
Catalytic enhancement on Ti–Zr complex oxide particles for electrochemical hydrogenation of oxalic acid to produce an alcoholic compound by controlling electronic states and oxide structures†
M.
Yamauchi,  *a
M.
Sadakiyo
a
and
*a
M.
Sadakiyo
a
and
*a
M.
Sadakiyo
a
and
Abstract
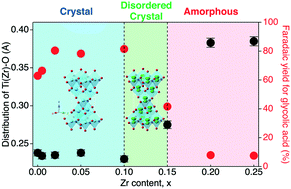
Ti1−xZrxO2 complex oxide particles with 0.02 ≤ x ≤ 0.1 show superior catalytic performances for the direct power storage into glycolic acid via electroreduction of oxalic acid. The atomic pair distribution function analysis of X-ray total scatterings suggested that structural periodicity is the key factor for the catalytic enhancement.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

