CO gas is produced by the oxygen from the carbonate species through a reaction with adsorbed C on the interface of the support and metal. The CO gas can also be produced from the remaining carbonate species. Meanwhile, carbon aggregates present on the nickel active sites in the absence of the La2O3 promoter cause the catalyst to be deactivated. This process highlights the fact that the reduction of coke formation and improvement of catalyst stability depend greatly on basic sites.
Moreover, cerium (Ce), zirconia (Zr), neodymium (Nd), and certain alkali earth metals can be used in catalysts as effective promoters.48 For instance, strong basicity appears to be stimulated by the use of Ce as a promoter on hydrotalcite derivatives, with CO2-TPD indicating the occurrence of CO2 desorption peaks at temperatures exceeding 280 °C. The intensification of direct methane breakdown is caused by the difficult nature of the reaction between CO2 adsorbed on strong basic sites and methane. Active carbonate species capable of reacting with methane via the DRM path are thus formed when CO2 is adsorbed on weak basic sites.49 In the case of Ni/Zr–Ce–Nd catalysts, numerous Lewis basic sites of mild strength are contributed by Nd, whereas strong and medium basic sites are contributed by Ni and Zr. Coking problems can be lessened by adding Nd, which enhances the oxidation rate of carbonaceous deposits by making the surface oxygen in Ce and Ce–Zr oxides more mobile. Therefore, better stability can be achieved by using a large quantity of Nd, whereby it increases the number of mildly strong Lewis basic sites.50
Dry reforming of methane has been undertaken with the use of mixed oxide catalysts such as perovskite-type oxides, which are usually made up of lanthanide or alkaline earth metals in conjunction with a transition metal (e.g. Cr, Mn, Fe, Ni, and Co). Lewis basicity of great strength is generally exhibited by catalysts such as perovskite oxides.51,52 The latter can also be formulated in a way that the pore size is efficiently regulated, which is essential for preventing coke from forming during the reaction.53–55 For example, the technique of homogeneous deposition (HDP) has been employed for the preparation of Ni-substituted CaZrNiOx, which subsequently leads to an increase in the number of basic sites.56 In contrast to Ca–ZrOx catalysts, CaZrNiOx catalysts have displayed a more extensive distribution of moderately strong basic sites compared to their basic sites of great strength. The reduced distribution of strong basic sites in the Ca–ZrOx catalyst may be explained by the possibility of Ni species covering a portion of the sites. Similarly, Pr promotion can help to improve perovskite-type oxide catalysts, such as Ni–Mg–Al mixed oxides. In DRM, the catalyst showing optimal performance is promoted with 6% wt. Pr. Moreover, it must be highlighted that conversion is unaffected by Pr, thus leading to better stability in relation to basic site thermal stability and Ni particle sizes.57
The catalyst type support determines the character of catalyst basicity in most cases. For example, MgO exhibits Lewis basicity of great strength and its use in methane dry reforming is helpful as its basic qualities increase the chemisorption of acidic CO2. A research conducted on dry reforming of methane using a Ni/MgO catalyst found that CO2 chemisorption is extensive on basic sites of low strength. Moreover, carbon deposition is minimised as CO formation is induced by the reaction between the chemisorbed carbon species and the generated carbon. MgO is thus concluded to be an appropriate support due to it minimising or completely preventing carbon from being deposited.58 This conclusion is consistent with another study, which has reported that CO2-TPD indicates the basic sites of the MgO support to be weak (100–350 °C) and moderate (350–570 °C). In contrast, the weak basic sites in MgO catalysts did not exhibit significant alteration when Ni and Pd are added, prompting a shift to the basic sites of even lower strength.45 Moreover, the adsorption of CO2 on the catalyst has been affected by the MgO facet. A Ni catalyst with MgO(111) nanosheets as a support has been investigated in the context of CO2 reforming of methane, thus revealing that Ni/MgO(111) not only displays more extensive activity, but is also stable for a longer period of time.59 In particular, the stable MgO(111) nanosheets have been prepared by using a Mg ribbon dissolved in methanol and mixed with 4-methoxybenzyl alcohol. The resulting mixture has been subjected to a hydrothermal treatment in an autoclave, followed by solvent removal and calcination at 500 °C.59 A direct correlation has been posited to exist between the enhanced catalyst performance and broad distribution of active Ni particles, which is due to the robust interplay between the metal and support. The notion is further justified by the enhanced catalyst performance and high number of MgO(111) basic sites, owing to its typical surface qualities. Zhang et al. studied the comparison between the density of the basic sites of MgO(111) and commercial MgO. Based on the CO2-TPD result, MgO(111) has shown to possess more basic sites than the commercial MgO. The reason behind the former having more surface basic sites compared to the latter is explained by it being covered mainly by Mg2+/O2− pairs of moderate basicity.59
Mutch et al. have reported that in the context of CO2 adsorption on MgO(111), positive binding toward CO2 can be exhibited by low coordinate corner/edge sites on the (100) facet with thermodynamic stability, leading to innately low capacity.34 In contrast, low coordinate sites are denser in the (111) facet. Furthermore, a high CO2 capacity has been exhibited by the MgO(111) nanosheets. Fig. 6A and B show the correlation between the structure and activity derived from spectroscopy, highlighting that the capacity activation of the polar MgO(111) facet depends significantly on elevated temperatures.
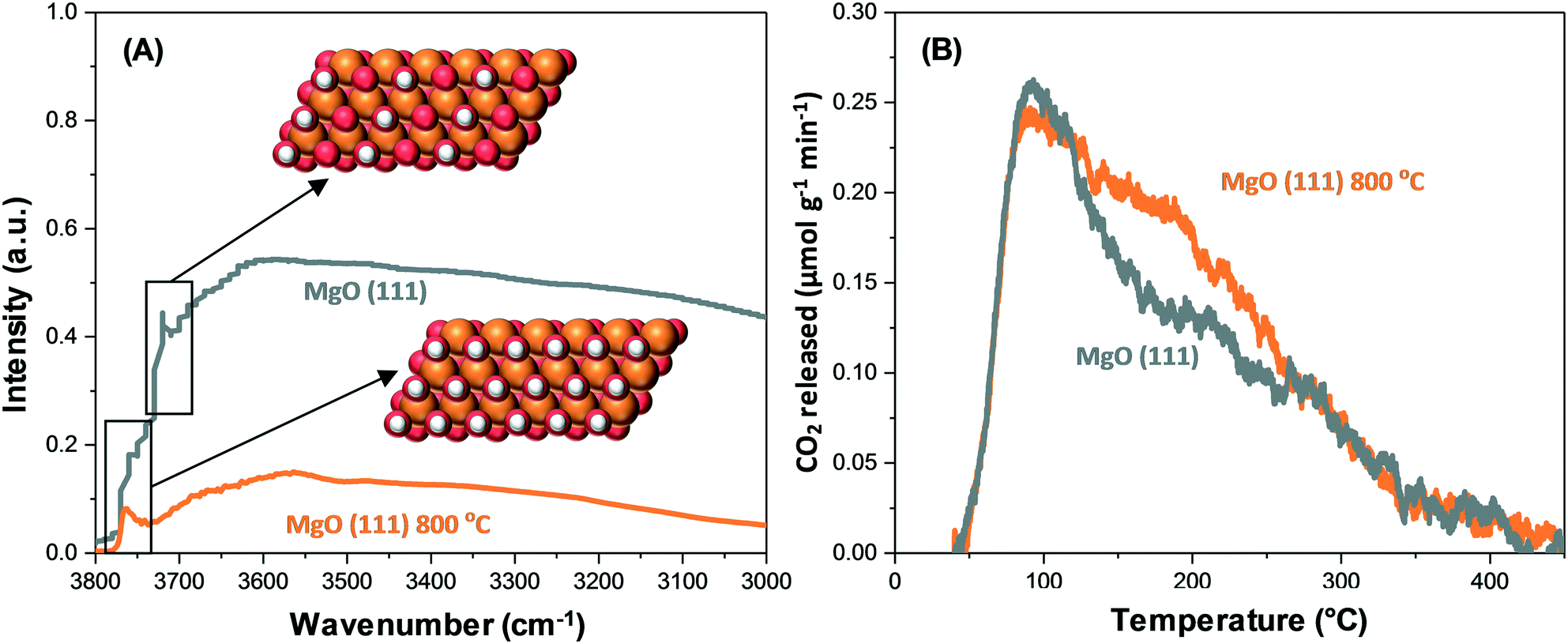 | ||
| Fig. 6 (A) DRIFTS spectra showing surface removal of hydroxyl of MgO(111) and MgO(111) at 800 °C.34 (B) CO2-TPD data showing the removal of surface hydroxyl of MgO(111) and MgO(111) at 800 °C, which leads to the formation of novel intermediate basic sites.34 Reproduced from ref. 34 with permission from the American Chemical Society. | ||
A promising method for high catalyst activity is the catalyst morphology design, whereby the support form impacts the catalyst basicity and helps catalysts to perform better, leading to hydrogen being generated sustainably. For example, CO2 can be more easily adsorbed and activated on the surface if a higher number of moderately strong basic sites are supplied by a Ni/La2O3 nanorod catalyst support.60 The discrepancies between the density of basic sites of nanorod La2O2CO3, 5Ni/La2O3-LOC (LOC = nanorod) and 5Ni/La2O3-C (C = conventional) are shown in Fig. 7. It can be observed that a larger number of moderately strong basic sites are associated with 5Ni/La2O3-LOC compared to 5Ni/La2O3-C. Furthermore, the XRD results have suggested that La(OH)3 formation on the reduced 5Ni/La2O3-LOC surface is stimulated by the La2O2CO3 nanorod, thus reinforcing CO2 adsorption via inducing the proliferation of hydroxyl groups on the support surface.
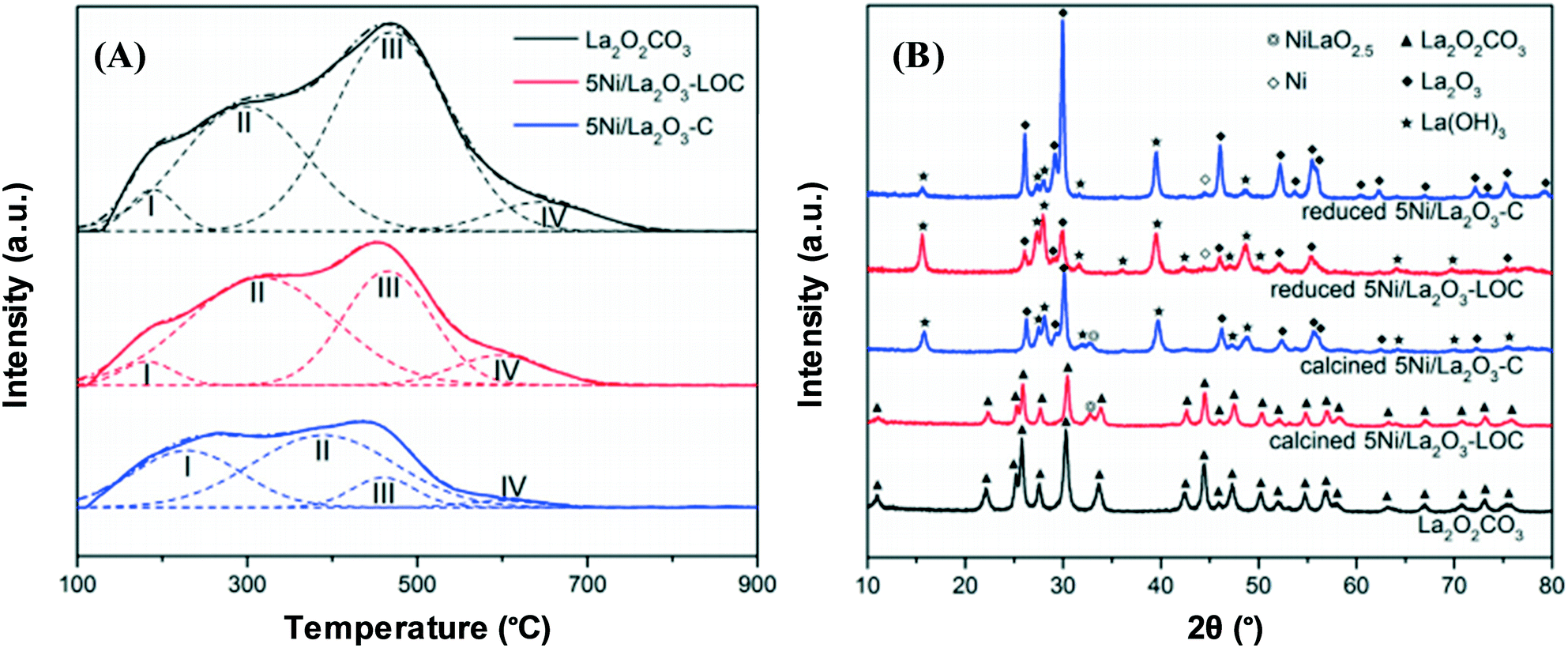 | ||
| Fig. 7 (A) CO2-TPD profiles of the reduced catalysts. (B) XRD patterns of the calcined and reduced catalysts.60 Reproduced from ref. 60 with permission from Elsevier. | ||
Huang et al. have focused on the impact of Ir/La2O2CO3 nanorods with selective exposure of the (110) facets in the catalytic steam reforming of glycerol. Their findings indicate that with regard to hydrogen production, significant activity, selectivity, and 100-hour stability are demonstrated by the Ir/La2O2CO3 nanorod catalyst.61 The development of hexagonal La2O2CO3 nanorods with selective exposure of the (110) facets may be a potential explanation for the high activity by the catalyst. Meanwhile, sintering of metal Ir particles has been inhibited by the potent interaction between the facets and Ir catalysts. The high-resolution transmission electron microscopy (HRTEM) of the La2O2CO3 nanorod catalyst is shown in Fig. 8, where the various facets of (001) and (110) are indicated in (a), (b) and (c). Furthermore, Fig. 8(d) shows the La2O2CO3 nanorod catalyst, whereas Fig. 8(e) shows the comparison between the nanorod catalysts (with different preparation methods denoted as La2O2CO3-r1, La2O2CO3-r2 and La2O2CO3-r3) and the commercial La2O2CO3 (Ir/La2O2CO3-c) in terms of basicity. The observation derived indicates that the La2O2CO3 crystallographic orientation has determined the number and types of basic sites. In contrast to the commercial Ir/La2O2CO3-c, intense basicity peaks of medium strength are exhibited by the nanorod catalyst. Moreover, the La3+–O2− pairs situated primarily on the (110) surfaces of La2O2CO3 are the main contributors for the high density of basic sites on the nanorod catalyst.62,63
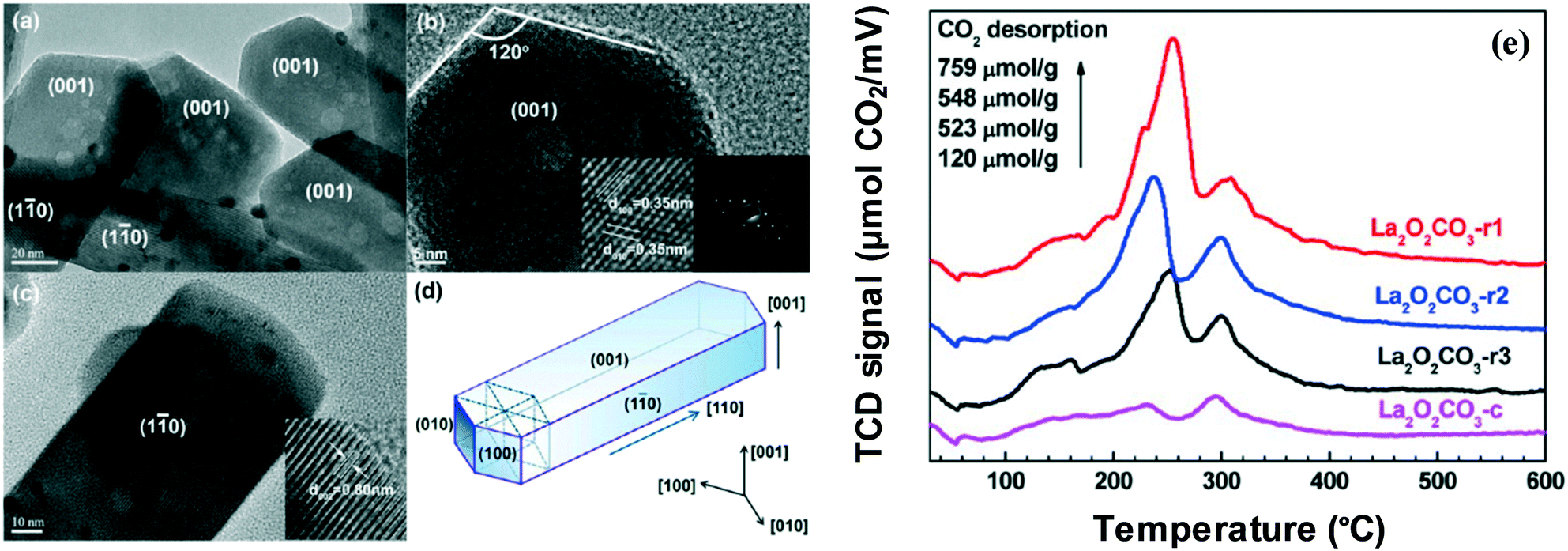 | ||
| Fig. 8 HRTEM (a–c) of the Ir/La2O2CO3 nanorod catalyst. The crystalline fringes (b) and electron diffraction patterns (c) are shown under magnification; (d) representation of the La2O2CO3 nanorod catalyst; (e) CO2-TPD profiles of Ir catalysts with different supports of La2O2CO3.61 Reproduced from ref. 61 with permission from the American Chemical Society. | ||
4. Remarks and conclusions
To date, the general consensus on the role of surface basic sites is to reduce or inhibit coke formation on catalysts. Since the crucial factor of methane dry reforming is dependent on the catalyst stability, the correlation between the basic sites with such activity may be determined. In view of this, the basic sites should be established thoroughly in order to find the optimum catalytic activity. One of the techniques to determine the surface basic sites is through in situ FTIR spectroscopy, namely by using a suitable probe molecule (e.g. pyrrole and carbon dioxide). The probe molecule is thus able to identify the active sites of catalysts so that the true active sites can further correlate with the catalytic activity. In addition, this review article comprehensively discussed the category of basic sites according to the formation of unidentate and bidentate carbonates during CO2 adsorption, which belongs to the corresponding strong and medium basic sites. The CO2-TPD approach was also highlighted to demonstrate the efficient determination of basic sites and categorise the types of basic sites depending on the CO2 desorption temperature. The surface basic sites can be enhanced by adding a suitable basic promoter, such as La2O3, MgO, and CeO2. In addition, these promoters can act as the main supports of metal catalysts. Apart from the types of supports and promoters, the different facets of metal oxide catalysts may further influence active basic sites. For instance, the facet MgO(111) is more active than MgO(100) in CO2 adsorption because of the higher density of low coordinate sites. Additionally, La2O2CO3 with the (110) facet possesses more active sites in CO2 adsorption. Therefore, it can be deduced that the activity of dry reforming can be enhanced via tailoring the facet of metal oxide supports by using suitable catalyst preparation techniques. In addition, the shape of the catalyst also influences the basic site's catalyst concentration. It has been shown that the nanorod shape of supports exhibits better CO2 adsorption compared to a conventional catalyst. This review thus revealed that the promoter, support structure, and support shape played a key role in yielding enhanced surface basic sites, which inhibited coke formation on catalysts. A thorough understanding of the abovementioned key factors in controlling the basic character of catalysts can significantly improve the catalyst design as it allows the selection of the right basic promoters and supports. It also results in the implementation of unique and advanced synthesis techniques for shape and structure manipulation. The strong correlation between basic sites and catalytic activity may also aid the approximate prediction of catalytic performance in methane dry reforming.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Nippon Sheet Glass Foundation for Materials Science and Engineering through Universiti Teknologi Malaysia under cost center numbers (4B371) and (01M42).References
- D. Barthomeuf, Catal. Rev.: Sci. Eng., 1996, 38, 521–612 CrossRef.
- A. Corma and S. Iborra, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Academic Press, 2006, vol. 49, pp. 239–302 Search PubMed.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- H. P. Boehm, Discuss. Faraday Soc., 1971, 52, 264–275 RSC.
- J. Rorrer, S. Pindi, F. D. Toste and A. T. Bell, ChemSusChem, 2018, 11, 3104–3111 CrossRef CAS PubMed.
- J. Quesada, L. Faba, E. Díaz and S. Ordóñez, ChemCatChem, 2018, 10, 3583–3592 CrossRef CAS.
- N. A. Sitte, M. Bursch, S. Grimme and J. Paradies, J. Am. Chem. Soc., 2019, 141, 159–162 CrossRef CAS PubMed.
- C. Jiang, Y. Wang, H. Zhang, N. Chang, L. Li, K. Xie and I. Mochida, Catal. Sci. Technol., 2019, 9, 5031–5044 RSC.
- A. Martínez, G. E. Mijangos, I. C. Romero-Ibarra, R. Hernández-Altamirano and V. Y. Mena-Cervantes, Fuel, 2019, 235, 277–287 CrossRef.
- L. Lin, E. Silva Gomes, F. Payan, M. Jaber, J.-M. Krafft, G. Laugel and H. Lauron-Pernot, Catal. Sci. Technol., 2019, 9, 6072–6084 RSC.
- J. Abou Rached, M. R. Cesario, J. Estephane, H. L. Tidahy, C. Gennequin, S. Aouad, A. Aboukaïs and E. Abi-Aad, J. Environ. Chem. Eng., 2018, 6, 4743–4754 CrossRef CAS.
- W. Bing, H. Wang, L. Zheng, D. Rao, Y. Yang, L. Zheng, B. Wang, Y. Wang and M. Wei, Green Chem., 2018, 20, 3071–3080 RSC.
- H. Liu, D. Wierzbicki, R. Debek, M. Motak, T. Grzybek, P. Da Costa and M. E. Gálvez, Fuel, 2016, 182, 8–16 CrossRef CAS.
- A. G. Bhavani, W. Y. Kim and J. S. Lee, ACS Catal., 2013, 3, 1537–1544 CrossRef CAS.
- J. W. Han, J. S. Park, M. S. Choi and H. Lee, Appl. Catal., A, 2017, 203, 625–632 CrossRef CAS.
- H. Hattori, Mater. Chem. Phys., 1988, 18, 533–552 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- W.-J. Jang, J.-O. Shim, H.-M. Kim, S.-Y. Yoo and H.-S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono, R. R. Mukti, Y. H. Taufiq-Yap and M. R. Sazegar, Appl. Catal., A, 2014, 147, 359–368 CrossRef CAS.
- A. Posada-Borbón and H. Grönbeck, Phys. Chem. Chem. Phys., 2019, 21, 21698–21708 RSC.
- S. Rhatigan and M. Nolan, J. Mater. Chem. A, 2018, 6, 9139–9152 RSC.
- L.-B. Sun, X.-Q. Liu and H.-C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- K. Bhattacharyya, A. Danon, B. K. Vijayan, K. A. Gray, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2013, 117, 12661–12678 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2015, 504, 103–109 CrossRef CAS.
- H. Hattori, Appl. Catal., A, 2001, 222, 247–259 CrossRef CAS.
- F. Song, Q. Zhong, Y. Yu, M. Shi, Y. Wu, J. Hu and Y. Song, Int. J. Hydrogen Energy, 2017, 42, 4174–4183 CrossRef CAS.
- O. W. Perez-Lopez, A. Senger, N. R. Marcilio and M. A. Lansarin, Appl. Catal., A, 2006, 303, 234–244 CrossRef CAS.
- M. Zhang, J. Zhang, Y. Wu, J. Pan, Q. Zhang, Y. Tan and Y. Han, Appl. Catal., A, 2019, 244, 427–437 CrossRef CAS.
- J. Jia, C. Qian, Y. Dong, Y. F. Li, H. Wang, M. Ghoussoub, K. T. Butler, A. Walsh and G. A. Ozin, Chem. Soc. Rev., 2017, 46, 4631–4644 RSC.
- Z. Zuo, S. Liu, Z. Wang, C. Liu, W. Huang, J. Huang and P. Liu, ACS Catal., 2018, 8, 9821–9835 CrossRef CAS.
- A. A. Marianou, C. M. Michailof, D. K. Ipsakis, S. A. Karakoulia, K. G. Kalogiannis, H. Yiannoulakis, K. S. Triantafyllidis and A. A. Lappas, ACS Sustainable Chem. Eng., 2018, 6, 16459–16470 CrossRef CAS.
- Z. Bai, Y. Zheng, W. Han, Y. Ji, T. Yan, Y. Tang, G. Chen and Z. Zhang, CrystEngComm, 2018, 20, 4090–4098 RSC.
- M. Setvín, M. Wagner, M. Schmid, G. S. Parkinson and U. Diebold, Chem. Soc. Rev., 2017, 46, 1772–1784 RSC.
- G. A. Mutch, S. Shulda, A. J. McCue, M. J. Menart, C. V. Ciobanu, C. Ngo, J. A. Anderson, R. M. Richards and D. Vega-Maza, J. Am. Chem. Soc., 2018, 140, 4736–4742 CrossRef CAS PubMed.
- M. D. Susman, H. N. Pham, A. K. Datye, S. Chinta and J. D. Rimer, Chem. Mater., 2018, 30, 2641–2650 CrossRef CAS.
- Y. Liu, Y. Wu, Z. Akhtamberdinova, X. Chen, G. Jiang and D. Liu, ChemCatChem, 2018, 10, 4689–4698 CrossRef CAS.
- H. Liu, L. Yao, H. B. Hadj Taief, M. Benzina, P. Da Costa and M. E. Gálvez, Catal. Today, 2018, 306, 51–57 CrossRef CAS.
- L. P. Teh, S. Triwahyono, A. A. Jalil, R. R. Mukti, M. A. A. Aziz and T. Shishido, Chem. Eng. J., 2015, 270, 196–204 CrossRef CAS.
- K. Chakarova, I. Strauss, M. Mihaylov, N. Drenchev and K. Hadjiivanov, Microporous Mesoporous Mater., 2019, 281, 110–122 CrossRef CAS.
- A. M. Ferrari, S. Huber, H. Knözinger, K. M. Neyman and N. Rösch, J. Phys. Chem. B, 1998, 102, 4548–4555 CrossRef CAS.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and S. M. Sidik, Appl. Catal., A, 2014, 486, 115–122 CrossRef CAS.
- S. M. Sidik, S. Triwahyono, A. A. Jalil, M. A. A. Aziz, N. A. A. Fatah and L. P. Teh, J. CO2 Util., 2016, 13, 71–80 CrossRef CAS.
- K. Coenen, F. Gallucci, B. Mezari, E. Hensen and M. van Sint Annaland, J. CO2 Util., 2018, 24, 228–239 CrossRef CAS.
- V. A. Tsipouriari and X. E. Verykios, J. Catal., 1999, 187, 85–94 CrossRef CAS.
- R. K. Singha, A. Shukla, A. Sandupatla, G. Deo and R. Bal, J. Mater. Chem. A, 2017, 5, 15688–15699 RSC.
- K. Bu, S. Kuboon, J. Deng, H. Li, T. Yan, G. Chen, L. Shi and D. Zhang, Appl. Catal., A, 2019, 252, 86–97 CrossRef CAS.
- A. S. Al-Fatesh, M. A. Naeem, A. H. Fakeeha and A. E. Abasaeed, Chin. J. Chem. Eng., 2014, 22, 28–37 CrossRef CAS.
- N. H. Elsayed, N. R. M. Roberts, B. Joseph and J. N. Kuhn, Appl. Catal., A, 2015, 179, 213–219 CrossRef CAS.
- R. Dębek, M. E. Galvez, F. Launay, M. Motak, T. Grzybek and P. Da Costa, Int. J. Hydrogen Energy, 2016, 41, 11616–11623 CrossRef.
- A. Pappacena, R. Razzaq, C. De Leitenburg, M. Boaro and A. Trovarelli, Inorganics, 2018, 6, 39 CrossRef.
- F. Polo-Garzon and Z. Wu, J. Mater. Chem. A, 2018, 6, 2877–2894 RSC.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- D. L. Trimm, Catal. Today, 1999, 49, 3–10 CrossRef CAS.
- S. Dama, S. R. Ghodke, R. Bobade, H. R. Gurav and S. Chilukuri, Appl. Catal., A, 2018, 224, 146–158 CrossRef CAS.
- R. C. Rabelo-Neto, H. B. E. Sales, C. V. M. Inocêncio, E. Varga, A. Oszko, A. Erdohelyi, F. B. Noronha and L. V. Mattos, Appl. Catal., A, 2018, 221, 349–361 CrossRef CAS.
- J.-H. Park, I. Heo and T.-S. Chang, Catal. Commun., 2019, 120, 1–5 CrossRef CAS.
- O. H. Ojeda-Niño, F. Gracia and C. Daza, Ind. Eng. Chem. Res., 2019, 19, 7909–7921 CrossRef.
- M. Usman and W. M. A. Wan Daud, RSC Adv., 2016, 6, 91603–91616 RSC.
- L. Zhang, L. Li, J. Li, Y. Zhang and J. Hu, Top. Catal., 2014, 57, 619–626 CrossRef CAS.
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Appl. Catal., A, 2017, 202, 683–694 CrossRef CAS.
- X. Huang, C. Dang, H. Yu, H. Wang and F. Peng, ACS Catal., 2015, 5, 1155–1163 CrossRef CAS.
- F. Wang, R. Shi, Z.-Q. Liu, P.-J. Shang, X. Pang, S. Shen, Z. Feng, C. Li and W. Shen, ACS Catal., 2013, 3, 890–894 CrossRef CAS.
- C. Chu, Y. Zhao, S. Li and Y. Sun, Phys. Chem. Chem. Phys., 2016, 18, 16509–16517 RSC.
| This journal is © The Royal Society of Chemistry 2020 |