
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyoxometalates as alternative Mo precursors for methane dehydroaromatization on Mo/ZSM-5 and Mo/MCM-22 catalysts†
Ignacio
Julian
 *abc,
José L.
Hueso
abc,
Nidya
Lara
a,
Albert
Solé-Daurá
d,
Josep M.
Poblet
d,
Scott G.
Mitchell
*bc,
Reyes
Mallada
abc and
Jesús
Santamaría
abc
*abc,
José L.
Hueso
abc,
Nidya
Lara
a,
Albert
Solé-Daurá
d,
Josep M.
Poblet
d,
Scott G.
Mitchell
*bc,
Reyes
Mallada
abc and
Jesús
Santamaría
abc
aInstitute of Nanoscience of Aragon (INA) and Department of Chemical and Environmental Engineering, University of Zaragoza, Campus Rio Ebro, I+D+i Building, 50018 Zaragoza, Spain. E-mail: ijulian@unizar.es
bInstitute of Materials Science of Aragon (ICMA), Consejo Superior de Investigaciones Científicas (CSIC) - University of Zaragoza, C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: scott@unizar.es
cNetworking Research Centre on Bioengineering, Biomaterials and Nanomedicine (CIBER-BBN), 28029 Madrid, Spain
dDepartment of Química Física i Inorgànica, Universitat Rovira I Virgili, Marcel·lí Domingo 1, 43007 Tarragona, Spain
First published on 1st October 2019
Abstract
The conversion of methane into higher molecular weight hydrocarbons of greater added value has emerged as one of the grand challenges of the 21st century. The non-oxidative methane dehydroaromatization (hereafter MDA) reaction is a promising methane valorisation reaction since it transforms methane into added-value aromatics and olefins, namely benzene, naphthalene and ethylene. Molybdenum-promoted ZSM-5 zeolite has proven to be one of the most effective catalysts for MDA providing a shape-selective environment for the conversion of methane into benzene. However, one of the principle disadvantages of using aluminosilicates in the presence of methane is that the catalyst suffers from rapid deactivation induced by coke formation, which ultimately leads to a decrease in activity and aromatics selectivity, making the process unsuitable for large-scale industrial applications. Better control of the metal dispersion on the surface of the aluminosilicate supports represents a crucial factor to partially suppress catalyst coking and improve stability. Here we show how different molecular polyoxomolybdate (POM) anions can be used as alternative Mo precursors to conventional Mo salts for the preparation of catalysts for the MDA reaction. Molecular dynamics simulations and experimental testing were conducted to characterize the interphase interactions between polyoxomolybdates and zeolite surfaces at the atomistic level and to evaluate the MDA performance of different POM-based catalysts supported on ZSM-5 and MCM-22, respectively. The catalysts prepared using hexamolybdate anions, [Mo6O19]2−, were found to be more active and selective towards benzene than those employing the commercial heptamolybdate, [Mo7O24]6−. The Mo loading and dispersion of MoOx species were found to be the key factors leading to enhanced catalytic stability on ZSM-5 and MCM-22-based supports for MDA where the 5% Mo6/MCM-22 catalyst provided a constant aromatics yield above 7% for more than 18 hours time-on stream operating at 700 °C with a diluted methane flow under atmospheric pressure. The zeolitic catalysts prepared with the Mo6 precursor were found to be amongst the most promising MDA catalysts in the literature and the results of this study pave the way for the selection and use of different POMs as innovative metal precursors to formulate new catalysts and further improve the MDA reaction process.
Introduction
The transformation of methane into heavy hydrocarbons of higher molecular weight and greater added value is a scientific and technological challenge that has been studied since the second half of the 20th century. Among the direct CH4 conversion routes, non-oxidative methane dehydroaromatization (hereafter MDA) into added-value aromatics and olefins, i.e. benzene, naphthalene and ethylene, is a promising route for methane valorisation.1,2 Since MDA catalysis was first proposed in 1993,3 a number of studies have indicated that aluminosilicate zeolite ZSM-5 is an effective catalyst to provide a shape-selective environment for the conversion of methane into benzene. Since then, several alternative zeotype structures have been developed and tested for this catalytic process, e.g. MCM-22, MCM-36, ITQ-2, IM-5, NTU-9, among others.4–7 However, from all of these catalysts, ZSM-5 and MCM-22 remain as the most promising shape-selective supports for methane dehydroaromatization.8,9 The main drawback of using these aluminosilicates in the presence of methane is the fast catalyst deactivation induced by coke formation, leading to a decrease in activity and selectivity towards light aromatics which makes the process unsuitable for large-scale industrial applications.10It is widely accepted that metal dispersion along the zeolite pores plays a key role in the catalyst stability and coke resistance.11–13 A standard catalyst preparation procedure is based on the incipient wetness impregnation of 3–6 wt% Mo salt, using ammonium heptamolybdate (NH4)6Mo7O24·4H2O, on a ZSM-5 support (Si/Al = 10–40) followed by calcination in air at 550 °C.8,9 For such metal loading, this preparation results in well-dispersed Mo oxide clusters within the zeolite pores as well as small Mo aggregates at the surface of the support. However, the coking rates and catalyst activity loss along the time on stream using this synthesis procedure are still high.
In order to enhance metal dispersion, several strategies have reported improvement using the addition of ammonia to the aqueous solution of ammonium heptamolybdate prior to the impregnation process14 or by the hydrothermal post-synthesis of commercial ZSM-5 in an aqueous solution of Al(NO3)3 to create a uniform porous network that facilitates the access of the Mo species into the channels.15 Additional strategies involve the selective silanization of the external sites of the fresh H-ZSM-5 with organosilane reagents to decrease the number of such sites and, thus, the number of Mo species was retained at the external surfaces during the subsequent physical cation exchange of MoO3 and pre-treated ZSM-5 powders.16
Some authors suggest that at calcination temperatures above 500 °C some fully oxidised Mo species of the benchmark Mo precursor ammonium heptamolybdate (“Mo7”) carry out solid exchange with the OH groups (Brønsted acid sites) of the zeolite framework leading to the generation of dimeric [Mo2O5]2+ species. These cationic species anchor to two cation exchange sites of the support whereas some other Mo species accumulate as MoO3 clusters at the external zeolite surface.1,16,17 The efficiency of metal dispersion and, thus, Mo anchoring depends on the support acidity (Si/Al ratio), however other factors such as metal loading as well as the impregnation and calcination procedures must also be considered. In fact, the initial arrangement of Mo species at the zeolite surface and their mobility through the migration process seem to play a key role in the final metal dispersion on the catalyst. Although Mo7 has been regularly employed as the standard Mo precursor due to its relatively high availability, the [Mo7O24]6− anion is just one member of a vast family of known polyoxomolybdates.18,19 Polyoxomolybdates belonging to the polyoxometalate (POM) class of inorganic molecular metal oxides span different structural types each possessing different tunable physicochemical properties.20,21 To the best of our knowledge, polyoxomolybdates have not been extensively exploited as potential Mo precursors for the preparation of highly coke-resistant Mo/ZSM-5 and Mo/MCM-22 catalysts for the MDA reaction.
In this regard, exploring the affinity of different polyoxomolybdates as molecular precursors for Mo deposition on zeolitic surfaces may help to gain insight into the mobility of Mo species and to predict the quality of metal dispersion in the calcined catalyst.
This work explores the use of two different polyoxomolybdate structures, namely hexa- and octamolybdate anions (which possess different charges and sizes), as Mo precursors for Mox/ZSM-5 and Mox/MCM-22 catalysts for the MDA reaction. Specifically, this evaluates the impact of the induced metal dispersion given by different Mo precursors (hexa-, hepta- and octamolybdates), zeotype supports (ZSM-5 and MCM-22) and metal loads (5–10 wt% Mo) on the catalytic MDA process at 700 °C and 1500 mL gcat−1 h−1.
Experimental procedure
Catalyst synthesis
The synthesized polyoxometalates (POMs) were [(n-C4H9)4N]2[Mo6O19] (from now on “Mo6”) and [(n-C4H9)4N]4[Mo8O26] (“Mo8”). In both cases, basic alkaline salts, i.e. Na2MoO4 and [(n-C4H9)4N]Br, were employed as POM precursors, according to the established literature procedures.22 Briefly, the synthesis of both POMs proceeds via the acidification of an aqueous solution of sodium molybdate dihydrate with diluted HCl followed by dropwise addition of tetrabutylammonium hexamolybdate under vigorous stirring at room temperature until the formation of a white precipitate.In the case of “Mo6”, the resulting slurry was heated (85 °C) and stirred for 45 min obtaining a yellowish solid that was filtered and washed thrice with 80 mL of water. Crystallization was accomplished by dissolving the dried crude product in 80 mL of hot acetone (60 °C) followed by cooling down to −20 °C. After 24 h, the yellow crystalline products were filtered, washed twice with 20 mL of diethyl ether and dried for 12 h in a vacuum.
In the case of “Mo8”, the slurry was stirred at room temperature for 15 min, followed by a set of centrifugation and washing steps with 30 mL water and acetone, respectively. After two more washing steps with 60 mL ethanol and 60 mL diethyl ether, the white powder obtained was freeze-dried.
For the sake of comparison, a third batch of catalysts was synthesized using the typical Mo precursor employed for the preparation of Mo/ZSM-5 by impregnation, i.e. ammonium molybdate tetrahydrate (NH4)6Mo7O24·4H2O, from now on “Mo7”, 99.98% Sigma-Aldrich.
Mox/ZSM-5 and Mox/MCM-22 were prepared by incipient wetness impregnation using the Mo6, Mo7 and Mo8 precursors, respectively. Commercial NH4-ZSM-5 zeolite (SiO2/Al2O3 = 23, Zeolyst) was purchased and MCM-22 (Si/Al = 20) was prepared following the procedure described by Corma et al. using hexamethyleneimine (HM) as a large molecule that acted as the structure-directing agent (SDA).4 Briefly, crystallization was carried out in a stainless steel Teflon-lined autoclave at 150 °C under agitation (60 rpm) and autogenous pressure for 7 days. The solid was recovered by filtration, washed repeatedly with deionized water and dried overnight at 120 °C. The resulting sample was calcined in air at 550 °C for 5 h to remove the occluded organic SDA in the inner pores. The zeotype material was refluxed in 1.0 M NH4NO3 solution to exchange Na+ with NH4+ and finally calcined 550 °C for 5 h. This procedure was repeated twice in order to obtain the sample in its acidic form.
The resulting impregnated materials were dried at 120 °C overnight and calcined at 550 °C for 6 h, using a heating rate of 1 °C min−1. The calcination temperature and time were carefully selected in order to promote the diffusion of MoOx species into the pores and to form [Mo2O5]2+ dimers that are able to anchor at the Brønsted sites of the zeolite. Table 1 lists the summary of the synthesized catalyst indicating the Mo precursor (Mo6, Mo7 or Mo8), metal load (5–10 wt% Mo) and support (ZSM-5 and MCM-22).
| Support | Precursor | Mo load (wt%) | Sample ID |
|---|---|---|---|
| ZSM-5 | [(C4H9)4N]2 Mo6O19 | 5 | 5Mo6/ZSM-5 |
| (NH4)6Mo7O24·4H2O | 5Mo7/ZSM-5 | ||
| [(C4H9)4N]4 Mo8O26 | 5Mo8/ZSM-5 | ||
| MCM-22 | [(C4H9)4N]2 Mo6O19 | 5 | 5Mo6/MCM-22 |
| 8 | 8Mo6/MCM-22 | ||
| 10 | 10Mo6/MCM-22 | ||
| (NH4)6Mo7O24·4H2O | 5 | 5Mo7/MCM-22 |
Catalyst characterisation
A number of characterization techniques were employed to identify and measure the physico-chemical and structural properties of the synthesized catalysts. Argon adsorption was employed to study the textural properties of the samples. Measurements were carried out using a Micrometrics ASAP 2020 analyzer, degassing the samples at 350 °C prior to analysis. Determination of the specific surface was performed using the equation proposed by Brunauer, Emmett and Teller (BET), considering the consistency criteria proposed by Rouquerol for microporous materials23 and the micropore volume was evaluated at P/P0 = 0.01. Thermogravimetric analysis (TGA) was used to determine the changes in the mass that the POMs and catalysts undergo when heated at a programmed temperature. The differential gravimetric (DTG) and TG data were studied with a Q5000SA thermal analyzer and the measurements were carried out in air at a heating rate of 10 °C min−1 from room temperature to 900 °C. Scanning electron microscopy (SEM) was used to study the morphology of catalysts and the distribution of molybdenum on the surface of the zeolite support. Analyses were carried out using an INSPECT-F50 (FEI Company). The equipment uses a high resolution Schottky field emission gun and a variety of detectors. A secondary electron detector (SED), back-scattered electron detector (BSED) and energy-dispersive X-ray detector (EDX) were used to examine the morphology, to study the dispersion of molybdenum and to determine the amount of molybdenum loaded, respectively. The quantification of wt% Mo by SEM-EDX was carried out by measuring the Mo content of four separated 100 × 100 μm areas of representative regions of the characterized samples. Transmission electron microscopy (TEM) and scanning-TEM were performed with a Tecnai F30 (FEI company) and Titan Low Base 60-300 (FEI Company), respectively, to visualise the catalyst structure in detail and to evaluate the dispersion of Mo species and the eventual formation of aggregates on the zeolite external surface. X-ray diffraction (XRD) was employed to identify changes in the crystalline structure of zeolites ZSM-5 and MCM-22 with the addition of molybdenum. Data were recorded on an Empyrean diffractometer (PANalytical) operating at 45 kV and 40 mA with CuKα radiation. Raman spectroscopy was used to identify the presence of molybdenum species in Mox/ZSM-5 and Mox/MCM-22 catalysts. Characterisation was carried out at room temperature using an Alpha 300 Raman spectrometer (WITec) using a 532 nm laser and a CCD camera as the detector. The input power and acquisition time were 1 mW and 5 s, respectively. Fourier transform infrared spectroscopy (FTIR) was employed to confirm the main groups present in the synthesized POMs based on their vibrational and rotational modes. FTIR spectroscopy was performed on a Jasco FT/IR-4100 spectrometer. X-ray photoelectron spectroscopy (XPS) was used to verify the oxidation state of molybdenum species at the surface level in the Mox/ZSM-5 and Mox/MCM-22 catalysts. XPS characterisation was carried out with an Axis Ultra DLD (Kratos Tech). The spectra were excited by a monochromatized AlKα source (1486.6 eV) run at 15 kV and 10 mA. The binding energies were referenced to the internal C1s (284.6 eV) standard of adventitious carbon. Microwave plasma coupled with atomic emission spectroscopy (MP-AES) analysis was performed to quantify the amount of molybdenum present on the zeolite support. The catalytic samples were digested in aqua regia (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v nitric acid/hydrochloric acid) at 200 °C for ten minutes under microwave heating and characterized using the Agilent 4100 MP-AES equipment. Up to six independent measurements were carried out for each sample. Al magic-angle spinning nuclear magnetic resonance (27Al MAS NMR) was conducted to evaluate the effect of Mo addition on the change of the zeolite structures. The 27Al MAS NMR spectra were recorded at 104.3 MHz using a 2.5 μs pulse with a 3 s recycle delay (2048 scans) on a 400 MHz solid-state Bruker Avance AV-400-WB NMR spectrometer using a 4 mm probe. Samples were spun at 8 kHz.
:3 v/v nitric acid/hydrochloric acid) at 200 °C for ten minutes under microwave heating and characterized using the Agilent 4100 MP-AES equipment. Up to six independent measurements were carried out for each sample. Al magic-angle spinning nuclear magnetic resonance (27Al MAS NMR) was conducted to evaluate the effect of Mo addition on the change of the zeolite structures. The 27Al MAS NMR spectra were recorded at 104.3 MHz using a 2.5 μs pulse with a 3 s recycle delay (2048 scans) on a 400 MHz solid-state Bruker Avance AV-400-WB NMR spectrometer using a 4 mm probe. Samples were spun at 8 kHz.
Molecular dynamics simulations
MD simulations were carried out with the GROMACS 4.5.4 code24–26 and within the scheme of the AMBER99 force field.27 This methodology has been successfully used to study aggregative and ion-pairing processes involving polynuclear metal–oxo clusters in solution.28–31 The potential energy of the system is empirically described by the sum of the bonding terms, i.e. bond, angle, and dihedral deformation energies, and the nonbonding terms, which include additive pairwise 1–6–12 electrostatic and van der Waals potentials. Nonbonding terms are used to reproduce interactions between atoms in different molecules or separated by more than three bonds within a molecule. Force-field parameters for the inorganic POMs were obtained following the procedure by Lopez et al.32 Further details of this procedure are found in the ESI.† The set of Lennard-Jones parameters for Mo was taken from UFF33 and for Si, O and H atoms of the zeolite, we used those reported by Leroch et al.34 The TIP3P water model35 was used to represent solvent H2O molecules.In all cases, three POM anions and the number of cations required to neutralize the system were embedded in a prismatic solvent (water) box of 150 × 40.2 × 26.8 Å with an embedded ZSM-5 structure of 58.5 × 40.2 × 26.8 Å. Simulations were performed under 3D periodic boundary conditions using an atom cut-off of 14 Å for van der Waals interactions and of 10 Å for coulombic interactions, correcting the long-range electrostatics with the particle–particle mesh Ewald (PME) summation method.36 Newton's equations of motion were integrated using the leapfrog algorithm37 with a time step of 1 fs. Bonds involving H atoms were restrained by the LINCS algorithm38 and for the zeolite, we assumed a semi-rigid model in which the SiO2 framework is fixed during the simulations and only the shallow OH groups were free to move according to the bond and angle parameters proposed in ref. 34. All of the simulations were performed at 300 K, starting with random velocities and the temperature was controlled by means of the velocity-rescaling algorithm39 with a relaxation time of 0.1 ps. Before the production runs, all systems were equilibrated by 1000 steps of energy minimization, followed by an initial 250 ps run at constant volume and temperature (NVT) fixing the solute. The system was re-filled with water molecules and re-equilibrated until a suitable density of water was achieved. A final 250 ps NVT run with the solute relaxed was carried out before the 100 ns production simulations, which were also performed within the canonical (NVT) ensemble. Data were collected from the trajectories every 2 ps.
Catalytic tests
Reaction tests were carried out in fixed beds of catalytic powders (500 mg) using a gas mixture of methane and nitrogen (CH4:N2 = 80:20), in which nitrogen was employed as the internal standard. The spatial velocity and reaction temperature were set to 1500 mLSTP gcat−1 h−1 and 700 °C, respectively, for all tests. A 20 °C min−1 heating ramp was used to heat the catalytic sample up to the reaction temperature in the presence of the above-mentioned CH4:N2 composition. As a result of the high sample temperature and the presence of a reducing atmosphere, the initial molybdenum oxide species present in the calcined Mo/MCM-22 and Mo/ZSM-5 samples were carburized into oxycarbides (MoOxCy) and Mo2C during the so-called induction period. It is widely accepted that these carburized species are the active species for methane dehydroaromatization.40,41
The employed quartz reactor vessel has an internal diameter of 1 cm. Three mass flow controllers (Bronkhorst 0–20 mLSTP min−1) were used to feed reactive, CH4/N2, or regenerative, N2/O2, gas mixtures. Catalyst regeneration tests were conducted at 580 °C using 5% O2 in N2 at a spatial velocity 2400 mLSTP gcat−1 h−1. The sample temperature was measured and controlled with a thermocouple placed within the core of the fixed bed. The outlet reaction gases were passed through a heated pipe (230 °C) into a gas chromatograph (Thermo Fisher Trace 1300 with an auxiliary oven Trace 1310), in which four different columns and three detectors allowed the identification and quantification of permanent gases (TCD), light hydrocarbons (FID1) and aromatics (FID2), respectively. The scheme of the experimental set-up is presented in Fig. S1.†
Methane conversion (xCH4) and hydrocarbon selectivity (SCxHy) were calculated using eqn (1) and (2), where ṅi, ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) i, Ai and RFi are the molar flow, volumetric flow, peak area and response factor of species i, respectively. The response factor represents the ratio between the response of a detector to a compound (peak area) and the concentration of that compound in a mixture of gases. The hydrocarbon yield (YCxHy) is defined as the product of conversion by selectivity.42
i, Ai and RFi are the molar flow, volumetric flow, peak area and response factor of species i, respectively. The response factor represents the ratio between the response of a detector to a compound (peak area) and the concentration of that compound in a mixture of gases. The hydrocarbon yield (YCxHy) is defined as the product of conversion by selectivity.42
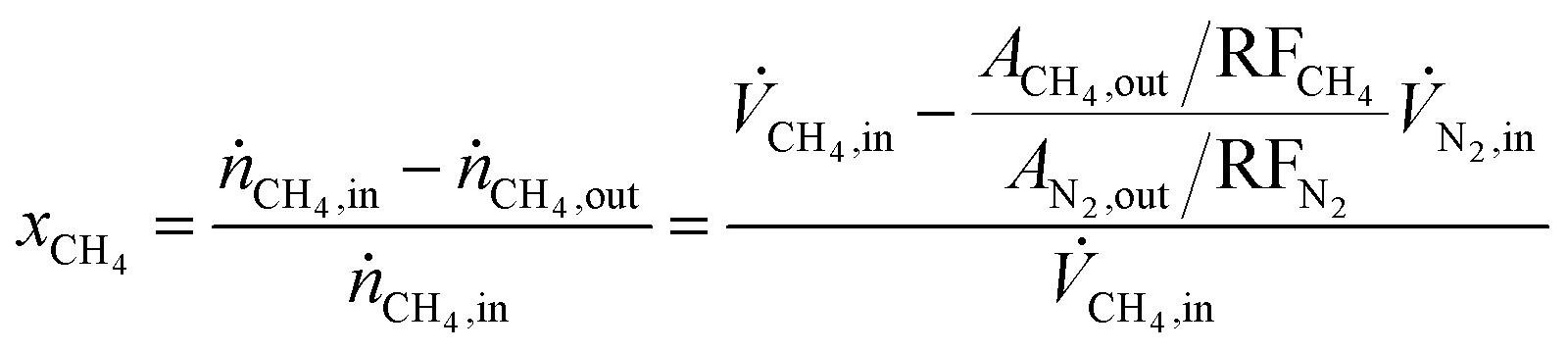 | (1) |
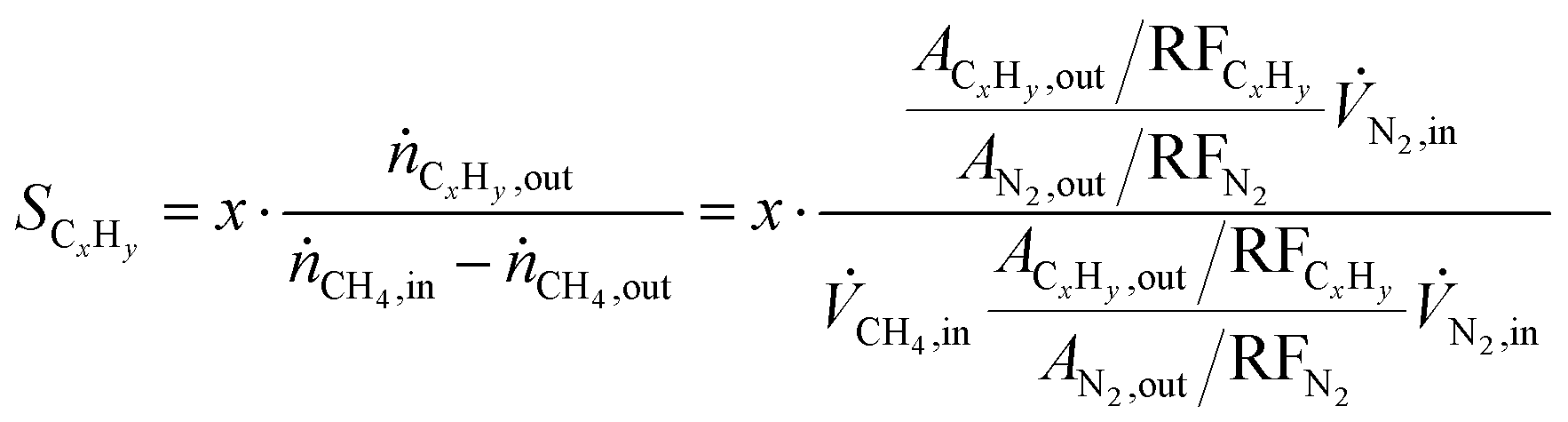 | (2) |
Results and discussion
The effect of the different molybdenum precursors (Mo6, Mo7 and Mo8) on the catalytic performance of 5% Mox/ZSM-5 for methane dehydroaromatization was evaluated. This metal load was selected for comparison, since it is widely reported that the Mo content in the range of 3–6 wt% promotes aromatics productivity in the MDA process on ZSM-5 supports with similar acidity.8,43,44 The role of each molybdenum precursor was explored using a theoretical perspective using molecular dynamics simulations on the interaction of the POM with the zeolite support. Next, the effect of the zeotype support (either ZSM-5 or MCM-22) on the catalytic MDA performance was evaluated using the most promising Mo precursors. Finally, the effect of metal loading (5–8–10 wt% Mo) on the best catalyst configuration, i.e. combination of the most suitable Mo precursor and zeotype support in terms of MDA performance, was studied. Fig. 1 shows a representation of this research work highlighting the geometry and dimensions of the different configurations of polyoxomolybdates employed as Mo precursors for the synthesis of Mo/ZSM-5 and Mo/MCM-22 species (Fig. 1a). The catalyst preparation procedure and calcination temperature at which Mo dimers are formed and anchored to the zeolite acid sites are shown in Fig. 1b. The geometry and pore size of the zeolitic catalyst supports are shown in Fig. 1c and the scheme on how the combination of Mo and the zeotype supports (acting as molecular sieves) promote methane conversion into benzene is shown in Fig. 1d.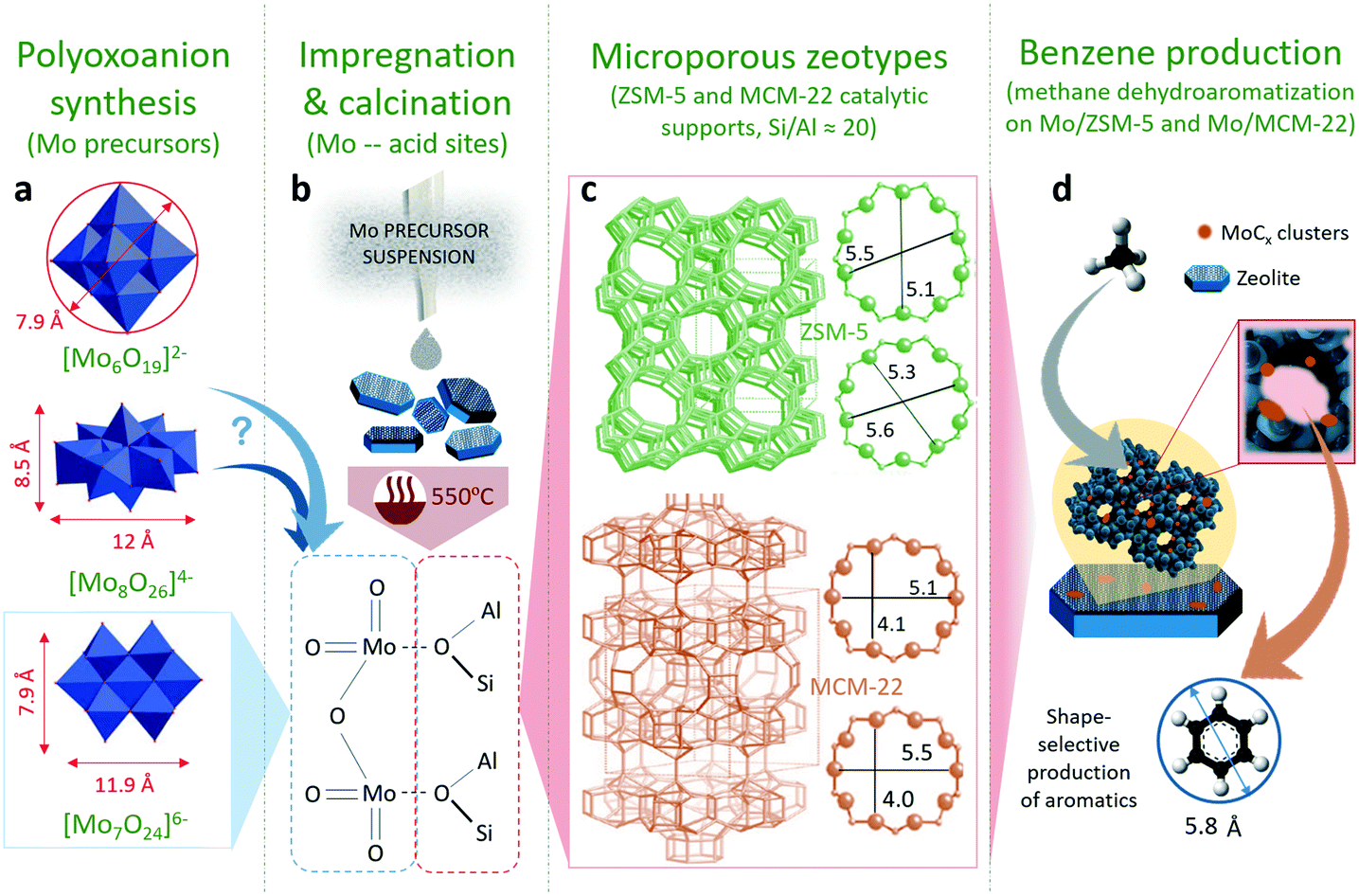 | ||
| Fig. 1 a) Geometry and dimensions of the polyoxomolybdate precursors, b) catalyst synthesis procedure: incipient wetness impregnation of the different precursors followed by calcination at 550 °C to potentially form dimeric [Mo2O5]2+ species at the acid sites of the support, c) microstructure of the employed zeotype supports, ZSM-5 and MCM-22, and characteristic pore dimensions, and d) molecular reaction drawing to illustrate the methane dehydroaromatization (MDA) process on the Mo/ZSM-5 catalyst. | ||
Role of the Mo precursor
A multidisciplinary approach involving catalyst characterization, molecular dynamics simulations and experimental testing under reaction conditions was utilized to evaluate the role of the Mo precursor on the catalyst MDA performance.The Raman spectra of the Mox/ZSM-5 samples calcined at 500 °C and 550 °C revealed that at 500 °C, Mo species still remain at the external surface of the zeolite in the form of MoO3 aggregates (Fig. S4†), whereas at 550 °C, they are able to migrate and anchor in the pores. The diffusion of Mo species at 550 °C is not instantaneous and, thus, the calcination time must be also carefully selected. Fig. S5–S7† show how the calcination time affects the MoOx crystal arrangement and size for each of the polyoxomolybdates. Interestingly, the TGA of the Mo precursors and fresh 5% Mox/ZSM-5 samples revealed noticeable differences in the thermal stability of both precursors and catalysts. In Fig. 2a, the TGA and DTG curves of Mo7 show four mass loss steps at 124, 171, 280, and 779 °C in air. The first mass loss is assigned to the loss of weakly bonded water molecules from the Mo7 structure, while the second and third mass loss steps correspond to the evolution of ammonium ions followed by structurally intercalated crystalline water, respectively. The sudden weight loss for the commercial salt Mo7 at 779 °C indicates its sublimation (commensurate with the appearance of the sharp peak in the DTG curve). In contrast, Mo6 and Mo8 showed excellent thermal stability below 900 °C and their weight loss in the temperature range of 250–360 °C is related to the loss of solvent molecules and counter cations. Interestingly, Fig. 2b reveals that the ZSM-5 based catalysts prepared with the three different Mo precursors and calcined at 550 °C show a different thermal stability above 700 °C. Initially, all samples dehydrate at low temperatures and remain stable up to the calcination temperature. However, above 700 °C, Mo sublimation becomes evident although its evolution with temperature differs between the three evaluated samples. The less stable sample is, again, the catalyst that uses the commercial Mo precursor. This suggests that the Mo species formed at the zeolite surface and/or within the zeolite pores and, thus, the interaction between Mo species and the support is somehow different for the three precursors. In this regard, the different coordination and/or oxidation state of molybdate species among the three pre-calcined samples may impact the effective formation of the active Mo2C during the induction period at the beginning of the reaction (MoxOy + CH4 ≥ MoxOyCz + H2O + CO), thus, leading to eventually different catalytic performance. Concerning the eventual Mo sublimation during MDA, all catalytic tests were performed at 700 °C where no metal loss was detected. To validate the previous hypothesis on the different coordination of Mo species and the ZSM-5 support, the samples were characterized by XPS (Table 2). This technique qualitatively confirmed Mo(VI) as the main oxidation state, i.e. MoO3, in all the fresh pre-calcined catalysts. Nevertheless, the presence of Mo(V) in the Mo6 sample suggests that the incorporation mechanism of Mo differs among the three precursors. In the spent catalysts after MDA, however, the metal was reduced to lower oxidation states indicating, for instance, the presence of Mo2C (related to Moδ+). Taking into account that the penetration on the samples is around 10 nm, the atomic composition obtained by XPS provides a good estimate of the metal distribution throughout the sample.
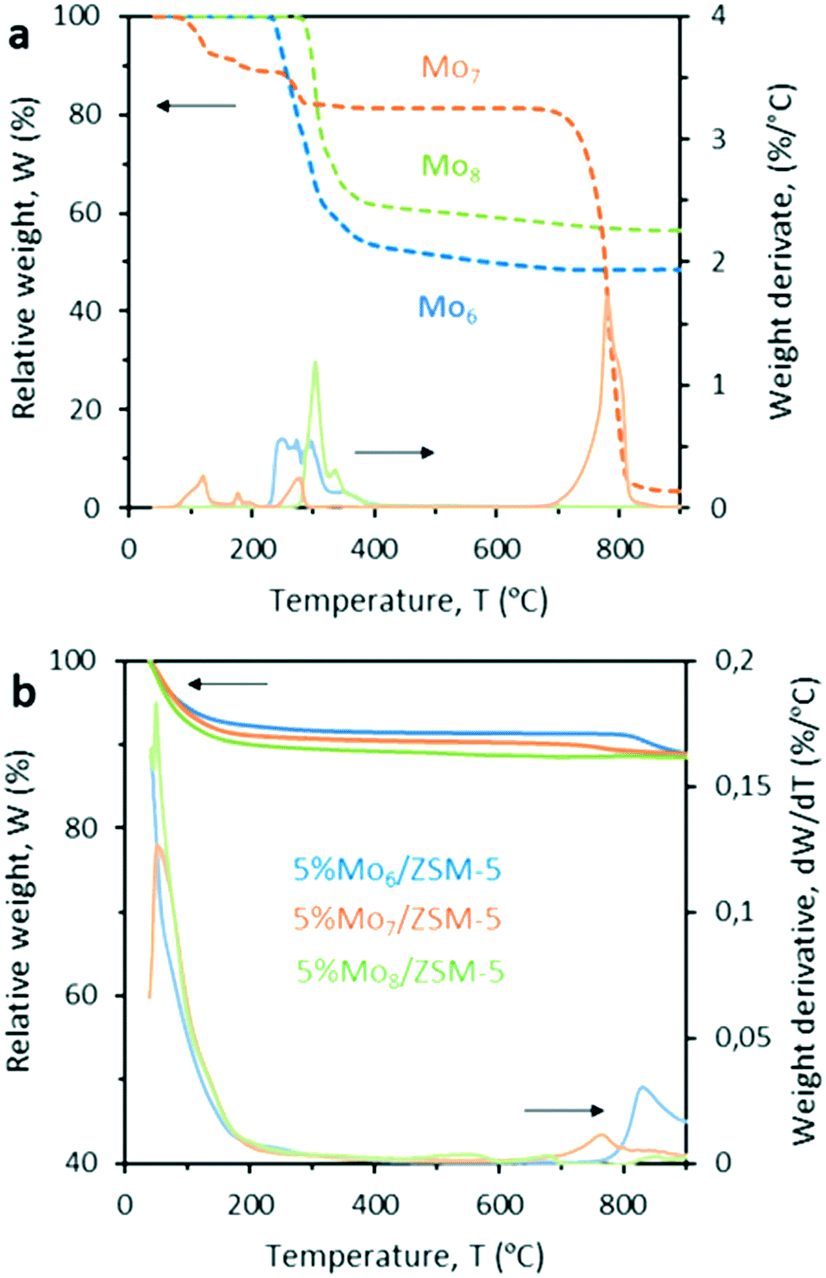 | ||
| Fig. 2 Thermogravimetric analysis of a) Mo precursors Mo6, Mo7 and Mo8 and b) fresh Mox/ZSM-5 catalysts calcined at 550 °C. | ||
| Fresh catalyst | Spent catalyst | Fresh catalyst | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Moδ+ | Mo4+ | Mo5+ | Mo6+ | Moδ+ | Mo4+ | Mo5+ | Mo6+ | Atomic Si/Mo | Surface % wt Mo | |
| a Binding energy (eV). b Abundance of the Mo oxidation state in the sample (%). | ||||||||||
| 5% Mo6/ZSM-5 | 232.1a | 233.3 | 229.3 | 230.7 | 233.7 | 34.6 | 3.7% | |||
| 22%b | 78% | 25% | 10% | 65% | ||||||
| 5% Mo7/ZSM-5 | 232.0 | 233.4 | 229.0 | 230.1 | 232.5 | 234.0 | 31.5 | 4.1% | ||
| 3% | 97% | 26% | 12% | 35% | 27% | |||||
| 5% Mo8/ZSM-5 | 233.0 | 229.3 | 230.4 | 234.2 | 16.2 | 6.5% | ||||
| 100% | 15% | 34% | 51% | |||||||
| 5% Mo7/MCM-22 | 233.5 | 229.2 | 230.4 | 233.7 | 37.7 | 3.4% | ||||
| 100% | 11% | 19% | 70% | |||||||
| 5% Mo6/MCM-22 | 232.4 | 233.6 | 228.9 | 230.9 | 233.2 | 48.4 | 2.9% | |||
| 38% | 62% | 21% | 33% | 46% | ||||||
| 8% Mo6/MCM-22 | 233.3 | 229.4 | 230.5 | 233.8 | 25.4 | 4.6% | ||||
| 100% | 18% | 42% | 40% | |||||||
| 10% Mo6/MCM-22 | 232.8 | 229.0 | 230.2 | 233.8 | 21.2 | 5.0% | ||||
| 100% | 25% | 35% | 40% | |||||||
Both Si/Mo ratio and weight content of Mo at the external surface of the fresh samples reveal that: a) the bigger the Mo precursor anion and the higher the bulk Mo load, the higher the concentration of the Mo species at the surface; b) the MCM-22 support is able to hold more Mo species within the pores compared to ZSM-5.
Raman spectroscopy was performed to gain insight into the MoOx structures formed in each of the different samples by detecting the metal-oxide vibrational modes in all of these powders (Fig. 3).
 | ||
| Fig. 3 Raman spectra of the catalysts with different Mo precursors (5% Mo loading). | ||
All the samples showed a wide band at 394 cm−1 corresponding to the reticular vibration of the zeolite. Additionally, the band at around 295 cm−1 is attributed to the bending mode of terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in crystalline MoO3 species and 1003 cm−1 to the MoO stretching mode in isolated molybdates (only slightly perceived in the catalyst prepared using the Mo8 octamolybdate precursor). The absence or very low intensity of bands at 680 and 820 cm−1 corresponding to the bending and stretching modes of the Mo–O–Mo bond suggests good metal dispersion and the absence of big crystalline MoO3 facets at the zeolite surface.45–47 It seems that all of the catalysts with 5% Mo show the characteristic dimeric [Mo2O5]2+ species anchored to the zeolite support, since all present the band at 960 cm−1. However, the greater intensity of this band in the Mo6 sample is attributed to the vibration of a greater number of molecular molybdate–support bonds. Analogously, the lower intensity of this band for the samples prepared with Mo7 and Mo8 precursors is attributed to the lower presence of anchored (MoOx)n+ species and, thus, is attributed to more Mo clustering and poorer dispersion. This hypothesis was further confirmed by TEM, STEM-EDX and 27Al MAS NMR. As can be observed in Fig. 4, the bigger the size of the initial polyoxoanion (Mo precursor), the bigger and less disperse the size of Mo oxide clusters on the zeolite support. The high metal dispersion in 5% Mo6/ZSM-5 is particularly remarkable. Accordingly, 27Al MAS NMR characterization suggested that the use of Mo8 as the precursor was detrimental for the synthesis of 5% Mo/ZSM-5. Mo8/ZSM-5 showed significant dealumination of the zeolite support and generation of both superficial and bulk Al2(MoO4)3 species in contrast to Mo6 or Mo7. Mo6 was the precursor that minimized both dealumination and generation of aluminum molybdate crystallites at the external surface (Fig. S8†). In agreement with the previous observation, N2 adsorption measurements indicate that the use of the Mo6 precursor leads to the smallest pore volume and a surface area decrease with respect to the fresh zeolite support and, thus, leads to greater metal dispersion (Table 3). The detailed pore size distribution of the pristine and impregnated zeotype supports with 5% Mo6 is observed in Fig. S9.†
O in crystalline MoO3 species and 1003 cm−1 to the MoO stretching mode in isolated molybdates (only slightly perceived in the catalyst prepared using the Mo8 octamolybdate precursor). The absence or very low intensity of bands at 680 and 820 cm−1 corresponding to the bending and stretching modes of the Mo–O–Mo bond suggests good metal dispersion and the absence of big crystalline MoO3 facets at the zeolite surface.45–47 It seems that all of the catalysts with 5% Mo show the characteristic dimeric [Mo2O5]2+ species anchored to the zeolite support, since all present the band at 960 cm−1. However, the greater intensity of this band in the Mo6 sample is attributed to the vibration of a greater number of molecular molybdate–support bonds. Analogously, the lower intensity of this band for the samples prepared with Mo7 and Mo8 precursors is attributed to the lower presence of anchored (MoOx)n+ species and, thus, is attributed to more Mo clustering and poorer dispersion. This hypothesis was further confirmed by TEM, STEM-EDX and 27Al MAS NMR. As can be observed in Fig. 4, the bigger the size of the initial polyoxoanion (Mo precursor), the bigger and less disperse the size of Mo oxide clusters on the zeolite support. The high metal dispersion in 5% Mo6/ZSM-5 is particularly remarkable. Accordingly, 27Al MAS NMR characterization suggested that the use of Mo8 as the precursor was detrimental for the synthesis of 5% Mo/ZSM-5. Mo8/ZSM-5 showed significant dealumination of the zeolite support and generation of both superficial and bulk Al2(MoO4)3 species in contrast to Mo6 or Mo7. Mo6 was the precursor that minimized both dealumination and generation of aluminum molybdate crystallites at the external surface (Fig. S8†). In agreement with the previous observation, N2 adsorption measurements indicate that the use of the Mo6 precursor leads to the smallest pore volume and a surface area decrease with respect to the fresh zeolite support and, thus, leads to greater metal dispersion (Table 3). The detailed pore size distribution of the pristine and impregnated zeotype supports with 5% Mo6 is observed in Fig. S9.†
 | ||
| Fig. 4 TEM images of the fresh catalyst synthesized with different polyoxomolybdate precursors and zeolite supports: a) 5% Mo6/ZSM-5, b) 5% Mo7/ZSM-5, and c) 5% Mo8/ZSM-5. d) STEM-HAADF images of 5% Mo7/ZSM-5 with EDX data to illustrate the presence of Mo nanoclusters, e) 5% Mo6/MCM-22, and f) 5% Mo7/MCM-22. STEM-HAADF micrographs of Mo6/MCM-22 with Mo loads: 5% (g), 8% (h) and 10% (i). | ||
| Sample | BET surface area (mb g−1 zeolite) | Max. pore volume (cm3 g−1 zeolite) | Metal loada (wt%) | Metal loadb (wt%) |
|---|---|---|---|---|
| a Determined by MP-AES. b Determined by SEM-EDX. | ||||
| H-ZSM-5 | 571 ± 4 | 0.198 | — | — |
| 5% Mo6/ZSM-5 | 452 ± 2 | 0.155 | 4.7 ± 0.1 | 4.9 ± 0.2 |
| 5% Mo7/ZSM-5 | 438 ± 2 | 0.151 | 4.7 ± 0.1 | 4.9 ± 0.7 |
| 5% Mo8/ZSM-5 | 436 ± 3 | 0.151 | 3.9 ± 0.2 | 4.0 ± 0.5 |
| MCM-22 | 768 ± 3 | 0.262 | — | — |
| 5% Mo6/MCM-22 | 685 ± 2 | 0.248 | 4.5 ± 0.5 | 4.4 ± 0.4 |
| 8% Mo6/MCM-22 | 551 ± 2 | 0.180 | 7.6 ± 0.3 | 7.7 ± 1.0 |
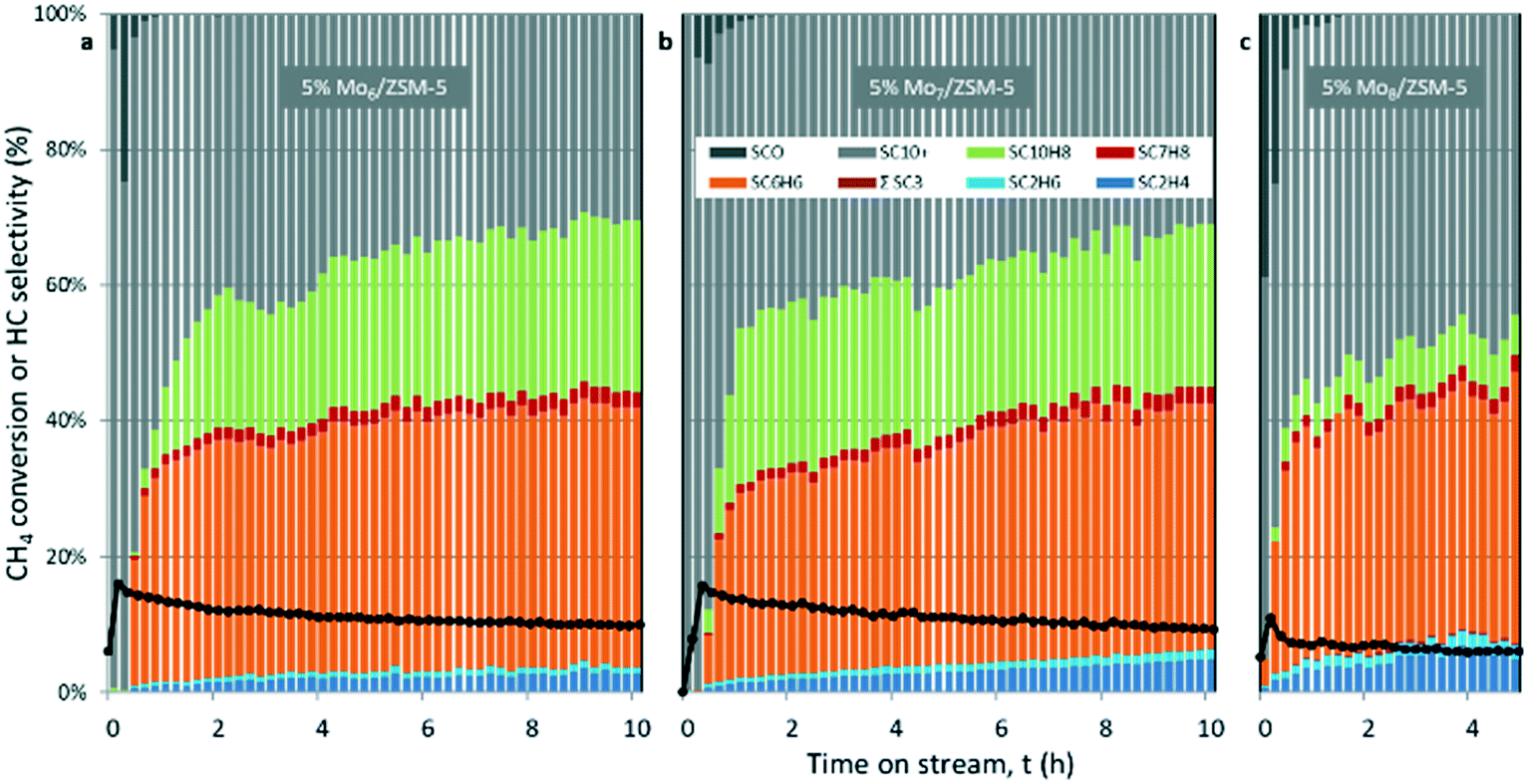 | ||
| Fig. 5 CH4 conversion and hydrocarbon product selectivity for MDA on 5% Mox/ZSM-5 using Mo6 (a), Mo7 (b) and Mo8 (c) as Mo precursors. Experimental conditions: 1500 mL gcat−1 h−1, 700 °C, 1 atm, CH4:N2 = 80:20. | ||
The Mo6/ZSM-5 catalyst exhibited the highest methane conversion among the three catalysts in our study. For instance, methane conversions for the three catalysts after 4 hours on stream are xCH4-4h (5% Mo6/ZSM-5) = 11.6%; xCH4-4h (5% Mo7/ZSM-5) = 11.2%; xCH4-4h (5% Mo8/ZSM-5) = 6.5%. These differences in the MDA performance may be attributed to the different accessibility of CH4 to the Mo sites, which also depends on the Mo dispersion on the support.
The Mo7/ZSM-5 catalyst showed the highest initial selectivity towards aromatics, i.e. benzene and naphthalene (Fig. 5b), however, it was not as stable as the Mo6 sample which, after 3 h of reaction, exhibited the highest stable production of benzene YC6H6 of ca. 4.5%.
In contrast, the catalyst containing the Mo8 precursor showed the lowest CH4 conversion and thus the lowest hydrocarbon productivity. This poor performance could be attributed to Mo remaining on the surface of the catalyst (as indicated by the comparatively larger MoO3 aggregates observed by STEM-HAADF). Moreover, this catalyst displayed a greater tendency for coke formation (Fig. 5c), with Scoke (4h) roughly 49% higher than that for the Mo6 catalyst. The spent catalysts after 15 hours on stream were subjected to regeneration under the conditions stated in the Experimental procedure section until CO2 was not detected in the outlet gas.
A second MDA reaction cycle was carried out with the regenerated catalysts to evaluate the effect of the reaction-regeneration cycles on metal dispersion and catalyst stability for the two most suitable candidates according to the previous results, i.e. Mo6/ZSM-5 and Mo7/ZSM-5.
The performance of the fresh and regenerated catalysts, together with the STEM-HAADF micrographs of the fresh, spent and regenerated catalyst samples is depicted in Fig. 6. During catalyst regeneration and coke burning, Mo species sintered and partial migration towards the outer surface of the zeolite occurred. Consequently, the easier access of methane molecules to the metal surface and partial pore blocking by the clusters, led to an increase of methane conversion and selectivity to C2. Interestingly, the sample prepared with Mo6 as the precursor formed smaller aggregates after regeneration (Fig. 6e), keeping higher metal dispersion and promoting benzene yield with respect to the conventional heptamolybdate impregnation catalyst (Fig. 6g and h).
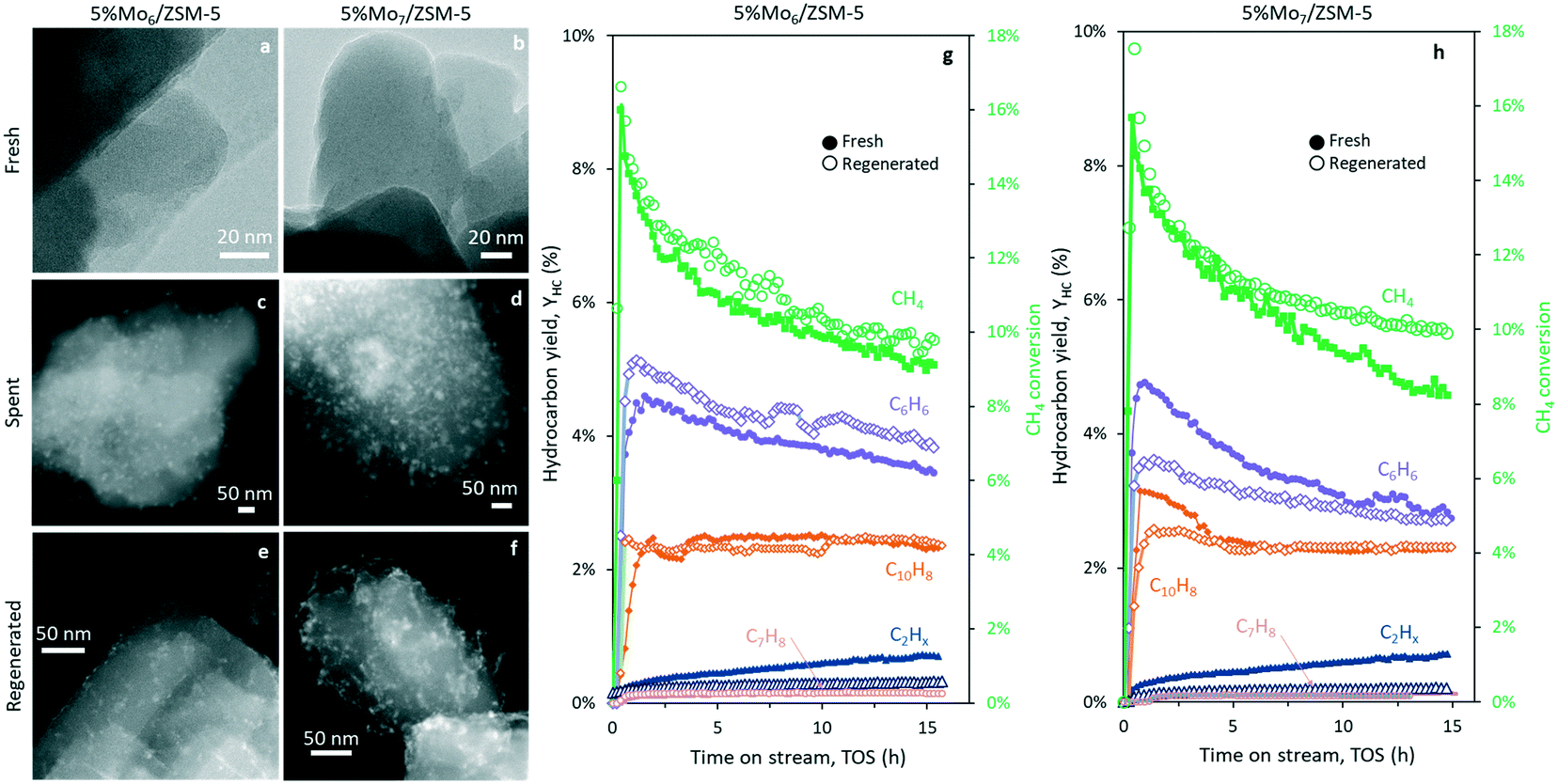 | ||
| Fig. 6 STEM-HAADF micrographs of the fresh (a and b), spent (c and d) and regenerated (e and f) 5% Mo6/ZSM-5 (a, c and e) and 5% Mo7/ZSM-5 (b, d and f) samples. MDA performance of the fresh and regenerated 5% Mo6/ZSM-5 (g) and 5% Mo7/ZSM-5 (h) catalysts. Operating conditions: 700 °C, 1500 mL gcat−1 h−1, CH4:N2 = 80:20. | ||
Fig. 6f reveals the partial accumulation of Mo species in the form of filaments at the zeolite surface of Mo7/ZSM-5, suggesting that the coking mechanism may proceed differently between Mo6- and Mo7-based samples. The observation of carbon nanotubes and fibers in spent Mo7/ZSM-5 and its absence in Mo6/ZSM-5 may explain the stronger methane conversion and benzene yield decay along the time on stream (Fig. 6h) using the fresh Mo7/ZSM-5 catalyst.
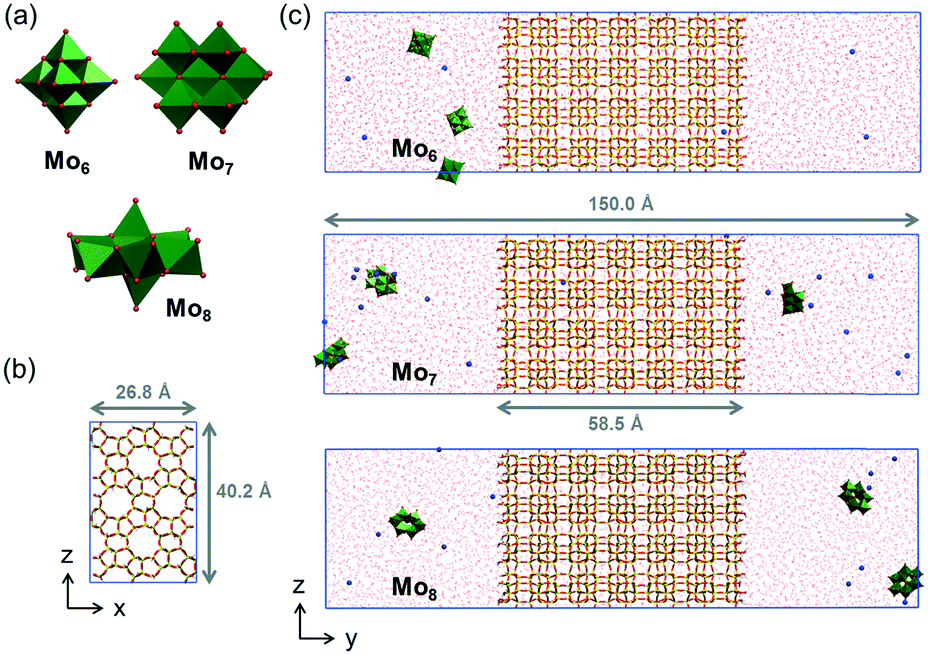 | ||
| Fig. 7 a) Polyhedral representation of the analyzed polyoxomolybdates, [Mo6O19]2− (Mo6), [Mo7O24]6− (Mo7) and [Mo8O26]4− (Mo8); b) view of the periodic cell employed in the simulations along the y axis, which is perpendicular to the zeolite surface; c) snapshot of the simulated systems with Mo6 (top), Mo7 (middle) and Mo8 anions (bottom) at the beginning of the simulation. | ||
The visual analysis of the trajectories revealed that the analysed anions behave rather differently over the zeolite surface. While Mo6 and Mo8 anions spend most of the time in the solvent bulk and approach the zeolite in scarce occasions, Mo7 anions appear to be more prone to interact with the surface. This is reflected in Fig. 8a, in which the density of Mo7 anions at the solvent–zeolite interface is much higher than those of Mo6 and Mo8. The latter two present a more homogeneous distribution in the range of distances covered by the solution, indicating a lower affinity to the surface. In fact, while only short-lived contacts lasting no more than 800 ps were observed between Mo6 and ZSM-5, Mo7 anions were found to be anchored on the ZSM-5 surface for dozens of nanoseconds. Mo8 anions are found in an intermediate situation, as their contacts with the surface can last up to 2.5 ns. In the case of Mo7, the first anion reaches the surface after 2 ns of simulation and interacts there until the end of the simulation. The same phenomenon was observed for the second Mo7 unit that comes in contact with the zeolite after ca. 14 ns of the MD run. The third unit keeps moving through the solvent subject to Brownian motion during the whole simulation, most likely because the presence of two polyanions already deposited on the surface makes the adsorption of the third one less favourable due to electrostatic repulsion. The trends in the residence time of each anion on the ZSM-5 surface can be correlated to the charge density of the anion, estimated as the charge per metal ratio (q/m). This parameter has been extensively used in POM chemistry as a descriptor of the charge density at the surface of the polyoxoanions and in fact, it nicely correlates with the Brønsted basicity of POMs52 or with their hydrophilic characteristics.31 Therefore, Mo7 (q/m = 0.86) is the anion among the series bearing the highest charge density, followed by Mo8 (q/m = 0.50) and finally Mo6 (q/m = 0.33). For the association of POMs to hydrophobic bodies such as proteins, it has been observed that high charge densities can result in structures that are too hydrophilic to provide persistent interaction. However, the polar characteristics of the ZSM-5 surface and its pores full of water molecules favour the association of highly charged anions such as Mo7.
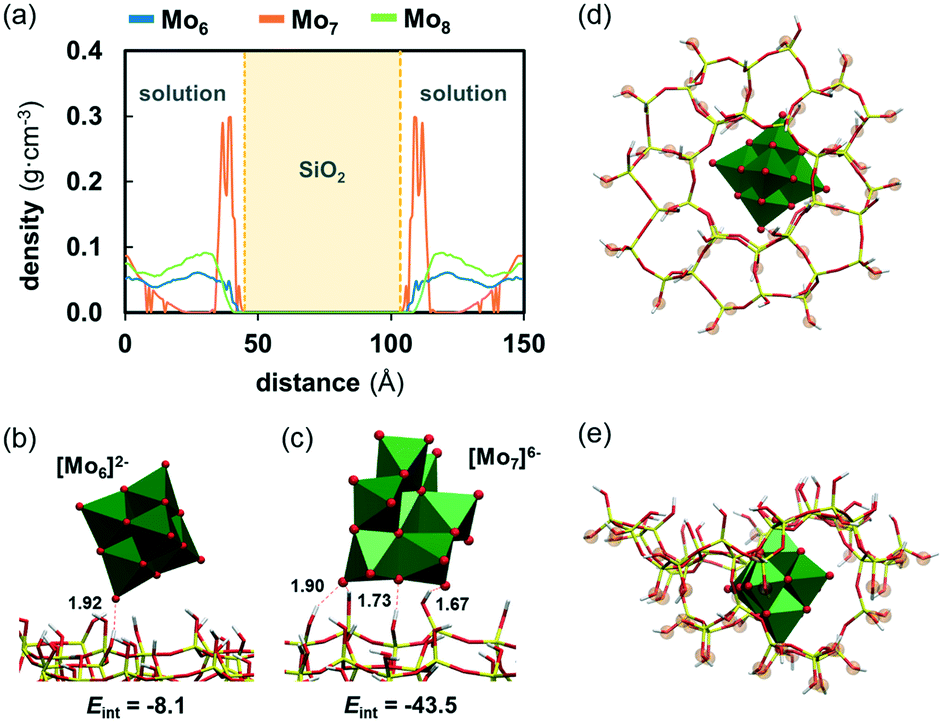 | ||
| Fig. 8 a) Density of Mo6 (blue line), Mo7 (orange line) and Mo8 (green line) along the y axis of the simulation box averaged over 100 ns of simulation; b) and c) representative snapshots of the interaction of Mo6 and Mo7 on the ZSM-5 surface. Solvent molecules were omitted for clarity. Distances are given in Å. Eint (kcal mol−1) represents the non-bonding interaction energy between an individual anion and the zeolite averaged over 3 POMs and over all the frames in which there is at least 1 hydrogen bond between the POM and the zeolite (H-bond criteria: d ≤ 3.5 Å; ∠(O–H⋯OPOM) ≤ 130°); optimised structure of the model system used to calculate the encapsulation energy of Mo6 inside a pore of ZSM-5: (d) top view and (e) side view. Highlighted oxygen atoms remained fixed during the geometry optimization at the position of the crystal, and they were capped with hydrogen atoms. | ||
Fig. 8b and c show the representative snapshots of the interaction of Mo6 and Mo7 on the zeolite surface. These interactions mainly involve hydrogen bonds between hydroxyl groups at the zeolite surface acting as donors and the basic oxygen atoms of the POM framework, being the terminal oxygens participating more actively in the contacts since they are more exposed. These hydrogen bonds lay within a range of distances from 1.6 to 2.4 Å and thus, they can be classified as moderate H bonds in which the interaction is mostly electrostatic. Accordingly, the strength of these attractive interactions was found to be strongly dependent on the charge of the cluster. Those concerning the least charged Mo6 are weak (Eint = −8.1 kcal mol−1 in average) and involve an average number of 1.3 hydrogen bonds, with a maximum number of 4. Oppositely, the Mo7 anion, which bears the highest charge density among the series, affords much stronger and consequently, more persistent interactions with ZSM-5 (Eint = −43.5 kcal mol−1 in average). In line with the fact that Mo8 behaves similarly to Mo6, it also provides weak interactions with the surface (Eint = −9.6 kcal mol−1 in average). Interestingly, we found that the topology of Mo7 does allow the formation of not only a larger number of H bonds with Si–OH groups (3.1 in average, with a maximum of 7), but also stronger ones (interaction energy per H bond is −6.2 kcal mol−1 for Mo6vs. −14.0 kcal mol−1 for Mo7). The nature of these interactions indicates that the adsorption of Mo7 on the surface of the zeolite has a less reversible character than that of Mo6 or Mo8. Unlike Mo7, Mo6 and Mo8 are freer to move through the surface and in and out the solvent. This fact could be related to the higher catalytic activity when using Mo6 as the precursor compared to Mo7, as the latter is not as likely to move from the surface into the pores as Mo6, which is less prone of becoming trapped at the surface by silanol groups. Therefore, it is reasonable to think that Mo-carbides resulting from calcination of Mo7 might be more prone to remain on the surface instead of inside the pores, which is detrimental for the catalytic activity.
On the other hand, the experimentally observed differences between Mo6 and Mo8 cannot be rationalized from their behaviour at the solution/zeolite interface. As the higher activity when using the Mo6 compared to Mo8 may arise from the fact that the smaller anion can get into the zeolite pores more easily, we evaluated the ability of Mo6 to move into the ZSM-5 structure using a cluster model for the zeolite (see Fig. 8d and e). Although Mo6 appears to have the right size and shape to fit inside a pore, we found that this process is highly electronically unfavourable (ΔE ≈ +100 kcal mol−1). This suggests that, as with the other precursors, the Mo6 cluster might have to break into smaller fragments to reach the internal part of the pores. In fact, Mukai et al. reported the synthesis of a Keggin-type phosphomolybdate with [PMo12O40]3− inside the supercages of a Y-zeolite53,54 which are connected by windows of 7.4 Å in diameter. They reported that most of the synthesized POM remained in the sample after several washing steps, which was attributed to the fact that Keggin anions are sterically trapped inside the pores, as they possess a very large diameter to move through the windows. Analogously, Chen et al.55 reported that, although the phosphomolybdate cannot penetrate the pore system of the ZSM-5 zeolite, molybdenum species coming from Keggin-type phosphomolybdate can be encapsulated in the secondary pores of nanocrystalline ZSM-5. Particularly, these authors found that the use of [PMo12O40]3− as the Mo precursor improved the activity of n-octane conversion on Mo–Ni/ZSM-5 catalysts. Considering that the diameter of Keggin anions is only about 2.3 Å larger than the Lindqvist (Mo6) one and that the diameter of ZSM-5 windows is ca. 5 Å, it might be reasonable to think that Mo6 is too large to cross the pore windows in one piece. This is further supported by the highly unstable nature of the anion inside the cage. By gathering all these results, we hypothesised that the thermal decomposition of Mo6 might lead to a higher and more disperse amount of cationic dimers [Mo2O5]2+ with the ability to anchor to the acid sites of the zeolite compared to that of Mo8, which might give a wider mixture of species upon calcination, some of them being useful and some others detrimental to the catalytic activity, such as MoO3. The intricate mechanisms of metal oxide decomposition are out of the scope of this work and in this regard, future work will be devoted to the characterisation of the intermediates and products in the thermal decomposition of polyoxomolybdates moving towards the rational selection of catalyst precursors for improved supported catalysis.
Role of the support
With the aim to increase the selectivity to benzene, we selected a support (MCM-22) with a smaller pore size than ZSM-5 and very similar to the size of the dynamic diameter of the benzene molecule (Fig. 1c).4 Therefore, by keeping the same metallic load (5 wt% Mo), the effect of changing the support to MCM-22 was evaluated with the two polyoxomolybdate precursors that exhibited higher benzene productivity: Mo6 and Mo7 (Fig. 9).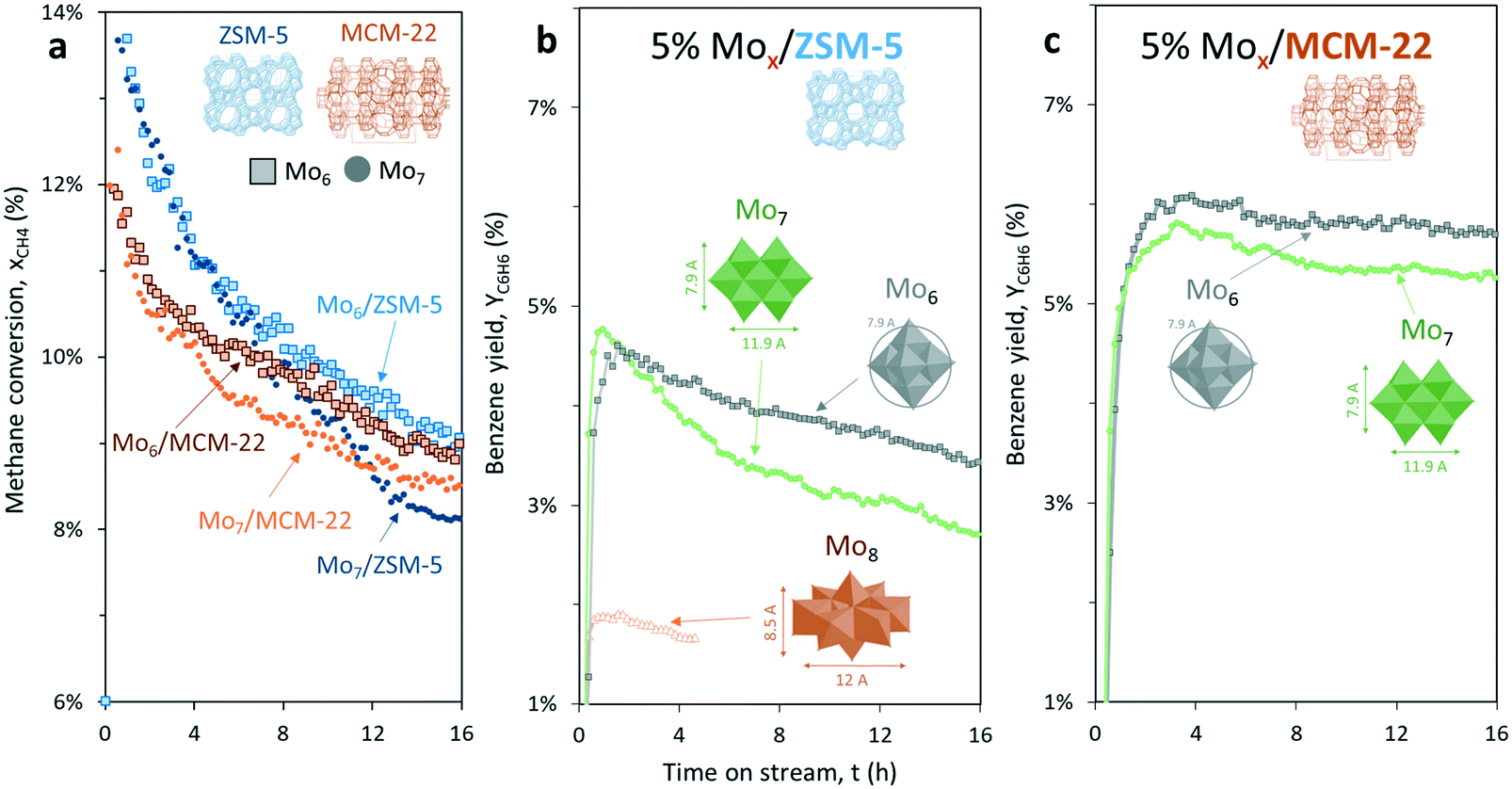 | ||
| Fig. 9 (a) Methane conversion, (b) benzene yield for 5% Mox/ZSM-5 and (c) 5% Mox/MCM-22 catalysts under MNOC at 700 °C and 1500 mL gcat−1 h−1 (CH4:N2 = 80:20), where x = 6 or 7. | ||
As a general observation, methane conversion on MCM-22 is slightly lower (e.g. roughly 10.1% in the case of ZSM-5, and 9.0% for MCM-22 after 5 hours on stream) but this support leads to less coking and higher stability with respect to the time on stream (Fig. 9a). In addition, MCM-22 is more selective to benzene than ZSM-5.
Regardless of the selected zeotype support, the catalysts prepared with Mo6 as a Mo precursor outperform the catalysts prepared with the commercial salt (Mo7), both in terms of stability and aromatics productivity.
For instance, benzene productivity after 5 hours on stream for each tested sample was 6.1% in the case of Mo6/MCM-22, 5.1% for Mo7/MCM-22, 4.1% in the case of Mo6/ZSM-5 and just 3.7% for Mo7/ZSM-5, respectively (Fig. 9b and c).
The STEM-HAADF micrographs of the spent and regenerated 5% Mox/MCM-22 samples also revealed the presence of bigger coke aggregates (spent) and superficial Mo-oxide clusters (regenerated) in the sample prepared using the commercial heptamolybdate species (Fig. S4†). This further supports the previous results on ZSM-5, illustrating the improved stability of the Mox/MCM-22 catalysts prepared using Mo6 as the precursor.
Up to this point, the combination of 5% wt Mo6 and MCM-22 has led to the highest and most stable benzene yield.
Role of the metal load
Following up the previous results, the catalytic performance of the most promising sample, i.e. Mo6/MCM-22, is evaluated for the MDA reaction process using different Mo loadings (5, 8 and 10 wt%).This behavior is related to the accumulation of Mo species formed during the calcination, either as oxides or molybdate dimers, that generate both saturation of the zeolite channels ([Mo2O5]2+) and partial blockage of the pores (MoO3). As the STEM-HAADF images indicate, the excess Mo accumulates in the form of Mo aggregates on the support surface (Fig. 4). These micrographs confirm the formation of large Mo clusters at 8 wt% Mo load (Fig. 4h) as well as large external crystals on the surface of the support containing 10 wt% Mo (Fig. 4i). Importantly, these clusters are thought to be responsible for both the very high initial catalytic activity and the fast deactivation due to coking of the 10% Mo6/MCM-22 sample (Fig. 10).
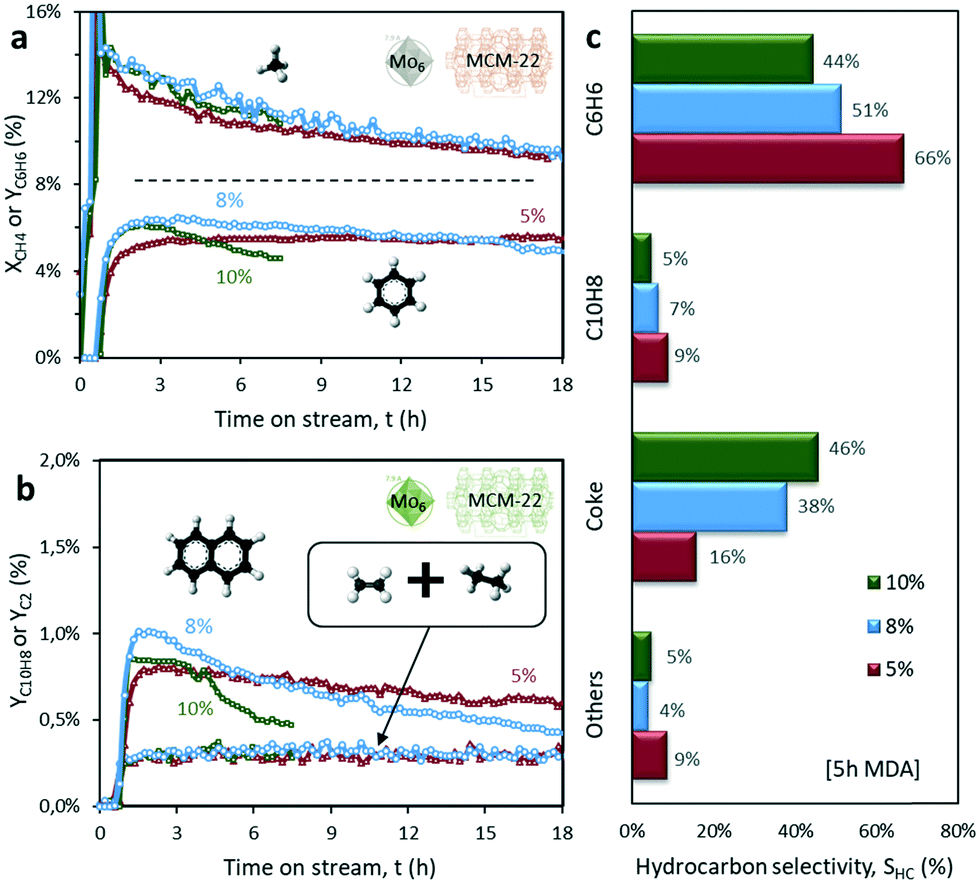 | ||
| Fig. 10 a) Transient methane conversion and benzene yield, b) naphthalene and C2 (ethane + ethylene) yield and c) hydrocarbon selectivity distribution after 5 hours on stream obtained for x% Mo6/MCM-22 catalysts using different Mo loadings (5, 8, 10 wt%). Operating conditions: 700 °C, 1500 mL gcat−1 h−1, CH4:N2 = 80:20. | ||
The XRD diffractograms of the three samples confirmed the formation of large external Mo clusters at 8% and 10% Mo loads, as revealed by the characteristic MoO3 peak at 23.3° (Fig. S10†). The 5% Mo/MCM-22 sample, however, does not present any peak at this diffraction angle, thus, indicating a good overall metal dispersion within the MCM-22 pores. In addition, the Raman spectra showed that new peaks related to crystalline MoO3 appeared at 670, 820, and 1000 cm−1 for the 8% Mo and 10% Mo samples, which supports the XRD results. The XRD and Raman results for the three considered MCM-22 based catalysts are discussed in detail in the ESI† (Fig. S10).
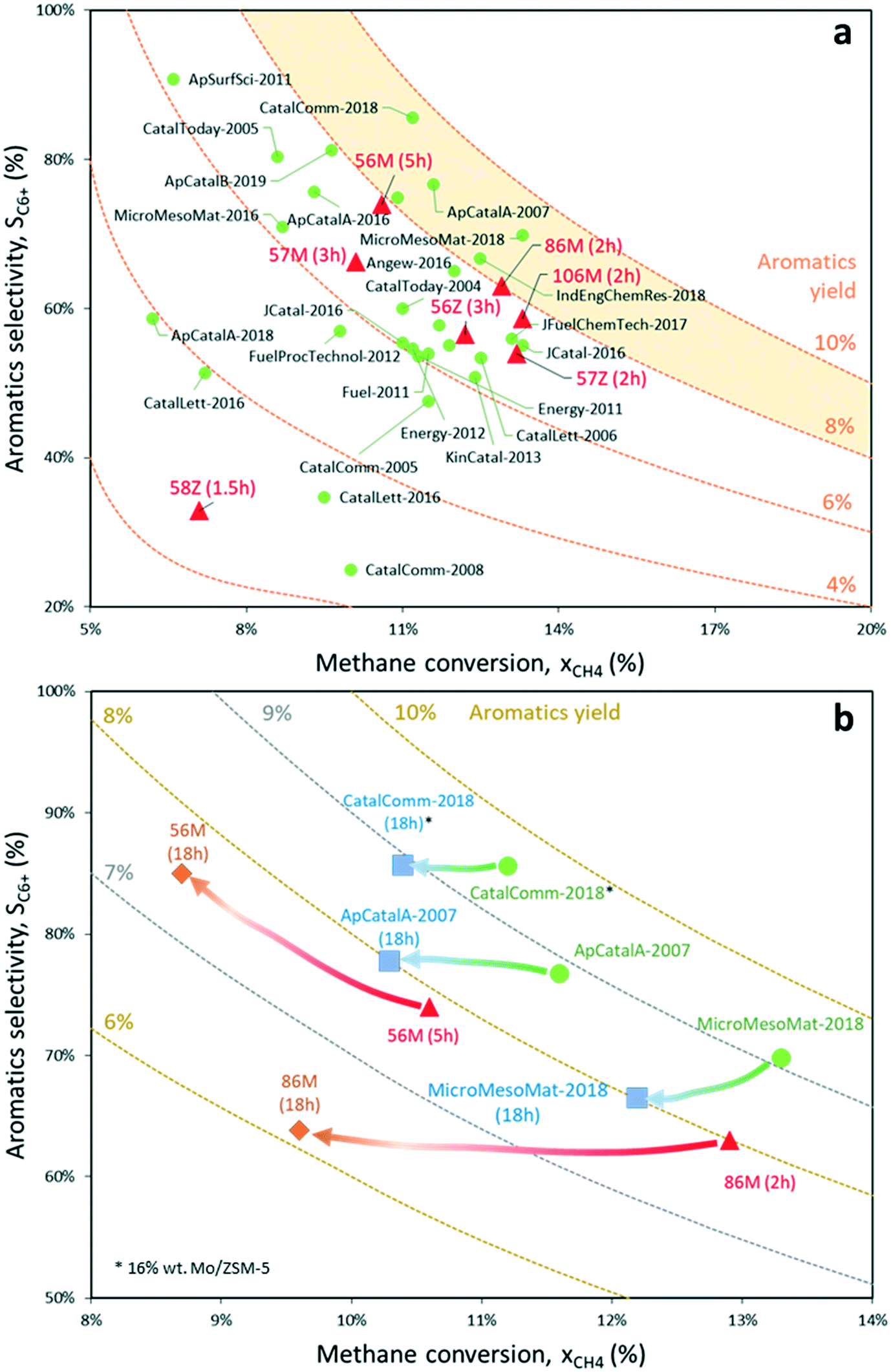 | ||
| Fig. 11 a) Overview of the highest transient aromatics yields reported in the literature for methane dehydroaromatization performed at 700 °C using Mo-based catalysts supported on different zeotypes.5,7,8,56–74 Red triangles represent experimental values reported in this work. Code: “% wt Mo – Mo precursor – Zeotype (Time on stream)”, e.g. “56M” corresponds to 5% Mo6/MCM-22; b) methane conversion and aromatics selectivity evolution after 18 hours on stream for the catalysts with the highest reported transient aromatics productivity15,56,57 and the two best catalysts of this work, i.e. 5% Mo6/MCM-22 and 8% Mo6/MCM-22. | ||
The following aromatic compounds present in the product gas were included in the calculation of the selectivity to aromatics for samples 5% Mo6/MCM-22 and 8% Mo6/MCM-22: benzene, toluene, naphthalene (main MDA product), o-, m- and p-xylene, ethylbenzene, anthracene and C12+ (traces). By plotting these results, it is clear that the main advantage of the 5% Mo6/MCM-22 catalyst is that it maintains a nearly constant aromatics yield for more than 18 hours on stream thanks to the joint effect of the MCM-22 support and hexamolybdate Mo precursor. Crucially, the excellent metal dispersion on this sample helps to minimize coke production and maintain stable aromatics productivity in comparison to the best Mo/ZSM-5 and Mo/MCM-22 samples reported in the literature for the MDA process,15,56,57 for which the yield drops faster. Nonetheless, except for the best Mo-based catalyst to date, which keeps an outstanding 9% aromatics yield after 18 hours on stream but contains a 16% wt Mo load,56 the rest provide similar long-term aromatics productivity to 5% Mo6/MCM-22, although keeping lower selectivity.
Conclusions
The dispersion of MoOx species was found to be the key factor to enhance the catalytic stability of Mo/ZSM-5 and Mo/MCM-22-based catalysts for the non-oxidative coupling of methane.A homogeneous metal load distribution was observed in the case of 5% wt loading, regardless of the zeotype support. Samples with higher metal loads (8 and 10 wt% Mo) did not improve the MDA process yield towards aromatics, but rather resulted in the formation of MoO3 aggregates at the surface of the support that led to rapid catalyst deactivation.
The characterization of the interaction between the different POM anions and ZSM-5 by means of MD simulations evidenced strong and highly persistent interactions between ZSM-5 and Mo7, whereas Mo6 and Mo8 showed much weaker and shorter interactions due to their lower charge density. The trapping of Mo7 at the surface could hamper the deposition of molybdenum oxide fragments inside the pores upon calcination, in line with the lower catalytic activity observed for this precursor.
In contrast, Mo6 exhibits weaker contacts with the surface that might result in higher preference for loading molybdenum oxide inside the zeolite structure rather than on the surface. Although the Mo8 precursor also interacts weakly with the zeolite surface, it shows lower catalytic activity than Mo6, presumably due to the formation non-catalytically active species during the calcination process.
The laboratory-synthesized MCM-22 support outperformed the commercial ZSM-5 zeolite in terms of stability (less coking) and benzene yield (main product of interest). The Mo precursor based on the polyoxomolybdate [(C4H9)4N]2[Mo6O19] “Mo6” was found to be more active and selective towards benzene than the commercial polyoxomolybdate salt (NH3)6[Mo7O24]·4H2O.
The 5% Mo6/MCM-22 catalyst displays superior stability (pseudo-steady-state benzene yield for 18 hours on stream) and high transient selectivity to aromatic compounds (>75%), working at 700 °C with a diluted methane flow (CH4:N2 = 80:20) under atmospheric pressure with a spatial velocity of 1500 mL gcat−1 h−1. Importantly, the catalysts prepared with the “Mo6” precursor are among the most promising catalysts for methane dehydroaromatization found in the literature and these results pave the way for the selection and use of other types of polyoxometalates as precursors to be used in formulations to further improve on the well-established (standard) precursor, heptamolybdate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the European Union's Horizon 2020 Research and Innovation Programme (ADREM project – Grant Agreement No. 680777) is gratefully acknowledged. The microscopy measurements were conducted at the Laboratorio de Microscopias Avanzadas, Instituto de Nanociencia de Aragon, Universidad de Zaragoza, Spain. The synthesis of the materials has been performed at the Platform of Production of Biomaterials and Nanoparticles of the NANOBIOSIS ICTS, more specifically at the Nanoparticle Synthesis Unit of the CIBER in BioEngineering, Biomaterials & Nanomedicine (CIBER-BBN). JMP thanks the Spanish Ministry of Science (CTQ2017-87269-P), the Generalitat de Catalunya (2017SGR629) and the URV for support. JMP also thanks the ICREA foundation for an ICREA ACADEMIA award.References
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, Stable Mo/HZSM-5 methane dehydroaromatization catalysts optimized for high-temperature calcination-regeneration, J. Catal., 2017, 346, 125–133 CrossRef CAS.
- Y. Xu, X. Bao and L. Lin, Direct conversion of methane under nonoxidative conditions, J. Catal., 2003, 216, 386–395 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Dehydrogenation and Aromatization of Methane Under Nonoxidizing Conditions, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- A. Corma, C. Corell and J. Perez-Pariente, Synthesis and Characterization of the Mcm-22 Zeolite, Zeolites, 1995, 15, 2–8 CrossRef CAS.
- P. Wu, Q. Kan, D. Wang, H. Xing, M. Jia and T. Wu, The synthesis of Mo/H-MCM-36 catalyst and its catalytic behavior in methane non-oxidative aromatization, Catal. Commun., 2005, 6, 449–454 CrossRef CAS.
- H. Liu, C. Zhou, Y. Zhang and Q. Kan, Facile preparation of hierarchically porous IM-5 zeolite with enhanced catalytic performance in methane aromatization, J. Fuel Chem. Technol., 2017, 45, 1074–1081 CrossRef CAS.
- A. Martinez, E. Peris and G. Sastre, Dehydroaromatization of methane under non-oxidative conditions over bifunctional Mo/ITQ-2 catalysts, Catal. Today, 2005, 107–108, 676–684 CrossRef CAS.
- K. Sun, D. M. Ginosar, T. He, Y. Zhang, M. Fan and R. Chen, Progress in Nonoxidative Dehydroaromatization of Methane in the Last 6 Years, Ind. Eng. Chem. Res., 2018, 57, 1768–1789 CrossRef CAS.
- J. J. Spivey and G. Hutchings, Catalytic aromatization of methane, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- P. Schwach, X. Pan and X. Bao, Direct Conversion of Methane to Value-Added Chemicals over Heterogeneous Catalysts: Challenges and Prospects, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- I. Vollmer, G. Li, I. Yarulina, N. Kosinov, E. J. Hensen, K. Houben, D. Mance, M. Baldus, J. Gascon and F. Kapteijn, Relevance of the Mo-precursor state in H-ZSM-5 for methane dehydroaromatization, Catal. Sci. Technol., 2018, 8, 916–922 RSC.
- I. Vollmer, A. Mondal, I. Yarulina, E. Abou-Hamad, F. Kapteijn and J. Gascon, Quantifying the impact of dispersion, acidity and porosity of Mo/HZSM-5 on the performance in methane dehydroaromatization, Appl. Catal., A, 2019, 574, 144–150 CrossRef CAS.
- I. Vollmer, B. van der Linden, S. Ould-Chikh, A. Aguilar-Tapia, I. Yarulina, E. Abou-Hamad, Y. G. Sneider, A. I. O. Suarez, J. Hazemann, F. Kapteijn and J. Gascon, On the dynamic nature of Mo sites for methane dehydroaromatization, Chem. Sci., 2018, 9, 4801–4807 RSC.
- C. Sun, S. Yao, W. Shen and L. Lin, Highly dispersed molybdenum oxide supported on HZSM-5 for methane dehydroaromatization, Catal. Lett., 2008, 122, 84–90 CrossRef CAS.
- Y. Song, C. Sun, W. Shen and L. Lin, Hydrothermal post-synthesis of HZSM-5 zeolite to enhance the coke-resistance of Mo/HZSM-5 catalyst for methane dehydroaromatization reaction: Reconstruction of pore structure and modification of acidity, Appl. Catal., A, 2007, 317, 266–274 CrossRef CAS.
- W. Ding, G. Meitzner and E. Iglesia, The effects of silanation of external acid sites on the structure and catalytic behavior of Mo/H-ZSM5, J. Catal., 2002, 206, 14–22 CrossRef CAS.
- J. Hu, S. Wu, Y. Ma, X. Yang, Z. Li, H. Liu, Q. Huo, J. Guan and Q. Kan, Effect of the particle size of MoO3 on the catalytic activity of Mo/ZSM-5 in methane non-oxidative aromatization, New J. Chem., 2015, 39, 5459–5469 RSC.
- M. Hutin, M. H. Rosnes, D. Long and L. Cronin, 2.10 - Polyoxometalates: Synthesis and Structure – From Building Blocks to Emergent Materials, Elsevier, Amsterdam, 2013 Search PubMed.
- D. Long, R. Tsunashima and L. Cronin, Polyoxometalates: Building Blocks for Functional Nanoscale Systems, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed.
- M. Lechner, R. Guettel and C. Streb, Challenges in polyoxometalate-mediated aerobic oxidation catalysis: catalyst development meets reactor design, Dalton Trans., 2016, 45, 16716–16726 RSC.
- C. Streb, New trends in polyoxometalate photoredox chemistry: From photosensitisation to water oxidation catalysis, Dalton Trans., 2012, 41, 1651–1659 RSC.
- A. P. Ginsberg, Inorganic Syntheses, John Wiley & Sons, Inc., 1990, vol. 27 Search PubMed.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Is the BET equation applicable to microporous adsorbents?, Stud. Surf. Sci. Catal., 2006, 160, 49–56 CrossRef.
- H. Berendsen, D. Vanderspoel and R. Vandrunen, Gromacs - a Message-Passing Parallel Molecular-Dynamics Implementation, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- D. Van der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. Mark and H. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- J. Wang, P. Cieplak and P. Kollman, How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules?, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- A. Chaumont and G. Wipff, Ion aggregation in concentrated aqueous and methanol solutions of polyoxometallates Keggin anions: the effect of counterions investigated by molecular dynamics simulations, Phys. Chem. Chem. Phys., 2008, 10, 6940–6953 RSC.
- A. Sole-Daura, A. Notario-Estevez, J. J. Carbo, J. M. Poblet, C. de Graaf, K. Y. Monakhov and X. Lopez, How Does the Redox State of Polyoxovanadates Influence the Collective Behavior in Solution? A Case Study with [I@V18O42](q-) (q=3, 5, 7, 11, and 13), Inorg. Chem., 2019, 58, 3881–3894 CrossRef CAS PubMed.
- R. E. Schreiber, L. Houben, S. G. Wolf, G. Leitus, Z. Lang, J. J. Carbo, J. M. Poblet and R. Neumann, Real-time molecular scale observation of crystal formation, Nat. Chem., 2017, 9, 369–373 CrossRef CAS PubMed.
- A. Sole-Daura, V. Goovaerts, K. Stroobants, G. Absillis, P. Jimenez-Lozano, J. M. Poblet, J. D. Hirst, T. N. Parac-Vogt and J. J. Carbo, Probing Polyoxometalate-Protein Interactions Using Molecular Dynamics Simulations, Chem. – Eur. J., 2016, 22, 15280–15289 CrossRef CAS PubMed.
- X. Lopez, C. Nieto-Draghi, C. Bo, J. Avalos and J. Poblet, Polyoxometalates in solution: Molecular dynamics simulations on the alpha-PW12O403- keggin anion in aqueous media, J. Phys. Chem. A, 2005, 109, 1216–1222 CrossRef CAS PubMed.
- A. Rappe, C. Casewit, K. Colwell, W. Goddard and W. Skiff, UFF, a Full Periodic-Table Force-Field for Molecular Mechanics and Molecular-Dynamics Simulations, J. Am. Chem. Soc., 1992, 114, 10024–10035 CrossRef CAS.
- S. Leroch and M. Wendland, Simulation of Forces between Humid Amorphous Silica Surfaces: A Comparison of Empirical Atomistic Force Fields, J. Phys. Chem. C, 2012, 116, 26247–26261 CrossRef CAS PubMed.
- W. Jorgensen, J. Chandrasekhar, J. Madura, R. Impey and M. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald - an N.Log(n) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- R. Hockney, S. Goel and J. Eastwood, Quiet High-Resolution Computer Models of a Plasma, J. Comput. Phys., 1974, 14, 148–158 CrossRef.
- B. Hess, H. Bekker, H. Berendsen and J. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, Canonical sampling through velocity rescaling, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- E. V. Matus, I. Z. Ismagilov, O. B. Sukhova, V. I. Zaikovskii, L. T. Tsikoza, Z. R. Ismagilov and J. A. Moulijn, Study of methane dehydroaromatization on impregnated Mo/ZSM-5 catalysts and characterization of nanostructured molybdenum phases and carbonaceous deposits, Ind. Eng. Chem. Res., 2007, 46, 4063–4074 CrossRef CAS.
- H. Liu, X. Bao and Y. Xu, Methane dehydroaromatization under nonoxidative conditions over Mo/HZSM-5 catalysts: Identification and preparation of the Mo active species, J. Catal., 2006, 239, 441–450 CrossRef CAS.
- I. Julián, H. Ramírez, J. L. Hueso, R. Mallada and J. Santamaría, Non-oxidative methane conversion in microwave-assisted structured reactors, Chem. Eng. J., 2018 DOI:10.1016/j.cej.2018.08.150 , In press.
- Y. Wu, L. Emdadi, Z. Wang, W. Fan and D. Liu, Textural and catalytic properties of Mo loaded hierarchical meso-/microporous lamellar MFI and MWW zeolites for direct methane conversion, Appl. Catal., A, 2014, 470, 344–354 CrossRef CAS.
- K. Velebna, M. Hornacek, V. Jorik, P. Hudec, M. Caplovicova and L. Caplovic, The influence of molybdenum loading on activity of ZSM-5 zeolite in dehydroaromatization of methane, Microporous Mesoporous Mater., 2015, 212, 146–155 CrossRef CAS.
- C. H. L. Tempelman and E. J. M. Hensen, On the deactivation of Mo/HZSM-5 in the methane dehydroaromatization reaction, Appl. Catal., B, 2015, 176, 731–739 CrossRef.
- W. Li, G. Meitzner, R. Borry and E. Iglesia, Raman and X-ray absorption studies of Mo species in Mo/H-ZSM5 catalysts for non-oxidative CH4 reactions, J. Catal., 2000, 191, 373–383 CrossRef CAS.
- A. Rzhevskii, P. Choi, F. Ribeiro, R. Gulotty and M. Olken, Monitoring of molybdenum H-ZSM5 catalyst preparation by in situ ultraviolet Raman spectroscopy, Catal. Lett., 2001, 73, 187–191 CrossRef CAS.
- D. Katsoulis, A survey of applications of polyoxometalates, Chem. Rev., 1998, 98, 359–387 CrossRef CAS PubMed.
- Y. Xu, Porous and low-dimensional molybdates, Curr. Opin. Solid State Mater. Sci., 1999, 4, 133–139 CrossRef CAS.
- F. Lefebvre, Synthesis, Characterization and Applications in Catalysis of Polyoxometalate/Zeolite Composites, Inorganics, 2016, 4, UNSP 13 CrossRef.
- L. S. Nogueira, S. Ribeiro, C. M. Granadeiro, E. Pereira, G. Feio, L. Cunha-Silva and S. S. Balula, Novel polyoxometalate silica nano-sized spheres: efficient catalysts for olefin oxidation and the deep desulfurization process, Dalton Trans., 2014, 43, 9518–9528 RSC.
- X. Lopez, J. J. Carbo, C. Bo and J. M. Poblet, Structure, properties and reactivity of polyoxometalates: a theoretical perspective, Chem. Soc. Rev., 2012, 41, 7537–7571 RSC.
- S. Mukai, T. Masuda, I. Ogino and K. Hashimoto, Preparation of encaged heteropoly acid catalyst by synthesizing 12-molybdophosphoric acid in the supercages of Y-type zeolite, Appl. Catal., A, 1997, 165, 219–226 CrossRef CAS.
- S. Mukai, I. Ogino, L. Lin, Y. Kondo, T. Masuda and K. Hashimoto, A stable “ship in the bottle” type 12-molybdophosphoric acid engaged Y-type zeolite catalyst for liquid phase reactions, React. Kinet. Catal. Lett., 2000, 69, 253–258 CrossRef CAS.
- L. Chen, X. Wang, X. Guo, H. Guo, H. Liu and Y. Chen, In situ nanocrystalline HZSM-5 zeolites encaged heteropoly acid H3PMo12O40 and Ni catalyst for hydroconversion of n-octane, Chem. Eng. Sci., 2007, 62, 4469–4478 CrossRef CAS.
- P. Tan, The catalytic performance of Mo-impregnated HZSM-5 zeolite in CH4 aromatization: Strong influence of Mo loading and pretreatment conditions, Catal. Commun., 2018, 103, 101–104 CrossRef CAS.
- Y. Liu, M. Zhao, L. Cheng, J. Yang, L. Liu, J. Wang, D. Yin, J. Lu and Y. Zhang, Facile synthesis and its high catalytic performance of hierarchical ZSM-5 zeolite from economical bulk silicon oxides, Microporous Mesoporous Mater., 2018, 260, 116–124 CrossRef CAS.
- D. Wang, Q. Kan, N. Xu, P. Wu and T. Wu, Study on methane aromatization over MoO3/HMCM-49 catalyst, Catal. Today, 2004, 93–95, 75–80 CrossRef CAS.
- L. Liu, D. Ma, H. Chen, H. Zheng, M. Cheng, Y. Xu and X. Bao, Methane dehydroaromatization on Mo/HMCM-22 catalysts: effect of SiO2/Al2O3 ratio of HMCM-22 zeolite supports, Catal. Lett., 2006, 108, 25–30 CrossRef CAS.
- H. Liu, S. Yang, S. Wu, F. Shang, X. Yu, C. Xu, J. Guan and Q. Kan, Synthesis of Mo/TNU-9 (TNU-9 Taejon National University No. 9) catalyst and its catalytic performance in methane non-oxidative aromatization, Energy, 2011, 36, 1582–1589 CrossRef CAS.
- H. Liu, J. Hu, Z. Li, S. Wu, L. Liu, J. Guan and Q. Kan, Synthesis of zeolite IM-5 under rotating and static conditions and the catalytic performance of Mo/H-IM-5 catalyst in methane non-oxidative aromatization, Kinet. Catal., 2013, 54, 443–450 CrossRef CAS.
- H. Liu, S. Wu, Y. Guo, F. Shang, X. Yu, Y. Ma, C. Xu, J. Guan and Q. Kan, Synthesis of Mo/IM-5 catalyst and its catalytic behavior in methane non-oxidative aromatization, Fuel, 2011, 90, 1515–1521 CrossRef CAS.
- Y. Xu, J. Lu, J. Wang and Z. Zhang, Mo-Based Zeolite Catalysts and Oxygen-Free Methane Aromatization, Prog. Chem., 2011, 23, 90–106 Search PubMed.
- D. Ma, D. Wang, L. Su, Y. Shu, Y. Xu and X. Bao, Carbonaceous deposition on Mo/HMCM-22 catalysts for methane aromatization: A TP technique investigation, J. Catal., 2002, 208, 260–269 CrossRef CAS.
- S. J. Han, S. K. Kim, A. Hwang, S. Kim, D. Hong, G. Kwak, K. Jun and Y. T. Kim, Non-oxidative dehydroaromatization of methane over Mo/H-ZSM-5 catalysts: A detailed analysis of the reaction-regeneration cycle, Appl. Catal., B, 2019, 241, 305–318 CrossRef CAS.
- A. Martinez and E. Penis, Non-oxidative methane dehydroaromatization on Mo/HZSM-5 catalysts: Tuning the acidic and catalytic properties through partial exchange of zeolite protons with alkali and alkaline-earth cations, Appl. Catal., A, 2016, 515, 32–44 CrossRef CAS.
- N. Kosinov, F. J. A. G. Coumans, E. Uslamin, F. Kapteijn and E. J. M. Hensen, Selective Coke Combustion by Oxygen Pulsing During Mo/ZSM-5-Catalyzed Methane Dehydroaromatization, Angew. Chem., Int. Ed., 2016, 55, 15086–15090 CrossRef CAS PubMed.
- A. A. Stepanov, L. L. Korobitsyna, Y. E. Barbashin and A. V. Vosmerikov, Effect of the conditions of thermal pretreatment on the properties of Mo/ZSM-5 catalyst of the nonoxidative conversion of methane, Russ. J. Phys. Chem. A, 2016, 90, 2364–2369 CrossRef CAS.
- C. H. L. Tempelman, M. Teresa Portilla, M. E. Martinez-Armero, B. Mezari, N. G. R. de Caluwe, C. Martinez and E. J. M. Hensen, One-pot synthesis of nano-crystalline MCM-22, Microporous Mesoporous Mater., 2016, 220, 28–38 CrossRef CAS.
- P. Tan, Active phase, catalytic activity, and induction period of Fe/zeolite material in nonoxidative aromatization of methane, J. Catal., 2016, 338, 21–29 CrossRef CAS.
- X. Yin, N. Chu, J. Yang, J. Wang and Z. Li, Synthesis of the nanosized MCM-22 zeolite and its catalytic performance in methane dehydro-aromatization reaction, Catal. Commun., 2014, 43, 218–222 CrossRef CAS.
- Y. Zhang, M. Kidder, R. E. Ruther, J. Nanda, G. S. Foo, Z. Wu and C. K. Narula, Promotional Effects of In on Non-Oxidative Methane Transformation Over Mo-ZSM-5, Catal. Lett., 2016, 146, 1903–1909 CrossRef CAS.
- T. H. Lim, K. Nam, I. K. Song, K. Lee and D. H. Kim, Effect of Si/Al-2 ratios in Mo/H-MCM-22 on methane dehydroaromatization, Appl. Catal., A, 2018, 552, 11–20 CrossRef CAS.
- P. Tan, Ammonia-basified 10 wt% Mo/HZSM-5 material with enhanced dispersion of Mo and performance for catalytic aromatization of methane, Appl. Catal., A, 2019, 580, 111–120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterisation of catalysts and computational details. See DOI: 10.1039/c9cy01490j |
| This journal is © The Royal Society of Chemistry 2019 |