
Controlling the secondary pollutant on B-doped g-C3N4 during photocatalytic NO removal: a combined DRIFTS and DFT investigation†

Abstract
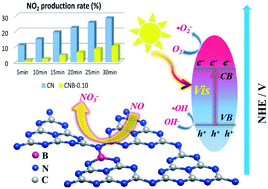
The accumulation of a toxic intermediate (NO2) during photocatalytic NO oxidation would lead to secondary pollution and largely limit the application of photocatalytic technology in air purification. Accordingly, the problems can be well tackled by employing a novel co-pyrolysis method to construct B-doped g-C3N4 (CNB), which could efficiently enhance the charge separation and dramatically inhibit the generation of the secondary pollutant (NO2) on CNB in contrast to the pristine CN. The effects of B-doping on the local electronic structure, reactant activation, and optical properties of CN were revealed. O2 activation was promoted on CNB owing to the localized electrons. With the combination of in situ DRIFTS verification and DFT simulation, it was revealed that the activation barrier of NO oxidation on CNB was significantly reduced, which facilitated the conversion of NO into the final product (NO3−). Subsequently, the mechanism for the inhibition of toxic NO2 was unraveled and the conversion pathways of photocatalytic NO oxidation on B-doped g-C3N4 have been proposed. The present work demonstrated a new strategy to optimize the photocatalytic performance for safer and efficient air purification.
- This article is part of the themed collection: 2019 Catalysis Science & Technology HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

