Towards coupling direct activation of methane with in situ generation of H2O2
Received
3rd July 2019
, Accepted 29th August 2019
First published on 2nd September 2019
Abstract
Due to the explosive nature of H2, O2 and CH4 mixtures, the concept of coupling in situ synthesis of H2O2 with low-temperature single-step methane conversion to methanol has not received sufficient attention. This study aimed to investigate this process using a microchannel reactor, which offers the opportunity to explore the process under a wide range of concentrations. Direct methane activation with in situ generation of H2O2 was successfully demonstrated in a microcapillary containing Au–Pd nanoparticles embedded on its silica-coated walls. The effect of H2, O2 and CH4 partial pressures, H2/O2 molar ratio, gas-to-liquid (G/L) ratio and liquid phase weight-hourly-space-velocity (WHSV) on the productivity and product distribution was investigated. CH4 partial pressure had the most significant effect on the productivity, while H2 and O2 partial pressures influenced the productivity less. The methane activation rate was found to be correlated with the H2O2 formation rate. With only O2 or pre-formed stabilized H2O2 methane activation was not found, in situ H2O2 synthesis was therefore essential. G/L affected neither the product distribution nor the productivity, however, lowering WHSV altered the product distribution favoring methanol formation.
1. Introduction
Natural gas with methane as its main component is considered to be a relatively clean source of fossil energy and a potentially valuable raw material. However, full exploitation of the enormous methane resources is yet to be realized mainly due to the challenges associated with transportation and storage. Therefore, it is of great interest to convert methane to products (chemical or fuel) that can be easily shipped.1,2 Indirect methane conversion, to syngas and then to alcohols or hydrocarbons, is an advanced and mature technology being widely practiced in industry. This route involves an energy-intensive thermochemical oxidation (e.g. steam reforming and autothermal reforming) to convert methane to CO and H2, which are then fed to a methanol synthesis process or to a Fischer–Tropsch synthesis process that involves production of a mixture of hydrocarbons including paraffins, olefins and alcohols.3–5 However, the costly nature of such multistep processes, mainly originating from the syngas production step, has triggered the development of an alternative low-temperature single-step approach for the direct selective oxidation of methane to methanol.6
Cleaving the C–H bonds of methane, which is relatively an inert molecule at low temperatures (ΔHC–H = 439.57 kJ mol−1), and activating the oxidant to form and regenerate the active sites are the major challenges of this approach.6,7 Hydrogen peroxide (H2O2) has been reported as a promising oxidant when used with catalysts such as CuFe–ZSM-5 (ref. 8 and 9) and Au–Pd nanoparticles.10,11 However, using pre-synthesized H2O2 is not only economically unviable, as it is more expensive than methanol, but it is also environmentally undesirable since the traditional anthraquinone process to produce H2O2 results in significant waste generation.12 In this view, in situ generation of H2O2 coupled with direct methane oxidation seems to be a strategy worthy of intensive investigation. In spite of an early work13 reporting higher selectivity towards methanol in presence of in situ generated H2O2, the concept of coupling H2O2 synthesis with methane oxidation has not received sufficient attention in the literature. This basically stems from the fact that H2, O2 and CH4 mixtures are explosive under a wide range of concentrations,14 and need to be diluted to guarantee safe operation, thus resulting in much lower methanol production rates.
This study aims to address this limitation by coupling the in situ generation of H2O2 with the direct methane conversion in an inherently safe washcoated microchannel reactor which offers the opportunity to explore the process under a wide range of concentrations. This catalytic washcoated microchannel reactor, which showed an outstanding performance for the direct H2O2 synthesis in our previous work,15 is obtained by embedding Au-Pd nanoparticles on the silica-coated walls of a microcapillary. In this context, the effects of CH4, O2 and H2 partial pressures, O2/H2 molar ratio, gas-to-liquid ratio and residence time on product yields were investigated.
2. Experimental
2.1. Catalyst preparation
Fused silica capillaries (320 μm ID) containing a 4 μm pre-coated layer of SiO2 on the wall (CP-SilicaPLOT, Agilent) were used as the microchannel reactor. Adopting the catalyst synthesis and coating from our previous work,15 the silica layer was first treated by a 1 M ammonium nitrate solution which was flushed through the capillary using a syringe pump. The capillaries were then dried and calcined at 120 and 300 °C, respectively. Prior to metal deposition, polyelectrolyte multilayers (PEMs) were built on the silica surface. It has been shown that PEMs enabled a facile control over metal ions loading and distribution by providing highly distributed charged surfaces.16 To form PEMs on the support, a layer-by-layer approach, in which 10 mg mL−1 polyacrylic acid (PAA, 35 wt% in H2O, Sigma-Aldrich) and 2 mg mL−1 polyethylene imine (PEI, 50 wt% in H2O, Sigma-Aldrich,) solutions were alternatively flushed through the microcapillary, was taken. A bi-layer polyelectrolyte was then obtained by repetition of the same procedure. Metal deposition was carried out upon formation of PEMs. For a desired metal loading of 5 wt%, considering coating weight and void fraction,15,17 a 5.7 mgmetal mL−1 (Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd = 1:2, molar ratio) solution in DI water was prepared using HAuCl4 (Aldrich, 30 wt% in HCl) and K2PdCl4 (Sigma, 99.99%) as the precursors. The solution was then flushed and held still in the capillary, to ensure metal ion biding on the surface before removing the excess by N2. Metal deposition was followed by a reduction step, in which NaBH4 solution was flushed until the capillary color turned into dark black, indicating a successful reduction of the deposited metal. Eventually, drying at 120 °C and calcination at 380 °C were carried out.
:Pd = 1:2, molar ratio) solution in DI water was prepared using HAuCl4 (Aldrich, 30 wt% in HCl) and K2PdCl4 (Sigma, 99.99%) as the precursors. The solution was then flushed and held still in the capillary, to ensure metal ion biding on the surface before removing the excess by N2. Metal deposition was followed by a reduction step, in which NaBH4 solution was flushed until the capillary color turned into dark black, indicating a successful reduction of the deposited metal. Eventually, drying at 120 °C and calcination at 380 °C were carried out.
2.2. Catalyst characterization methods
Formation of PEMs on the silica support was examined using XL30-ESEM-FEG scanning electron microscope (SEM) operated at 5 kV with a working distance of 10 mm from the sample. Metal particles distribution on the surface was studied by a FEI Tecnai G2 Sphera transmission electron microscope (TEM). The capillaries for TEM analyses were crushed and suspended in drops of ethanol. The X-ray photoelectron spectroscopy (XPS) measurements of the powder catalyst were carried out with a Kratos AXIS Ultra spectrometer, equipped with a monochromatic X-ray source and a delay-line detector (DLD). Spectra were obtained using the aluminium anode (Al Kα = 1486.6 eV) operating at 150 W.
2.3. Catalytic activity measurement
The schematic representation of the flow set-up is shown in Fig. 1. To keep a constant reaction temperature at 40 °C, the microcapillary was put in a thermostatic oven. Prior to the experiments, formation of a Taylor flow regime with alternating gas bubbles and liquid slugs under reaction conditions was confirmed using a transparent empty fused silica capillary. The liquid phase contained 0.05 M H2SO4 and 10 ppm NaBr (to inhibit H2O2 decomposition to H2O (ref. 18)) in DI water, and was sent to the microreactor using a syringe pump (Teledyne ISCO 500D). To avoid any explosion risk in the downstream, the outlet gas mixture was diluted by N2. The pressure was kept constant in the system by a back-pressure regulator (BPR) placed in the downstream after the gas–liquid separation unit. The gas flow rates were adjusted by mass flow controllers before the capillary inlet. The pressure was measured and monitored before and after the capillary. As presented in Table 1, the reaction was studied at different H2, O2 and CH4 partial pressures, gas-to-liquid ratios (G/L, v./v.) and liquid phase weight-hourly-space-velocities (WHSV, kgLiq. kgcat.−1 h−1). Gas samples were analyzed with an online compact GC (column: molsieve plot 5 m 0.32 mm) equipped with a TCD detector. As CH4 conversion was typically low (≲0.1%), it was not used for interpretation of the results. Liquid samples were collected every 45 min of reaction time. Part of the sample was used for the determination of H2O2 by titration using standard solution of cerium(IV) sulfate (Sigma Aldrich) and ferroin (VWR Chemicals) as an indicator. The other part of liquid sample was taken to quantify the formation of oxygenates. The amount of methanol in the sample was determined using an offline GC (Varian CP-3800). Aggregated concentration of methanol and methylhydroperoxide was measured by adding NaBH4 to the liquid sample to convert the latter to methanol which is then readily detectable on the GC.19 The liquid samples were also quantified for formic acid formation using an HPLC 155 (Shimadzu Sil-20AC). Oxygenate productivity was then calculated using following expression:| |  | (1) |
 |
| | Fig. 1 Scheme of the setup used for conducting the experiments. | |
Table 1 Set of reactions conditions employed for the parametric study
| Entry |
P
O2
|
P
H2
|
P
CH4
|
Total P (bar) |
WHSV |
G/L |
| 1 |
1 |
1 |
18 |
20 |
4200 |
2.5 |
| 2 |
1 |
1 |
28 |
30 |
4200 |
2.5 |
| 3 |
1 |
1 |
8 |
10 |
4200 |
2.5 |
| 4 |
0.5 |
0.5 |
8 |
9 |
4200 |
2.5 |
| 5 |
3 |
3 |
8 |
14 |
4200 |
2.5 |
| 6 |
6 |
6 |
8 |
20 |
4200 |
2.5 |
| 7 |
1 |
0.5 |
28 |
29.5 |
4200 |
2.5 |
| 8 |
0.5 |
1 |
28 |
29.5 |
4200 |
2.5 |
| 9 |
1 |
1 |
28 |
30 |
4200 |
5 |
| 10 |
1 |
1 |
28 |
30 |
2100 |
5 |
3. Results and discussion
3.1. Catalyst characterization
The formation of PEMs layers of ∼1 μm thickness was confirmed by SEM as presented in Fig. 2a. Successful deposition of metal nanoparticles on silica support was also evident as shown in TEM image (Fig. 2b). TEM indicated a narrow size distribution of metal nanoparticles over the support, yielding small particle size of ∼2.9 ± 1.2 nm. XPS was employed to evaluate the elemental composition and the oxidation state of Au and Pd at the surface of an AuPd/SiO2 powder catalyst prepared in a similar way as the AuPd wall-coated capillary (Fig. 3). The Au 4f7/2 and Pd 3d5/2 binding energies (BE) along with XPS-derived atomic ratios are shown in Table 2. The Pd 3d spectra characterized by two spin–orbit components of 3d5/2 and 3d3/2 exhibit three doublets attributed to three different Pd species-PdO2, PdO and Pd0.20,21 The reduced sample predominantly consists of metallic Pd, with small amount of PdO2, most likely formed due to oxidation in air during sample handling.21 The Au 4f spectra was typical of metallic Au. The BE of Au (4f7/2) was found to be 82.8 eV, exhibiting a prominent negative shift when compared to bulk Au (84 eV) or small Au nanoparticles (83.5–84 eV).22 This was proposed to be due to charge transfer from Pd to Au and was evident of Au–Pd alloying.22 Upon calcination, the PdO phase dominated but 25% of the Pd was still present in its metallic form. This is in agreement with our previous finding,23 where it was also found that during in situ generation of H2O2, the PdO phase present in the calcined catalyst was readily reduced and stayed at its metallic state. XPS also revealed no contamination from Cl, ensuring effectiveness of the washing step during synthesis.
 |
| | Fig. 2 (a) SEM showing the formation of PEMS, (b) TEM image of calcined Au–Pd capillary. | |
 |
| | Fig. 3 Experimental and fitted (a) Au 4f and (b) Pd 3d XPS for reduced and calcined AuPd/SiO2 powder catalysts. | |
Table 2 XPS binding energies for Au 4f7/2 and Pd 3d5/2, relative amount of each Pd species and surface Au/Pd atomic ratios of AuPd/SiO2 powder catalyst before and after calcination
| Sample |
Binding energy Au 4f7/2 (eV) |
Pd 3d5/2 |
Au/Pd |
| Binding energy (eV) |
Peak designation (relative %) |
| Reduced |
82.8 |
338.1 |
PdO2 (19%) |
0.74 |
| 334.8 |
Pd0 (81%) |
| Calcined |
83.3 |
338.2 |
PdO2 (10%) |
0.41 |
| 336.4 |
PdO (65%) |
| 334.8 |
Pd0 (25%) |
3.2. Catalytic activity
The catalytic microcapillary reactor was first tested at 20 bar, gas phase composition of H2/O2 = 50/50, WHSV of 4200 kgliq. kgcat.−1 h−1 and G/L of 2.5 to ensure in situ generation of H2O2. Upon reaching a stable concentration of ∼0.9 wt% H2O2 in the liquid phase, a methane activation experiment was initiated by introducing CH4 to the reactor, thus changing the gas phase composition to H2/O2/CH4 = 5/5/90 (Table 1, entry 1). It is believed that CH4 activation in the presence of H2O2 over Au–Pd catalyst proceeds through generation of methyl hydroperoxide (CH3OOH), an intermediate which then is converted to methanol.11 In addition to that, formation of formic acid and CO2, originating from methanol and methyl hydroperoxide over-oxidation has also been reported in the literature, especially when pre-formed H2O2 is utilized as the oxidant.24 In this study, the formation of neither formic acid nor CO2 was evident. The absence of over-oxidation reactions may be attributed to the significantly shorter residence time (13 s) employed in this work compared to that of batch systems (>5 min). Of course, trace amounts (<1 ppm) of formic acid and CO2 below the detection limit of the utilized instruments might be present. As shown in Fig. 4, a stable catalytic activity towards both H2O2 formation and CH4 activation was observed over a period of 9 h. H2O2 concentration at the liquid phase reached a stable amount of ∼0.2 wt%, and an oxygenate productivity of 18.1 mol kgmetal h−1 was obtained. The product distribution was also stable over time, resulting in a methanol-to-oxygenates molar ratio (MeOH/OX) of ∼70%. Upon removal of CH4 from the reaction medium it was observed that the catalyst was able to retain its original activity towards H2O2 formation after being subjected to CH4 oxidation for 5.25 h. The obtained oxygenate productivity was about one order of magnitude higher that that reported by Ab Rahim et al.25 who used a batch system using a gas composition of 0.86% H2, 1.72% O2, 75.86% CH4 and 21.55% N2 over a TiO2 supported Au–Pd bimetallic catalyst at total pressure of 32 bar. The very dilute H2 and O2 concentrations in the latter case leads to much lower (18–20 times) H2O2 formation rates compared to the microreactor system. Hence, the higher oxygenate productivity obtained here can be attributed to the higher rates of H2O2 generation in the microcapillary, suggesting that an effective coupling of the two reactions, is strongly dependent on the rate of peroxide formation. To further elucidate this conclusion, a parametric study was carried out, taking advantage of the inherently safe conditions of the microchannel reactor.
 |
| | Fig. 4 Oxygenate productivity, MeOH/OX ratio and outlet H2O2 wt% over time at a total pressure of 20 bar. Gas composition was switched from H2/O2 = 50/50 to H2/O2/CH4 = 5/5/90 after 90 min (see Table 1, entry 1). Gas composition was switched back to H2/O2 of 50/50 after 405 min. | |
The effect of CH4 partial pressure was studied by keeping H2 and O2 partial pressures at 1 bar, resulting in a roughly constant H2O2 (0.2 wt%) concentration in the reactor outlet. Although MeOH/OX remained unaffected, increasing CH4 partial pressure from 8 to 18 bar resulted in a significant improvement in oxygenate productivity as it was raised from 6.3 to 18.1 mol kgmetal−1 h−1. Further CH4 pressure increase did not have such a pronounced effect on the productivity (Fig. 5a), as the catalyst active sites seemed to be saturated at pressures above 18 bar. This behavior is in agreement with past studies where H2O2 was used as the oxidant.8
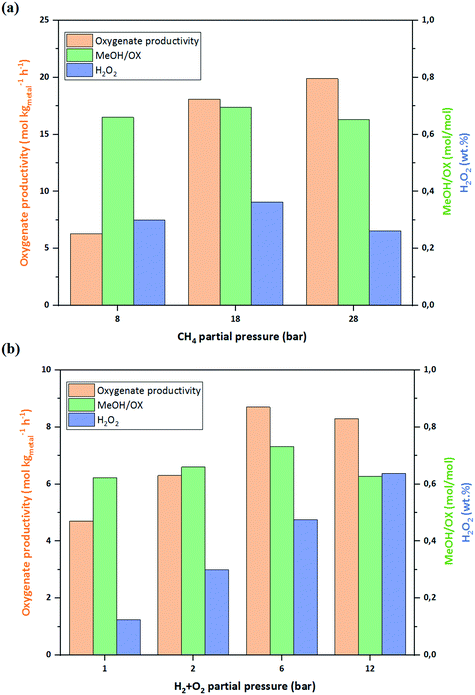 |
| | Fig. 5 (a) Effect of CH4 partial pressure at PO2(= PH2) of 1 bar, G/L of 2.5 and WHSV of 4200 kgliq. kgcat.−1 h−1. (b) Effect of H2 + O2 partial pressure at PCH4 of 8 bar, G/L of 2.5 and WHSV of 4200 kgliq. kgcat.−1 h−1. See Table 1, entries 1–6. | |
To study the effect of H2 + O2 pressure (H2/O2 = 1), CH4 pressure was kept at 8 bar and H2 + O2 was raised from 1 bar to 12 bar (Fig. 5b). By raising H2 + O2 pressure, the concentration of accumulated H2O2 in the reactor outlet was elevated up to 0.36% wt%. Higher pressures of H2 + O2, and therefore higher H2O2 formation rates, had a positive effect on oxygenate productivity, although to a lesser extent than the CH4 partial pressure. This suggests that H2 and O2 are also involved in the rate-determining step. As it can be seen in Fig. 5b, a H2 + O2 pressure of 12 bar slightly reduces the oxygenate productivity. At such a gas phase gas composition (H2/O2/CH4 = 0.3/0.3/0.4) there is possibly a competition for the active sites on the catalyst surface making initiation of CH4 activation controlled by the number of available sites. Hence an optimum is required between H2O2 formation rate and its role in subsequent methane activation. Based on the obtained results, it can be inferred that the most favorable gas phase composition to obtain high oxygenate productivity is a CH4-rich medium with an optimal H2 + O2 pressure.
In order to investigate the influence of oxidative conditions, the experiments were conducted at different H2/O2 ratios of 0, 0.5 and 2, see Fig. 6. The highest oxygenate productivity was achieved at H2/O2 of 0.5, suggesting that a more oxidative condition is favorable for methane activation as the productivity dropped from 15.3 to 11.5 mol kgmetal−1 h−1 when H2/O2 ratio was raised to 2. Completely removing H2 from the reaction medium (H2/O2 = 0) led to no methane activation. Remarkably, this was also the case when using pre-formed H2O2 (0.25 wt%) as the oxidant. This implies species generated during in situ H2O2 formation are essential and the intermediates for methane activation. It can be explained by the mechanism of methane activation (Fig. 7) over Au–Pd catalysts, which has been suggested to be a radical mechanism starting from formation of methyl radicals (˙CH3) through H abstraction from methane by hydroxyl radicals (˙OH).19 The primary oxygenates are then generated by having ˙CH3 radicals going through a termination step which is either a reaction with an ˙OOH radical or a molecular O2.8 The ˙OOH and ˙OH radicals have been reported to be formed during in situ generation H2O2,19,26 and it can be the reason why in absence of H2 (H2/O2 = 0) no methane activation was evident. However, the observation that in the simultaneous presence of the H2 and O2, an O2-rich gas phase was more beneficial can be explained by considering that O2 plays a dual role in the reaction, being involved both in generation of ˙OH, needed for ˙CH3 formation, and also in ˙CH3 termination step. This finding is in line with Agarwal et al.,19 who demonstrated O2 significant incorporation in the methanol product using an isotopic labeling technique. Another source of O2 and, also reactive intermediates including ˙OH and ˙OOH, can be the H2O2 decomposition.8,19 However, decomposition was observed to be well-suppressed (<5% decomposition) under the employed reaction conditions with the liquid phase containing 0.05 M sulfuric acid and 10 ppm NaBr, explaining why methane was not activated by using pre-formed H2O2 as the oxidant in these experiments.
 |
| | Fig. 6 Effect of H2/O2 ratio at PCH4 of 28 bar, G/L of 2.5 and WHSV of 4200 kgliq. kgcat.−1 h−1. See Table 1, entries 7 and 8. | |
Keeping G/L constant (= 5), the WHSV of the liquid phase was reduced from 4200 to 2100 kgliq. kgcat.−1 h−1, see Fig. 8a. The lower WHSV increased the concentration of H2O2 in the aqueous phase from 0.28% to 0.34% wt%, while the oxygenate productivity was unaffected, as expected from the near-differential operation conditions of the reactor. The proportional increase of oxygenates concentration with residence time also shows that the over-oxidation of oxygenates was not significant, since otherwise a drop in the productivity towards methanol and methylhydroperoxide would have been observed with decreasing WHSV. On the other hand, reducing WHSV resulted in an increase of MeOH/OX ratio from 0.65 to 0.84, suggesting a series reaction and confirming the reaction mechanism with methane first being converted to methylhydroperoxide that is further converted to methanol (Fig. 7).
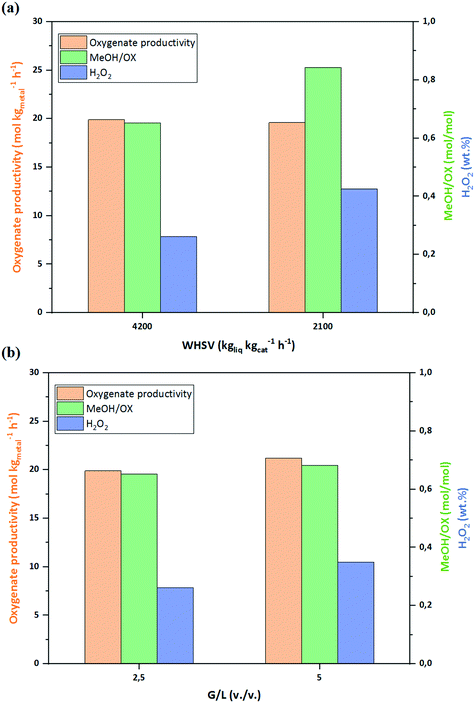 |
| | Fig. 8 (a) Effect of WHSV at G/L of 5 and (b) effect of G/L at WHSV of 4200 kgliq. kgcat.−1 h−1. PO2 (= PH2) = 1 bar and PCH4 = 28 bar. See Table 1, entries 2, 9 and 10. | |
The effect of G/L ratio on the performance was studied by changing gas flow rate at a constant liquid phase WHSV of 4200 kgliq. kgcat.−1 h−1. By increasing G/L from 2.5 to 5, peroxide concentration in the outlet slightly increased from 0.21 to 0.28 wt%, which is in agreement to our previous study on H2O2 synthesis.27 However, neither the oxygenate productivity nor the MeOH/OX ratio (Fig. 8b) were affected by G/L ratio. Varying WHSV and G/L can alter the size of gas bubbles and liquid slugs, and consequently gas–liquid–solid mass transfer rates.27 The unaffected productivities in both cases confirms that with the very slow kinetic of methane activation, mass transfer is not the limiting factor in the process.
4. Conclusions
Single-step selective methane oxidation was successfully coupled with in situ formation of H2O2 in a wall-coated microcapillary reactor, achieving an enhanced oxygenate productivity compared to what has been reported in the literature for supported Au–Pd nanoparticles. This improvement was mainly attributed to a significantly increased H2O2 formation rate in the microcapillary. The Au–Pd/SiO2 catalyst also showed a good stability in terms of productivity and product distribution. In order to have a better understanding of the process, the operational parameters that could affect the performance were identified and evaluated. CH4 partial pressure turned out to have a more significant effect compared to that of H2 and O2 partial pressures, suggesting that a CH4-rich medium and an optimal H2 + O2 partial pressures were favorable. Oxygenate productivity improved under a more oxidative medium (lower H2/O2), while in the presence of only O2, no activity was observed. Using pre-formed H2O2 also did not result in methane activation as it was highly stabilized. As a result, ˙OOH and ˙OH radicals which are either formed during H2O2 synthesis or during H2O2 decomposition were found to be essential for methane activation. A decrease in WHSV favored methanol formation over methylhydroperoxide formation, indicating that methanol is formed out of methylhydroperoxide. Over-oxidation towards formic acid and CO2 was not observed. The G/L ratio changed neither product distribution nor productivity of oxygenates.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (NWO), Project (10017587). We acknowledge the support of Carlo Buijs for SEM and TEM measurement.
References
- A. I. Olivos-Suarez, A. g. Szécsényi, E. J. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965–2981 CrossRef CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- B. Wang, S. Albarracín-Suazo, Y. Pagán-Torres and E. Nikolla, Catal. Today, 2017, 285, 147–158 CrossRef CAS.
- A. Delparish and A. K. Avci, Fuel Process. Technol., 2016, 151, 72–100 CrossRef CAS.
- A. Delparish, S. Koc, B. S. Caglayan and A. K. Avci, Catal. Today, 2019, 323, 200–208 CrossRef CAS.
- R. Palkovits, M. Antonietti, P. Kuhn, A. Thomas and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6909–6912 CrossRef CAS.
-
H. Gesser and N. H. E. Wolf, in Methane Conversion by Oxidative Processes, ed. N. H. E. Wolf, Van Nostrand Reinhold, New York, 1992, pp. 403–425 Search PubMed.
- C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, M. H. Ab Rahim, M. M. Forde, A. Thetford, D. M. Murphy, H. Hagen and E. E. Stangland, Chem. – Eur. J., 2012, 18, 15735–15745 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer and D. M. Murphy, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS.
- M. H. Ab Rahim, M. M. Forde, C. Hammond, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock and G. J. Hutchings, Top. Catal., 2013, 56, 1843–1857 CrossRef CAS.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor and D. J. Willock, Angew. Chem., Int. Ed., 2013, 52, 1280–1284 CrossRef CAS.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- Y. Wang and K. Otsuka, J. Catal., 1995, 155, 256–267 CrossRef CAS.
-
B. Lewis and G. Von Elbe, Combustion, flames and explosions of gases, Academic Press, New York, 1987 Search PubMed.
- S. Kanungo, V. Paunovic, J. C. Schouten and M. F. Neira D'Angelo, Nano Lett., 2017, 17, 6481–6486 CrossRef CAS.
- M. Logar, B. Jancar and D. Suvorov, Nanotechnology, 2007, 18, 325601 CrossRef.
- M. Sankar, Q. He, M. Morad, J. Pritchard, S. J. Freakley, J. K. Edwards, S. H. Taylor, D. J. Morgan, A. F. Carley and D. W. Knight, ACS Nano, 2012, 6, 6600–6613 CrossRef CAS.
- V. Paunovic, J. C. Schouten and T. A. Nijhuis, Appl. Catal., A, 2015, 505, 249–259 CrossRef CAS.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock and S. H. Taylor, Science, 2017, 358, 223–227 CrossRef CAS.
- A. Venezia, V. La Parola, V. Nicolı and G. Deganello, J. Catal., 2002, 212, 56–62 CrossRef CAS.
- A. Venezia, V. La Parola, G. Deganello, B. Pawelec and J. Fierro, J. Catal., 2003, 215, 317–325 CrossRef CAS.
- F. Liu, D. Wechsler and P. Zhang, Chem. Phys. Lett., 2008, 461, 254–259 CrossRef CAS.
- S. Kanungo, L. van Haandel, E. J. Hensen, J. C. Schouten and M. F. N. d'Angelo, J. Catal., 2019, 370, 200–209 CrossRef CAS.
- R. McVicker, N. Agarwal, S. J. Freakley, Q. He, S. Althahban, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.12.017.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins and J. A. Lopez-Sanchez, Catal. Sci. Technol., 2016, 6, 3410–3418 RSC.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- V. Paunovic, J. C. Schouten and T. Nijhuis, Catal. Today, 2015, 248, 160–168 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Shamayita
Kanungo
,
John
van der Schaaf
and
M. Fernanda
Neira d'Angelo
,
Shamayita
Kanungo
,
John
van der Schaaf
and
M. Fernanda
Neira d'Angelo








