
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic halogenation of methane: a dream reaction with practical scope?
Vladimir
Paunović
 * and
Javier
Pérez-Ramírez
*
* and
Javier
Pérez-Ramírez
*
Institute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1, CH-8093, Switzerland. E-mail: vladimir.paunovic@chem.ethz.ch; jpr@chem.ethz.ch
First published on 31st May 2019
Abstract
Development of catalysts that could surpass the activity and selectivity constraints of the non-catalytic radical-mediated halogenation of methane constitutes a long-standing challenge, which exhibits great potential to valorise this readily available resource for the production of commodities. This study presents comprehensive performance maps of a large library of materials, comprising carriers (quartz, SiO2, SiC, α-Al2O3, γ-Al2O3 and carbon), noble metals (Pt, Pd and Ru), metal oxides (Fe2O3 and CeO2), chlorides (PdCl2 and CuCl2) and oxyfluorides (TaOF3) supported on SiO2, γ-Al2O3, carbon or H-ZSM-5 carriers, sulfated systems (S-ZrO2, S-ZrO2-SBA-15, S-TiO2, S-Nb2O5, S-Ta2O5 and Nafion) and zeolites (3A, H-USY, H-MOR, H-SAPO-34, H-BETA and H-ZSM-5), in the chlorination and bromination of methane under practically relevant conditions and gains insights into the nature of the catalytic effects as a function of the catalyst and halogen of choice. The chlorination activity of different catalyst beds at low temperatures (473–523 K) was 2–5.5 times higher compared to that of the empty reactor of identical volume, while the bromination rate was almost unaffected by the solids in the whole temperature range (643–723 K). Except for zeolites and Pt/carbon, which promoted polyhalogenation, selectivities to halomethanes over most of the catalysts were similar to those in the non-catalytic reactions and were higher in bromination (SCH3Br = 80–95% versus SCH3Cl = 52–90% at XCH4 = 5–18%). The formation of carbon oxides (SCOx = 2–28%) over several materials in chlorination and virtually all systems in bromination implied the decomposition of halomethanes, which at higher temperatures led to coking, particularly in the latter reaction. The kinetic fingerprints along with the marginal impact of the Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al ratio, counter ions and extraframework aluminium species on the performance of the most active H-ZSM-5 catalyst indicated that methane chlorination over various materials is governed by the radical-chain mechanism, which limits the scope for breaking the selectivity–conversion relationships by tailoring the catalyst acidity. Nonetheless, the enhancement of chlorination activity over zeolites that followed a volcano-like dependence on their micropore size coupled with a more significant impact of the intracrystalline mesoporosity and crystallite size on the product distribution revealed the important role of confinement effects in this reaction, which may pave the way for advancements in the production of chloromethanes.
:Al ratio, counter ions and extraframework aluminium species on the performance of the most active H-ZSM-5 catalyst indicated that methane chlorination over various materials is governed by the radical-chain mechanism, which limits the scope for breaking the selectivity–conversion relationships by tailoring the catalyst acidity. Nonetheless, the enhancement of chlorination activity over zeolites that followed a volcano-like dependence on their micropore size coupled with a more significant impact of the intracrystalline mesoporosity and crystallite size on the product distribution revealed the important role of confinement effects in this reaction, which may pave the way for advancements in the production of chloromethanes.
Introduction
The reactions of methane with molecular halogens (X2, X = F, Cl, Br or I, eqn (1)–(4)) constitute the earliest known routes to functionalize this astonishingly inert hydrocarbon, the history of which dates back to 1840 when the French chemist Jean Baptist Dumas reported the conversion of marsh gas by the action of chlorine (Fig. 1).1,2 The extensive kinetic studies of these thermally or photo-activated halogenation processes performed between the 1910s and the 1940s led to the postulation of the radical-chain mechanism that was verified for all halogens and paraffins (Fig. 1).3–11 The halogen radicals (X˙) initially formed via the homolytic bond dissociation of X2 and regenerated through the reaction of alkyl radicals with X2 play a central role in this reaction network. The differences in stability between the X˙, which increase in the order F˙ < Cl˙ < Br˙ < I˙, cause methane fluorination to be extremely exothermic and often explosive, and chlorination and bromination to display medium and mild exothermicity, respectively, and moderate activity, while iodination is endothermic and equilibrium limited (eqn (1)–(4)).1 The differences in radicals' reactivity induce substantial variations in the product distribution as a function of the halogen, since mono- (CH3X), di- (CH2X2) and trihalomethanes (CHX3) could react in an analogous fashion to methane leading to polyhalogenation. In contrast to fluorination which inevitably leads to a complete hydrogen substitution, chlorination could provide a high selectivity to monohalomethanes if operated at a relatively high methane-to-chlorine ratio (typically molar CH4:Cl2 ratio > 10), although CH2Cl2 and CHCl3 are formed in significant amounts at close to the stoichiometric feed composition.12,13 In contrast, methane bromination yields a high selectivity to CH3Br (>90%) at smaller methane excess (CH4:Cl2 > 5), while polybromination is generally limited to the formation of CH2Br2.1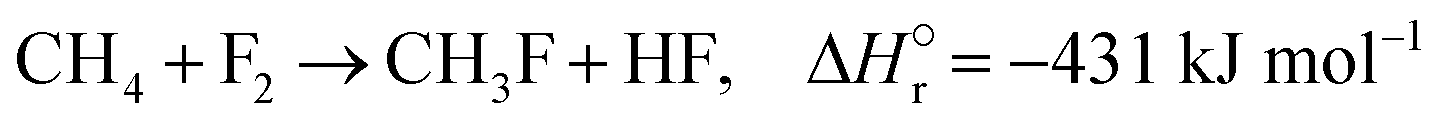 | (1) |
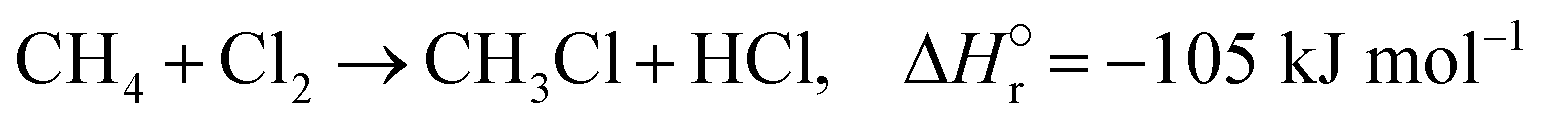 | (2) |
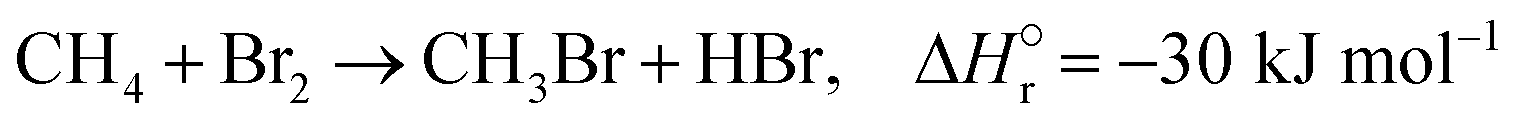 | (3) |
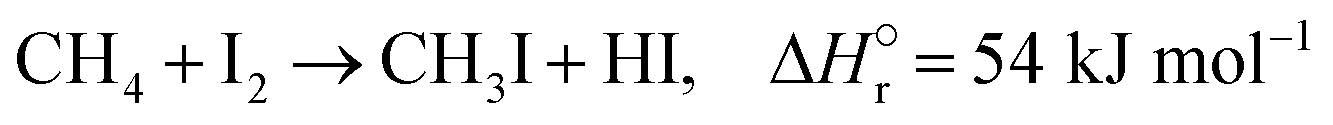 | (4) |
 | ||
| Fig. 1 Chronological overview of the major developments in methane halogenation presented along with the evolution in production of different carbon resources (X = Cl, Br). | ||
The industrial interest in methane halogenation already ignited in the early 20th century, when following a series of patents, the first industrial process based on non-catalytic thermal chlorination of methane was established in 1923 by Hoechst.2 After incorporating several refinements during its lifespan, this remains the state-of-the-art technology for the manufacture of chloromethanes (mostly CH2Cl2 and CHCl3), the world-wide production of which is estimated to exceed 1.4 Mt per year, with a market size of ca. 1000 M$ (Fig. 1).2 Although the latter chemicals are mostly used as solvents and precursors in synthesis of polymers and fine chemicals, as early as the 1920s CH3Cl was recognized as an attractive intermediate to produce methanol and ethers by reacting it with oxides such as CaO.14 This concept was advanced in the 1980s, when George A. Olah proposed a disruptive halogen-mediated route to valorise methane, the main constituent of highly abundant and inexpensive natural gas, by mitigating the costly and energy-intensive syngas-based processes.15 The concept is based on a selective C–H bond activation using Cl2 or Br2 to yield CH3X, which are versatile platform molecules that can be readily coupled into value-added chemicals and liquid fuels via catalytic pathways that are similar to those of methanol (Fig. 1).15–19 The implementation of this technology is contingent on the full recycling of the hydrogen halide (HX) formed in the halogenation and coupling steps and the ability to maximize the productivity of CH3X over polyhalomethanes and its coupling efficiency.1,20 The recent decade demonstrated significant progress in HX recovery via cataloreactant systems,21,22 electrochemical and thermochemical HX oxidation,23–26 and methane oxyhalogenation,26–28 as well as CH3X coupling,16–19 which brought this technology closer than ever to practical implementation. In contrast, the selective monohalogenation was tackled in a much less depth.
| CH4−nXn + CnH2n+2 → CH5−nXn−1 + CnH2n+1X | (5) |
| CH4−nXn + (n − 1)H2 → CH3X + (n − 1)HX | (6) |
One approach to enhance the selectivity to CH3X is based on the use of a substantial methane excess in the non-catalytic halogenation, which, however results in a large CH4 recycle stream.1,12,13 Alternatively, polyhalomethanes can be reformed by reproportionation with another alkane (eqn (5)),28–30 or hydrodehalogenation (eqn (6)).31 Still, the key to selective monohalogenation relies on mitigating the radical steps via an alternative reaction pathway that is facilitated by the catalyst. Although the early reports on halogenation describe the enhancement of the reaction rate in the presence of different “inert” solids (e.g., silica and active carbons) as well as CuCl2-based melts and fluidized beds,11,32 the fundamental landmark to this goal was set by Olah, who devised the concept of electrophilic C–H bond activation enabled through the polarization of the X2 molecule by the action of super-acid catalysts (Fig. 1).15,33 This pathway displays an inherent propensity to CH3X as the addition of strong electron withdrawing halogen substituents destabilizes the intermediate carbonium ion complex and hinders further substitution. The proof of concept was demonstrated using the homogeneous SbF5 catalyst, but aiming to achieve more practical space–time yields, Olah et al. studied supported noble metals as well as solid-acid catalysts based on oxyfluorides (Fig. 1).15,33 The former systems were found active at low temperatures (≤543 K), providing unprecedented selectivity to CH3Cl (80–99%) and CH3Br (99%) even when a substantial excess of halogen was applied. The same group also reported the catalytic effect of sulfated zirconia and several zeolites in methane chlorination.13,34 The high selectivity to CH3Cl (99%) can be obtained over mordenite using a molar ratio of CH4:Cl2 ≥ 4 at >623 K, while lower temperatures and small methane excess favour the radical pathways and consequent polyhalogenation. Nevertheless, the zeolite catalysts displayed a fast deactivation that was associated with excessive dealumination. The interest in selective halogenations has been rekindled in the early 2000s as a response to a shale gas surge. GRT Inc. described sulfated Nb2O5 and ZrO2 as selective bromination catalysts (≤95% CH3Br),21 while Degirmenci et al.35 reported high CH3Br selectivity (>99%) over sulfated ZrO2 included in SBA-15 structures (S-ZrO2-SBA-15) at large bromine excess. Joo et al.36 showed that methane chlorination over H–Y, Pt/H–Y and Pt/Na–Y leads to a slightly higher activity compared to the non-catalytic reaction, although the selectivity patterns remain almost unaffected. Batamack et al.37 reported the one-pot conversion of methane to valuable hydrocarbons by integrating methane halogenation and CH3X coupling over a SAPO-34 catalyst under a high CH4:X2 molar ratio of ca. 10.
In spite of these encouraging results, the previous reports were typically limited to the reaction of one halogen (i.e., Cl2 or Br2) and few catalytic systems, which were investigated under very different regimes, precluding thus the assessment of the catalytic over non-catalytic effects and derivation of the generalized performance trends as a function of the catalyst family and type of halogen, which are essential for catalyst development. Besides, the stability of halomethanes over different materials and the propensity of the latter towards coking or reactions with halogens in a corrosive halogenation environment were rarely discussed.
Herein, aiming to set the basis that will revitalize the research in this field, we conducted the first comprehensive evaluation of a large library of catalytic materials comprising inorganic and carbon carriers, supported noble metals, metal oxides, chlorides and oxyfluorides, as well as sulfated systems and zeolites in both methane chlorination and bromination under identical reaction conditions and provided a general overview on their potential to promote these reactions. In addition, the impact of the framework structure and topology on the performance of zeolite catalysts in methane chlorination was investigated, which along with the kinetics analysis uncovered the significant role of confinement effects on C–H bond activation with chlorine.
Experimental
Catalyst preparation
000 h−1) at 673 K for 5 h (5 K min−1). TaOF3/Al2O3 was prepared by gradually dissolving TaF5 (ABCR, 99%) in cold (195 K) methanol (Acros, 99.8%) followed by the addition of the resulting solution (0.48 M) to γ-Al2O3 (2.0 cm3 gAl2O3−1). Thereafter, the material was calcined at 523 K (1.5 K min−1).
:Al ratio of which are denoted by the prefix and suffix in the respective codes, were calcined prior to their use in the catalytic tests or as precursors in the further modification of the parent material. The zeolites containing ammonium ions were thus transformed into the protonic (H) form.
Li-ZSM-5-25, Na-ZSM-5-25, Cs-ZSM-5-25, Ca-ZSM-5-25 and Fe-ZSM-25 were prepared by triple ion exchange of H-ZSM-25 zeolite using a 0.1 M aqueous solution of the respective salts, LiNO3 (FLUKA, 98%), NaNO3 (Acros, ≥99%), CsNO3 (Acros, 99.99%), Ca(NO3)2·4H2O (Sigma-Aldrich, ≥99%) and FeCl2·4H2O (Sigma-Aldrich, 98%), at room temperature (100 cm3 gzeolyte−1) for 12 h, followed by drying and calcination. Pt-ZSM-5-25 (1 wt% Pt) was obtained by incipient wetness impregnation of the H-ZSM-5-25 material with a solution of [Pt(NH3)4]Cl2·nH2O using the analogous procedure detailed for supported metal catalysts. Mg-ZSM-5-140 (2 wt% MgO) was prepared following the method of Liu et al.18 Herein, a 0.16 M solution of Mg(NO3)2·6H2O (Sigma-Aldrich, 99%) was mixed with H-ZSM-5-140 zeolite for 2 h (3.2 cm3 gzeolite−1). The suspension was dehydrated at 353 K for 4 h, dried and calcined at 723 K for 8 h.
H-ZSM-25-ox was prepared by refluxing the parent H-ZSM-5-25 sample with 1 M H2C2O4 aqueous solution (50 cm3 gzeolyte−1) for 13 h. Thereafter, the material was recovered by filtration, washed with deionized water, dried and calcined. H-ZSM-5-25-st was derived by steaming (100 mol% H2O) the parent H-ZSM-5-25 material in a fluidized bed reactor (GHSV = 5350 h−1) for 12 h. H-ZSM-5-40-at zeolite was prepared using the well-established alkaline treatment protocol.39 In brief, the parent H-ZSM-5-40 zeolite was exposed to a 0.2 M NaOH (Acros, 98.5%) aqueous solution (3.75 cm3 gzeolyte−1) and the resulting suspension was stirred at 338 K for 30 min in a Mettler Toledo Easymax 102 reactor system. The treatment was quenched by diluting the mixture with a twenty-fold excess of deionized water, followed by solid separation by filtration and washing until neutral pH. The material was ion exchanged three times with 0.1 M NH4NO3 (Acros, 99%) aqueous solution (100 cm3 gzeolyte−1) at 338 K for 12 h, and finally dried and calcined. Nanocrystalline H-ZSM-5-40-nc zeolite was synthesized according to the procedure of Van Grieken et al.40 Aluminium isopropoxide (Al-iPr, Aldrich, ≥98%), NaOH, TEOS and ethanol (Acros, 99.8%) were added in the as-listed order to a 1 M TPAOH aqueous solution (molar ratio of TEOS:Al-iPr:NaOH:ethanol:TPAOH = 40:1:0.16:100:25) and stirred at room temperature for 12 h. The clear solution was transferred into a Teflon-lined autoclave and heated under static conditions at 438 K for 4 days. The final product was collected by filtration, washed with deionized water, dried and calcined. H-SnBETA-220 was synthesized using alkaline-assisted stannation,41 which is equivalent to the above detailed alkaline-treatment in all aspects, except that the parent BETA-220 zeolite (Tosoh Corp.) was exposed to an aqueous solution comprising 0.3 M NaOH, 0.04 M SnSO4 (ABCR, 95%) and 0.2 M tetrapropylammonium hydroxide (TPAOH, Alfa Aesar, 20 wt%).
Unless stated otherwise, the standard drying conditions involve a pressure of 5 kPa (vacuum), a temperature of 373 K and a treatment time of 12 h, while the standard calcination conditions involve a static air atmosphere, a temperature of 823 K, a time of treatment of 5 h and a heating rate of 5 K min−1.
 | ||
| Fig. 2 Flowsheet of the continuous-flow fixed-bed reactor setup for methane halogenation. The red-coloured sections of the linings were heated at 393 K to prevent condensation. | ||
Prior to testing, the solids were heated to 723 K for 2 h in flowing He (GHSV = 13700 h−1) to eliminate the traces of moisture, O2 and other possible gas impurities. The only exceptions were PdCl2/SiO2, CuCl2/SiO2, TaOF3/Al2O3 and Nafion catalysts, which were treated at 573 K to avoid their potential thermal decomposition or volatilization. Thereafter, the bed was cooled down to the desired temperature and left to stabilize for at least 30 min before the catalytic test. Unless stated otherwise, the reactant mixture comprising CH4:Cl2 in a molar ratio of 8.5:3.5 was fed at GHSV = 267 h−1 or 50 h−1 in methane chlorination and the mixture containing CH4:Br2 in a molar ratio of 8.5:3.2 ± 0.1 was introduced at GHSV = 260 h−1 or 48.5 h−1 in methane bromination. The reactor effluent was diluted with a He–Ar mixture (He:Ar = 71.5:4.5) and directed towards the analysis section. Carbon-containing compounds (CH4, CH3Br, CH2Br2, CH3Cl, CH2Cl2, CO and CO2) and Ar were quantified on-line via a gas chromatograph equipped with a GS-carbon PLOT column coupled to a mass spectrometer (GC-MS, Agilent GC 6890, Agilent MSD 5973N). Quantification of Cl2 and Br2 at the reactor outlet was performed by their absorption in an impinging bottle filled with 0.1 M KI aqueous solution (X2 + 3I− → I3− + 2X−) followed by iodometric titration (Mettler Toledo G20 Compact Titrator) of the formed triiodide (I3− + 2S2O32− → 3I− + S4O62−) with 0.01 M Na2S2O3 (Aldrich, 99.99%).
The conversion of methane, XCH4, the conversion of halogen, XX2, the reaction rate expressed with respect to methane, rCH4, the selectivity to product j, Sj, the space–time yield of carbon oxides, STYCOx, and the error of the carbon mass balance, εC, were calculated using eqn (7)–(12), respectively, in which ninleti and noutleti are the molar flows of reactant i at the reactor inlet and outlet, respectively, noutletj is the molar flow of product j at the reactor outlet and Wcat is the catalyst weight. To minimize the errors, parameter k in eqn (7) was set to 0, except for those measurements for which the error of the carbon mass balance systematically showed negative deviation indicative of coke formation, where it was set to 1/2. The absence of mass and heat transfer limitations in the catalytic tests was confirmed by the evaluation of the dimensionless moduli based on the criteria of Carberry, Mears and Weisz-Prater.42 The reported values of conversion, selectivities and reaction rates were determined as an average of 3–5 consecutive measurements performed during 3 h of operation under specified reaction conditions, with the first sample taken after 40 min of stabilization, while the standard deviation of the data set is indicated by the error-bars where appropriate. Unless stated otherwise, the error of the carbon mass balance was less than 3% in all experiments.
 | (7) |
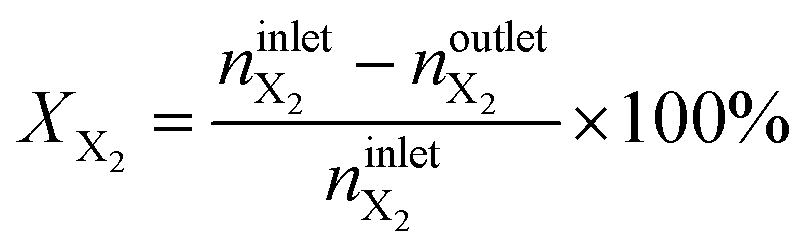 | (8) |
 | (9) |
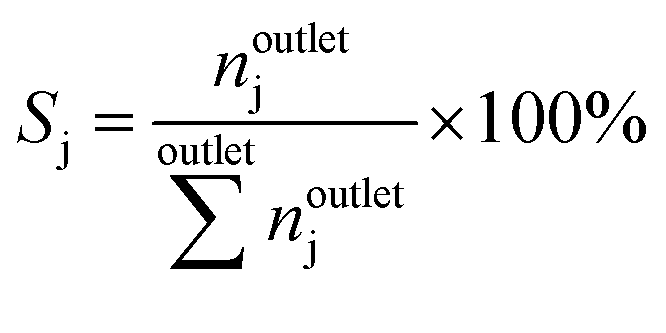 | (10) |
 | (11) |
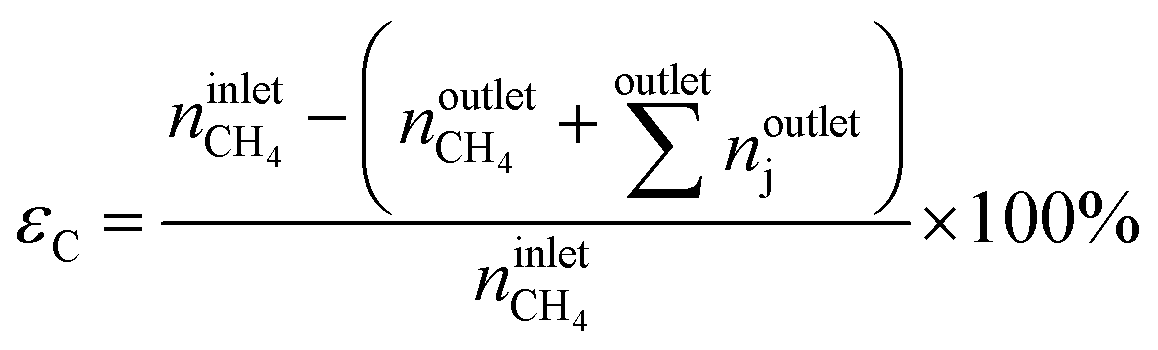 | (12) |
Results and discussion
Catalyst performance in methane halogenation
The preliminary insights into the catalytic effects in methane halogenation were acquired by analysing both the chlorination and bromination performances of Pt/SiO2, TaOF3/Al2O3, S-ZrO2 and H-SAPO-34 (Fig. 3), which are the main representatives of the previously reported catalyst families (Fig. 1).13,15,34,37 The activity and selectivity profiles of the materials were recorded by using the undiluted reactant mixtures comprising methane and halogen in a molar ratio of CH4:X2 = 3.5 in the chlorination and CH4:X2 = 3.2 ± 0.1 in the bromination reaction. Moderate methane excess simulates the practically relevant operating regime, which strives at full halogen consumption that facilitates the downstream product separation while minimizing the size of the hydrocarbon recycle. Moreover, the selected ratios were smaller than those at which free-radical halogenations display an inherently high selectivity to CH3X, enabling thus the resolution of the catalytic from the non-catalytic effects.15,34 With regard to the latter problems, the catalyst activities were compared at constant GHSV and constant temperature (GHSV = 267 h−1 and T = 523 K in methane chlorination, GHSV = 260 h−1 and T = 643 K in methane bromination) and benchmarked to the activity patterns of the empty glass reactor and reactor accommodating the bed of quartz particles, whereby the values of the latter two reaction parameters are consistent with the typical conditions applied in previous studies.13,15,34,35,37 This approach, which was practiced in the work of Olah et al.,13,15,34 enables the decoupling of the catalytic from the non-catalytic gas-phase contributions, since the volume of the interparticle voids does not differ significantly between the different catalyst beds. Besides, it also has practical relevance as it shows the level of productivity enhancement of a given reactor in the presence of catalysts compared to the inherent activity of its void volume or bed composed of inert particles.
 | ||
| Fig. 3 a) Conversion of reactants in methane chlorination at T = 523 K and GHSV = 267 h−1 (left panel) and bromination at T = 643 K and GHSV = 260 h−1 (right panel) over representative catalysts. b) Selectivity to CH3X in methane chlorination (left panel) and bromination (right panel) over representative catalysts as a function of methane conversion, which was adjusted by varying the temperature or GHSV. The catalysts are identified according to the shape and colour code presented in a), while the pattern of the symbol interior defined in b) provides the information on the reaction temperature and GHSV at which the data point was recorded. The green- and pink-coloured areas in b) outline the range of CH3X selectivities in methane chlorination and bromination, respectively, achieved in the void reactor and over various materials. Other conditions: CH4:Cl2 = 8.5:3.5, CH4:Br2 = 8.5:3.2 ± 0.1 and P = 100 kPa. | ||
The assessment of both methane and halogen conversion in methane chlorination at 523 K indicated that the bed of quartz particles displays an almost identical performance to an empty reactor of the same volume (Fig. 3a). Considering the fact that the effective void volume of the bed is ca. 2 times smaller than that of the empty reactor (bed porosity, ε ≈ 0.4),43 this result suggests that the chlorination reaction is promoted even in the presence of inert and low-surface area solids. The conversion of methane and Cl2 over Pt/SiO2, TaOF3/Al2O3, S-ZrO2 and H-SAPO-34 catalysts was ca. 2–2.5 times higher compared to that over quartz, suggesting the further activity promotion over these solids. The onset of methane bromination was shifted to ca. 120 K higher temperature with respect to chlorination. The activity of quartz particles was comparable to that of an empty reactor, but in contrast to chlorination, methane conversion in bromination was almost unaffected by the catalytic materials. Still, the conversion of bromine was higher over the catalysts, especially H-SAPO-34, which can be associated with a more pronounced polybromination, oxidation and reactions with a catalyst (vide infra).
In addition to the conversion profiles, the differences in the selectivity to CH3X were evaluated as another indicator of the potential catalytic effects (Fig. 3b). In general, the operation under comparable CH4:X2 molar ratios and GHSVs led to a higher fraction of CH3X in bromination compared to that in chlorination (SCH3Br = 80–95% versus SCH3Cl = 52–90% at XCH4 = 5–18%). Besides, the selectivity to CH3X declined less steeply upon increasing the methane conversion in bromination, corroborating the higher inherent propensity to selective monohalogenation in this reaction. Nevertheless, the selectivity to CH3X in both reactions decreased in a similar order, empty reactor ≈ quartz ≈ Pt/SiO2 > TaOF3/Al2O3 ≈ S-ZrO2 > H-SAPO-34, indicating the prominent impact of the catalyst on the product distribution.
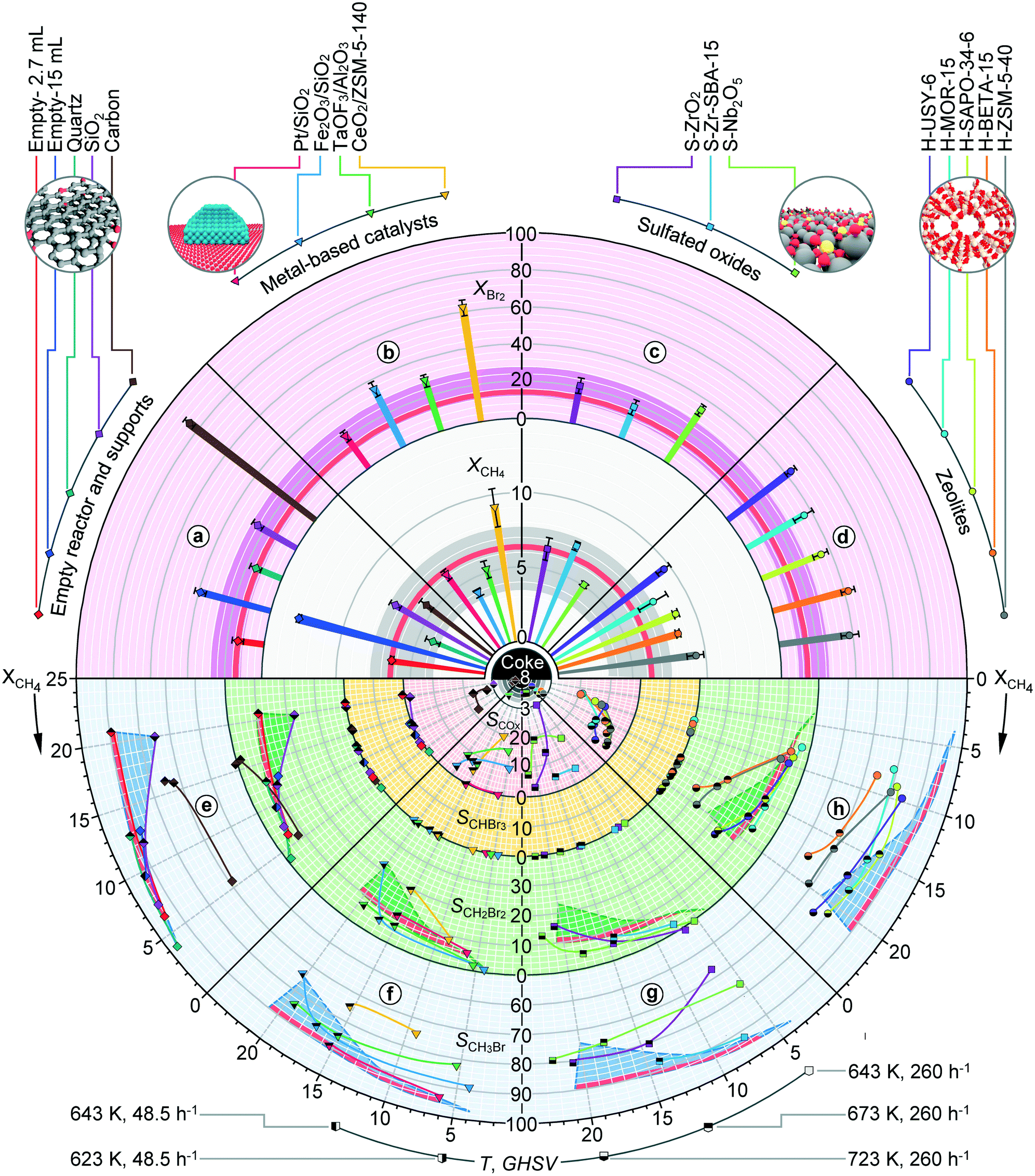
In order to generalize the performance trends, the analysis was extended to a significantly broader range of materials which can be classified into four distinct groups, supports, metal-based materials, sulphated oxides and zeolites. Aiming to provide a comprehensive overview of their catalytic behaviours and the facile benchmarking of the activity and selectivity patterns of the materials with respect to the void reactors, supports, other material families and their individual members, the circular plots are devised, as presented in Fig. 4 and 5. These are composed of the top semicircle, which provides the information of the catalyst activity, and the bottom semicircle, which outlines the product distribution patterns. The semicircles are further divided into four circular sectors, a–d (top) and e–h (bottom), which present the performances of the materials within a specific family. The activity sectors provide the information on the halogen and methane conversion at constant temperature and GHSV, which both increase in the direction from the circle centre. The bottom sectors present the selectivities to CH3X, CH2X2, CHX3 and COx as a function of methane conversion, which increases in a clockwise direction, and indicate the formation of coke deposits. The red- and dark-coloured background areas of the subsections denote the performance regions of empty reactors and inorganic carriers, respectively, serving thus as the reference for the evaluation of the catalytic contributions.
 | ||
| Fig. 4 Conversion of reactants in methane chlorination at T = 523 K and GHSV = 267 h−1 a) in empty reactors and over supports, b) supported metal-based catalysts, c) sulfated oxides and d) zeolites (top semicircle). Selectivity to different products in methane chlorination e) in empty reactors and over supports, f) supported metal-based catalysts, g) sulfated oxides and h) zeolites as a function of methane conversion which was adjusted by varying the temperature or GHSV (bottom semicircle). Conversions and selectivities are expressed in %. The catalysts are identified according to the shape and colour code presented above the top semicircle, while the pattern of the symbol interior defined below the bottom semicircle provides the information on the reaction temperature and GHSV at which the data point was recorded. The central part of the bottom semicircle displays the errors of the carbon mass balance in % for those catalytic points for which the consistent negative deviation of this parameter was observed, indicative of coke formation. The red- and darker-coloured areas outline the benchmark range of conversions and selectivities displayed by the empty reactor and different supports, respectively. More information regarding the plot is provided in the main text. Other conditions: CH4:Cl2 = 8.5:3.5 and P = 100 kPa. | ||
 | ||
| Fig. 5 Conversion of reactants in methane bromination at T = 643 K and GHSV = 260 h−1 a) in empty reactors and over supports, b) supported metal-based catalysts, c) sulfated oxides and d) zeolites (top semicircle). Selectivity to different products in methane bromination e) in empty reactors and over supports, f) supported metal-based catalysts, g) sulfated oxides and h) zeolites as a function of methane conversion, which was adjusted by varying the temperature or GHSV (bottom semicircle). Conversions and selectivities are expressed in %. The catalysts are identified according to the shape and colour code presented above the top semicircle, while the pattern of the symbol interior defined below the bottom semicircle provides the information on the reaction temperature and GHSV at which the data point was recorded. The central part of the bottom semicircle displays the errors of the carbon mass balance in % for those catalytic points for which the consistent negative deviation of this parameter was observed, indicative of coke formation. The red- and darker-coloured areas outline the benchmark range of conversions and selectivities displayed by the empty reactor and different supports, respectively. More information regarding the plot is provided in the main text. Other conditions: CH4:Br2 = 8.5:3.2 ± 0.1 and P = 100 kPa. | ||
The conversion levels attained over various inorganic supports, i.e., SiO2, SiC, α-Al2O3 and γ-Al2O3 are similar to or higher than that of the empty reactor, even when its volume exceeds the volume of the carrier bed by a factor of 5.5 (Fig. 4, sector a), corroborating thus the beneficial effect of solids on the low-temperature methane chlorination. Thereby, mesoporous SiO2 and γ-Al2O3 carriers displayed ca. 2 times higher conversion of reactants as compared to their nonporous counterparts, quartz, SiC and α-Al2O3, suggesting the positive impact of the surface area on methane chlorination. Nonetheless, the selectivity profiles of the former carriers proved the lower fraction of CH3Cl as compared to those of the latter group, which exhibited a similar product distribution pattern to the empty reactor (Fig. 4, sector e). Notably, while CH2Cl2 and to a small extent CHCl3 constituted the only by-products over quartz, SiO2 and SiC, the evolution of COx along with CH2Cl2 was very prominent over alumina supports. Given the oxygen-free inlet feed and the absence of reaction between methane and α-Al2O3 and γ-Al2O3 in the temperature range of interest, the formation of COx could only arise from the decomposition of chloromethanes over these carriers (vide infra, Fig. 7b).44 Another peculiar behaviour was observed for the carbon support, which although exhibiting ca. 1.5–2 times higher methane conversion than SiO2 and γ-Al2O3 and a selectivity profile that closely matched that of quartz and the empty reactor led to an immense Cl2 conversion. The latter significantly exceeded the stoichiometric level calculated on the basis of chloromethane production (ca. 30%) as well as the Cl2 conversion levels achieved over the materials displaying similar methane consumption (e.g., H-ZSM-5-40, vide infra), indicating that a part of Cl2 might react with the carbon support. This hypothesis was verified by exposing the carbon carrier to a pure chlorine stream which was fed at a similar temperature and GHSV to those applied in chlorination, whereby a halogen uptake of ca. 50% was measured.
The recorded performance profiles of various carriers define the benchmark regions (denoted as darker-coloured areas in Fig. 4) which enabled the potential catalytic contributions of the supported metal-based active phases to be decoupled. Specifically, the conversion of methane over all the studied systems was of similar magnitude to that of the respective carriers (Fig. 4, sector b). Pt/carbon, CuCl2/SiO2 and Fe2O3/SiO2 displayed a higher chlorine conversion, which can be associated with the carrier halogenation for the first (vide supra) and active phase chlorination for the latter two catalysts. In addition, CuCl2/SiO2 and Fe2O3/SiO2 catalysts exhibited the formation of deposits on the reactor walls and glass-beads located below the catalyst bed, which are indicative of the volatilization of the metal-halide species. The product distribution patterns of most of the metal, metal halide and metal oxide catalysts paralleled those of the respective supports and empty reactors, with PdCl2/SiO2 and Fe2O3/SiO2 exhibiting the highest selectivity to CH3Cl (Fig. 4, sector f). The most prominent deviations from these trends were displayed by Pt/carbon and TaOF3/Al2O3, which favoured COx production, while the first system also displayed a marked polyhalogenation activity.
The conversion levels of methane and Cl2 over sulfated catalysts were comparable to those of SiO2 and γ-Al2O3, with S-TiO2 displaying the highest activity (Fig. 4, sector c), while the selectivity to CH3Cl for most of these systems followed almost identical patterns to those recorded over quartz and in an empty reactor (Fig. 4, sector g). More peculiar deviations from the general selectivity trends were displayed by S-ZrO2 and S-Nb2O5 catalysts. Specifically, in the case of S-ZrO2, CH3Cl was the only chlorinated product at low-to-moderate methane conversions (≤11%). However, this behaviour was linked with high selectivity to COx (SCOx = 2–27%, mostly CO), likely produced through the decomposition of polychlorinated products (vide infra, Fig. 7b). The evolution of COx was also pronounced over the S-Zr-SBA-15 catalyst. In turn, S-Nb2O5 provided the highest selectivity to CH3Cl, which was ca. 2% higher than that of the empty reactor. The supporting of the latter active phase on SiO2 did not enhance the activity, while the CH3Cl selectivity curve was shifted to slightly lower values that are still above those of SiO2. Nevertheless, a small fraction of COx (SCOx = 2%) was also formed over the supported system.
More significant performance differences with respect to the previously discussed catalyst families were observed when the chlorination reaction was performed over zeolites. In particular, while the methane and Cl2 conversions achieved over zeolite 3A, H-USY-6, H-MOR-15 and H-SAPO-34 fall in the similar range to those of SiO2 and γ-Al2O3 supports, H-BETA-15 and H-ZSM-5-40 provided ca. 2–2.5 times higher conversion levels, enabling thus up to 5.5 times higher activity with respect to the benchmark quartz and empty reactor systems. Another peculiar feature of zeolites is related to the increased fraction of CH2Cl2 and CHCl3 with respect to CH3Cl. In addition, similar to the alumina carriers, all zeolite catalysts except H-ZSM-5-40 favoured the production of COx. This was especially pronounced for H-SAPO-34, which was also the only system leading to coke deposition at 573 K, as inferred both from more significant deviations of the carbon balance and visual inspection of the used catalyst. The evaluation of the H-ZSM-5-40 zeolite in a 10 h test using a CH4:X2 ratio of 8.5:3.5 and GHSW = 267 h−1 at 573 K indicated no performance deterioration, whereby the X-ray diffraction pattern and N2-isotherm of the used catalyst do not show significant variations with respect to the fresh material.
In analogy to methane chlorination, the performance analysis was further extended to additional supports and catalytic systems in methane bromination (Fig. 5). The conversions of methane and Br2 over the porous SiO2 support were ca. 2 times higher as compared to those over quartz, but still comparable to those of the empty reactor of identical volume. This suggests that the external surface has a smaller impact on the bromination activity, which appears to be primarily governed by the available void volume. This is also corroborated by the significantly higher conversion increase upon expanding the void reactor volume as compared to methane chlorination. In view of the product distribution, the SiO2 carrier displayed a similar behaviour to quartz and the empty reactor in the low temperature regime. However, a prominent drop in selectivity to CH3Br (ca. 15%) coupled with the evolution of small amounts of COx and coking occurred at higher temperatures (ca. 723 K), suggesting the interaction of brominated products with the SiO2 carrier. The fraction of COx as well as the propensity to coke was particularly high for the carbon support, which also displayed a marked bromine conversion in analogy to its behaviour in methane chlorination.
Compared to the empty reactor and inorganic carriers, supported metal-based catalysts (Fig. 5, sector b) and sulfated oxides (Fig. 5, sector c) generally led to comparable conversion levels, while their selectivity patterns were characterized by a lower fraction of CH3Br coupled with pronounced COx production. The exceptions to these trends were Pt/SiO2 and to some extent S-ZrO2-SBA-15, whose selectivity curves did not deteriorate substantially from the benchmark systems. Similar to methane chlorination, the volatilization of Fe2O3/SiO2 was also observed. In the case of sulfated oxides, SO2 was detected in the outlet feed, indicating their limited stability in a bromination environment. In addition, coke deposits were formed on S-ZrO2 at 723 K. Zeolite catalysts with the exception of H-MOR yielded slightly higher conversions of methane (ca. 1–2%) compared to SiO2. However, the product pool of all the zeolite catalysts was negatively affected by the increased fraction of COx as well as coking, which was prominent at temperatures ≥723 K.
Impact of zeolite structure on methane chlorination
The noticeable activity promotion of the H-ZSM-5-40 catalyst with respect to the empty reactor and various inorganic carriers in methane chlorination coupled with the considerable compositional and modification flexibility of the MFI framework makes this catalytic system highly suitable to study the structure–performance relationships that could cast light on the origins of its catalytic action (Fig. 6). In this respect, the activity of H-ZSM-5 materials was first analysed as a function of their Si:Al ratio, which is the central parameter controlling the acid properties and polarity of zeolites (Fig. 6, sectors a and e).45 The conversion of reactants was slightly lower over H-ZSM-5-15 as compared to the other H-ZSM-5 catalysts. Nonetheless, the overall variations in activity and product distribution were of limited extent in a broad range of Si:Al ratios (15–1000), which along with the similar behaviour of the TS-1-25 catalyst implies a weak correlation between the zeolite performance and the concentration and strength of Brønsted acidic sites. In addition, the introduction of various cations in the ZSM-5-25 framework had almost no influence on the activity and selectivity patterns (Fig. 6, sectors b and f). The only exception to the latter trend were Li-ZSM-5-25 and Na-ZSM-5-25, which led to a more significant conversion enhancement compared to the benchmark H-ZSM-5-40 and H-ZSM-5-25. This result might be the sign of the additional catalytic effect that could arise from the strong Lewis acidic (electrophilic) character of the relatively small and solvent-free Li+ and Na+ cations that has been found beneficial for the chlorination of aromatic compounds.46 Nevertheless, the selectivity–conversion profiles of both of these catalysts did not deflect significantly from those recorded over other ZSM-5 systems, except that similar to Cs-ZSM-5-25 it led to a slightly lower fraction of CH3Cl compared to CH2Cl2 and CHCl3 (Fig. 6, sector f). Consistent with the above observations, the treatments of the H-ZSM-5-40 catalyst with steam or oxalic acid, which cause the formation of extraframework aluminium species, loss of acidity and moderate increase in mesoporosity,47,48 did not affect the performance (Fig. 6, sectors c and g). Still, the alkaline-assisted desilication of this zeolite, which introduces a more significant fraction of intracrystalline mesopores that are also more accessible from the outside surface,39 led to a slight increase in the fraction of monochlorinated over polychlorinated products at essentially unaltered activity. A similar change in the selectivity profile at almost unaltered activity was observed for H-SnBETA-220 (prepared by alkaline-assisted stannation) with respect to the reference H-BETA-15 catalyst (Fig. 6, sectors c and g). Given the weak sensitivity of the chlorination reaction to the nature and strength of acid sites, the shift in the selectivity pattern observed in the samples with more developed mesoporosity can be associated with a decrease in the characteristic length of the microporous domains that could reduce the extent of polyhalogenation by facilitating the escape of CH3Cl. This hypothesis is further corroborated by the behaviour of H-ZSM-5-40-ns comprising crystals of smaller size with respect to H-ZSM-5-40, which also induced a slight decrease in the fraction of CH2Cl2 and CHCl3 with respect to CH3Cl (Fig. 6, sector g).
 | ||
| Fig. 6 Conversion of reactants in methane chlorination at T = 523 K and GHSV = 267 h−1 over ZSM-5 and BETA zeolites with different a) Si:Al ratios, b) counter ions, c) mesoporosities and crystallite sizes and d) at various feed compositions (top semicircle). Selectivity to different products in methane bromination over ZSM-5 and BETA zeolites with different e) Si:Al ratios, f) counter ions, g) mesoporosities and crystallite sizes and h) at various feed compositions as a function of methane conversion which was adjusted by varying the temperature (bottom semicircle). Conversions and selectivities are expressed in %. The catalysts are identified according to the shape and colour code presented above the top semicircle, while the pattern of the symbol interior defined below the bottom semicircle provides the information on the reaction temperature at which the data point was recorded. The central part of the bottom semicircle displays the errors of the carbon mass balance in % for those catalytic points for which the consistent negative deviation of this parameter was observed, indicative of coke formation. The gray-coloured areas outline the benchmark range of conversions and selectivities displayed by the H-ZSM-5-40 catalyst. More information regarding the plot is provided in the main text. Other conditions: CH4:Cl2 = 8.5:3.5 and P = 100 kPa. | ||
In addition to the variations in the framework structure, the impact of the reaction conditions on the chlorination performance of ZSM-5 zeolites was investigated (Fig. 6, sectors d and h). In particular, the impact of water co-feeding was evaluated as it was previously devised as a strategy to boost the selectivity to CH3Cl and minimize coking over H-SAPO-34.37 In fact, the addition of water to the chlorination feed decreased the fraction of CH2Cl2 and almost eliminated the formation of CHCl3 at virtually unaltered conversion levels. Nevertheless, it provoked a significant COx (mostly CO) evolution, indicating the preferential hydrolysis of the polychlorinated products, which was not considered before. This result also corroborates that the evolution of the COx that was observed over other zeolite frameworks in chlorination and was especially pronounced in bromination stems from the interaction of halomethanes with bridging oxygen sites, which could be replenished by water. Finally, the CH4:Cl2 ratio was increased to explore the scope for the enhancement of selectivity to CH3Cl. Notably, the operation with ten-fold methane excess caused only a small increase in selectivity to CH3Cl (ca. 6%) with respect to the use of the reference CH4:Cl2 ratio of 8.5:3.5 (Fig. 6, sectors d and h). Comparatively, the use of 10-fold methane excess in the case of SiO2 led to ca. 15% increase in selectivity to CH3Cl with respect to the above stated reference feed.
Mechanistic insights into methane halogenation over solid surfaces
The evaluation of a large number of materials in methane halogenations revealed their positive impact on the chlorination activity, but limited effect on the bromination rate as compared to the respective non-catalysed reactions (Fig. 3–5). Nonetheless, metal-based and sulfated catalysts displayed similar conversion levels to the supports in methane chlorination, while their CH3Cl selectivity profiles were comparable to those achieved in the empty reactor and over the bare carriers. Notably, the variations of the Si:Al ratio and counter cation as well as the formation of the extraframework aluminium species, which are expected to alter the polar intermediates and transition states, had almost no effect on the performance over the most active ZSM-5 zeolite. These results provide strong indication that the radical-chain mechanism dominates in the overall process, since the alternative activation pathways would induce more significant deviations in activity and selectivity patterns. To further support these findings, the apparent kinetic fingerprints of methane chlorination acquired over the H-ZSM-5-40 catalyst were compared to those recorded over SiO2 (Fig. 7a). The apparent activation energy of the zeolite catalyst was ca. 20 kJ mol−1 lower as compared to that of SiO2, in agreement with the observed activity enhancement. Still, the apparent reaction orders with respect to methane and Cl2 have very similar values for the two materials, suggesting the similarity in the mechanism of reactant activation. Additional experimental evidence supporting the involvement of radical intermediates in the chlorination over zeolite catalysts stems from a prominent decrease of the chlorination rate over H-ZSM-5-40 upon the addition of O2, which is a well-known inhibitor of radical processes.3,6,11 Moreover, the O2 inhibition constants over both H-ZSM-5-40 and SiO2 are essentially identical (ca. −0.6), whereby their absolute value closely matches the apparent reaction order measured with respect to Cl2. This is consistent with the mechanism of O2 inhibition involving its reaction with methyl radicals, which hampers the regeneration of chlorine radicals from Cl2 and hence chain propagation (Fig. 1).6
 | ||
| Fig. 7 a) Rate of methane chlorination as a function of reciprocal temperature and partial pressure of methane, chlorine and oxygen over SiO2 and H-ZSM-5-40. b) Selectivity to COx in methane chlorination at 523 K versus space–time yield of COx in the reaction of CH2Cl2 with different solids at 553 K. c) Selectivity to COx in methane bromination at 643 K versus space–time yield of COx in the reaction of CH2Br2 with different solids at 693 K. Other conditions: a) CH4:Cl2:O2:Ar:He = 17:7:0:4.5:71.5 (temperature variation), 17–47.5:7:0:4.5:71.5–41 (methane variation), 17:3.5–14:0:4.5:75–64.5 (chlorine variation) or 17:7:0–20:4.5:71.5–51.5 (oxygen variation). The impact of temperature on the reaction rate was studied at T = 485–523 K (H-ZSM-5-40) or 518–594 K (SiO2) and the impact of the partial pressures of reactants and oxygen was evaluated at T = 523 K (H-ZSM-5-40) or 594 K (SiO2) using GHSV = 2220 h−1 (H-ZSM-5-40) or 4440 h−1 (SiO2). The conversions of reactants were in the range of XCH4 = 1–9% and XCl2 = 1–37%. b and c) Methane halogenation: CH4:Cl2 = 8.5:3.5, CH4:Br2 = 8.5:3.2 ± 0.1 and GHSV = 267 h−1 (chlorination) or 260 h−1 (bromination). CH2X2 reaction with solids: CH2X2:Ar:He = 2:4.5:93.5, GHSV = 1110 h−1. All tests were performed at P = 100 kPa. | ||
The promotional effect of the solid surfaces in the catalytic chlorination may stem from the evolution of methyl radicals, as postulated in other methane functionalization processes such as oxidative coupling.49 Nonetheless, considering the significantly lower temperature window of methane chlorination (<623 K) with respect to oxidative coupling (>873 K), the surface-mediated formation of methyl radicals is expected to be less favored in the former reaction. This is corroborated by the negligible rate of methane oxidation over the most active H-ZSM-5 catalyst. Hence, the higher rates of methane chlorination over solid catalysts are likely associated with the enhancement of chlorine dissociation by the surfaces, which has the major contribution to the overall reaction barrier in this reaction at low temperatures (Fig. 8a).1,29 Namely, the unimolecular dissociation of Cl2 necessitates the interaction with the second molecular entity, referred to as the mediator (M), which excites the halogen molecule.50 In this context, one part of the promotional effect could come from the non-specific chlorine–surface collisions, generally known as “wall effects”.11,50 In addition, theoretical studies pointed out that Cl2 interaction with the defect sites on metal and metal-oxide surfaces (Fig. 8a, inset) may lead to the transfer of the electron density to the antibonding orbitals of the adsorbate.51,52 This weakens the Cl–Cl bond and might induce practically barrierless dissociation of Cl2 into atomic Cl˙. The dominant impact of the solid surfaces on the chlorine dissociation step is also corroborated by the observation that the rate difference between methane chlorination over the solids and methane chlorination in the empty reactor diminishes at higher reaction temperatures, since the latter reaction parameter increases the effectiveness of Cl2–M collisions and shifts the equilibrium towards dissociation, yielding thus sufficiently high concentration of Cl˙ radicals that can initiate the reaction.
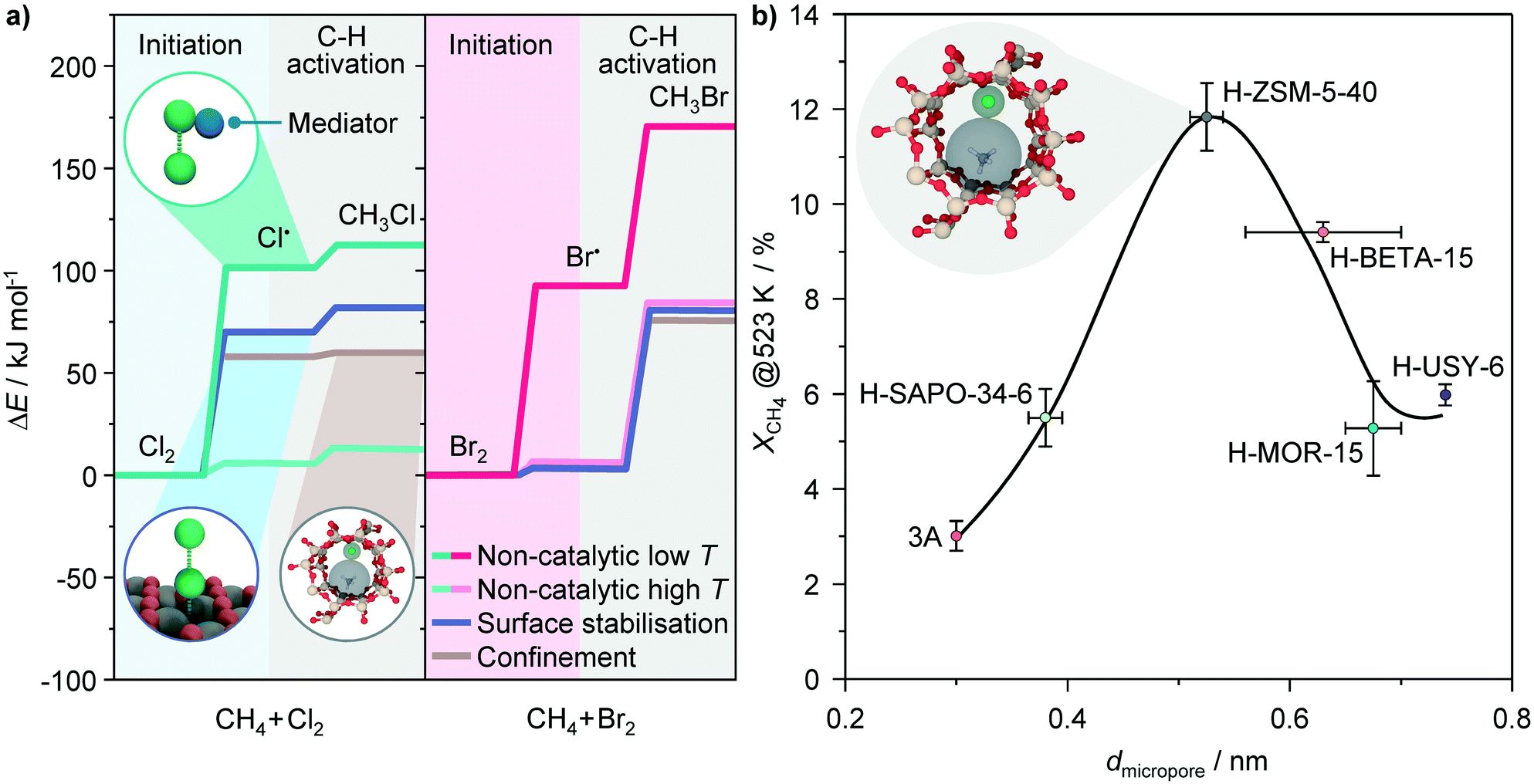 | ||
| Fig. 8 a) Rate determining steps involved in non-catalytic free-radical methane chlorination (left) and bromination (right) at low and high temperatures, along with the proposed impact of the catalysts on their energy barriers. The energy profiles of the initiation and C–H activation in non-catalytic processes are taken from literature.29,56 The insets depict the interaction of halogens with a mediator and defect sites on catalyst surfaces facilitating halogen dissociation, and confinement within the zeolite pores favoring C–X bond formation. b) Conversion of methane in methane chlorination as a function of the micropore diameter of various zeolites. Conditions: CH4:Cl2 = 8.5:3.5, T = 523 K, GHSV = 267 h−1 and P = 100 kPa. | ||
The radical nature of halogenation reactions over zeolite catalysts raises the question about the origin of activity enhancement over the H-ZSM-5 and BETA materials with respect to the other zeolites as well as other catalytic systems. The results discussed so far suggest that these are not correlated with their electronic, but rather geometric properties. The latter could have an impact on the confinement of the reactants, radical intermediates or transition states, facilitating thus the reaction. In fact, in addition to their acidity, the ability of zeolites to stabilize the reactive intermediates and transition states by the action of the dispersive van der Waals interactions within the well-defined system of micropores, generally referred to as shape selectivity, represents another important aspect of their catalytic action, particularly in the reactions involving hydrocarbons.53 Notably, if the activities of various zeolites attained in the low-temperature regime are plotted against their characteristic pore openings,54 a volcano-like dependence is obtained, which exhibits a maximum for micropore diameters of 0.56–0.6 nm (Fig. 8b). Strikingly, the latter micropore size range is comparable to the sum of the kinetic diameters of methane (0.38 nm) and chlorine radicals (0.16 nm),55 which are the intermediates participating in the second step that has a smaller contribution to the overall reaction barrier (Fig. 8a). These results indicate that although the chlorination of methane over zeolite catalysts preserves its radical character, the appropriate size of BETA and particularly ZSM-5 structures enables the optimal confinement of the reactive species, enabling thus a higher activity (Fig. 8b).
The above reasoning might also explain the lack of a significant promoting effect of the catalysts in methane bromination. Owing to weaker Br–Br and C–Br bonds as compared to the respective Cl–Cl and C–Cl bonds, the major part of the activation barrier in bromination is determined by C–H bond scission with the Br˙ radical (Fig. 8a).29 This leads to a higher reaction temperature of methane bromination as compared to chlorination, at which the concentration of Br˙ radicals is high enough to initiate the reaction, so that the promoting effect of the surface on the generation of these species becomes practically irrelevant. As the barrier for CH4 activation is not substantially affected by the solid, the catalytic and non-catalytic reactions display a comparable rate.
Another peculiar aspect of the catalytic halogenation relates to the formation of COx, which was not discussed in previous studies in this field. As shown by the experiments in which CH2X2 was taken as a model halogenated product and fed over the representative catalysts and carriers under conditions comparable to those applied in methane halogenation (Fig. 7b and c), the formation of COx primarily originates from the interaction of the halomethanes with the active oxygen sites of supports. In particular, the rate of carbon oxide evolution was high over S-ZrO2 for both CH2X2 and CH2Br2 and was generally higher for CH2Br2, consistent with the selectivity patterns observed in methane halogenations (Fig. 4 and 5).
In general, our results provide strong hints that radical-pathways play the major role in methane halogenation over solid surfaces. In this regard, they are coherent with our previous studies on methane oxyhalogenation, which showed that the catalysts primarily act as the source of reactive halogen radicals that activate methane in the gas phase.28,56
Conclusions
The evaluation of a large number of materials comprising supports, noble metals, halides, oxyhalides, and oxides supported on different carriers, sulfated oxides and zeolites in methane halogenations revealed their positive impact on the chlorination activity, but limited effect on the bromination rate as compared to the respective non-catalysed reactions. Particularly high conversion levels in methane chlorination were attained over the beds of ZSM-5 and BETA zeolites, displaying ≥5 times higher productivity than the void reactor of equivalent volume. Still, the fraction of CH3X over most of the studied catalysts was similar to that achieved in the void reactor or over the bare carriers and was generally higher in methane bromination than in methane chlorination. The major deviations from these trends were displayed by Pt/carbon and zeolite catalysts in methane chlorination, and BETA and ZSM-5 in bromination, which favoured polyhalogenation. Nonetheless, the formation of halomethanes over alumina carriers, alumina-supported catalysts, sulfated oxides, Pt/carbon and most of the zeolites in methane chlorination and virtually all catalytic systems in methane bromination was negatively affected by the evolution of COx. In addition, higher operating temperatures led to coke deposition over multiple catalysts in methane bromination as well as H-SAPO-34 in methane chlorination, while the carbon support displayed substantial halogen uptake in both reactions. These side processes stemming from the interaction of halomethanes and halogens with catalysts were typically not considered in previous studies, which in turn could lead to false conclusions on the catalytic performances. The variations of the Si:Al ratio and counter cation as well as the formation of the extraframework aluminium had almost no effect on the activity and selectivity patterns in methane chlorination over the most active ZSM-5 zeolite, which along with significant kinetic similarity between this material and SiO2 as well as the pronounced O2 inhibition effect indicates that the reaction primarily proceeds via a non-polar radical-chain mechanism. The promotion of activity is likely caused by the confinement of the radical intermediates and/or transition states, which is strongly supported by the volcano-like dependence of the chlorination activity on the size of zeolite micropores. Thereby, the maximum is achieved for the ZSM-5 channels, the diameter of which corresponds to the sum of kinetic diameters of methane and chlorine radicals. Overall, our results suggest that the radical mechanism prevails in the halogenation over a broad range of catalysts, limiting thus the scope for improving the selectivity to CH3X. Nonetheless, marked enhancement of the chlorination activity over ZSM-5 and BETA catalysts along with an increased fraction of CH2Cl2 and CHCl3 might exhibit interesting potential for advancements in the production of these important chemicals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Ms. S. Kaiser, Mr. G. Manzocchi and Dr. R. Lin. for their help in the preparation of Pt/carbon and H-SnBETA-220 catalysts.References
- R. Lin, A. P. Amrute and J. Pérez-Ramírez, Chem. Rev., 2017, 117, 4182–4247 CrossRef CAS PubMed.
- M. Rossberg, W. Lendle, G. Pfleiderer, A. Tögel, T. R. Torkelson and K. K. Beutel, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, vol. 9, pp. 15–42 Search PubMed.
- R. N. Pease and G. F. Walz, J. Chem. Soc., 1931, 53, 382 CrossRef CAS.
- S. Yuster and L. H. Reyerson, J. Phys. Chem., 1934, 39, 859–876 CrossRef.
- C. F. Reed and C. L. Horn, US Pat., 2046090, 1936 Search PubMed.
- H. B. Hass, E. T. McBee and P. Weber, Ind. Eng. Chem., 1936, 28, 333–340 CrossRef CAS.
- H. B. Hass, E. T. McBee and L. F. Hatch, Ind. Eng. Chem., 1937, 29, 1335–1338 CrossRef CAS.
- W. E. Vaughan and F. F. Rust, J. Org. Chem., 1940, 7, 449–471 CrossRef.
- E. T. McBee, H. B. Hass and J. A. Pianfetti, Ind. Eng. Chem., 1941, 33, 185–188 CrossRef CAS.
- G. B. Kistiakowsky and E. R. Van Artsdalen, J. Chem. Phys., 1944, 12, 469–478 CrossRef CAS.
- A. S. Bratolyubov, Russ. Chem. Rev., 1961, 30, 602–612 CrossRef.
- E. T. McBee, H. B. Hass, C. M. Neher and H. Strickland, Ind. Eng. Chem., 1942, 34, 296–300 CrossRef CAS.
- I. Bucsi and G. A. Olah, Catal. Lett., 1992, 16, 27–38 CrossRef CAS.
- R. H. McKee, US Pat., 1688726, 1928 Search PubMed.
- G. A. Olah, B. Gupta, M. Farina, J. D. Felberg, W. M. Ip, A. Husain, R. Karpeles, K. Lammertsma, A. K. Melhotra and N. J. Trivedi, J. Am. Chem. Soc., 1985, 107, 7097–7105 CrossRef CAS.
- S. Svelle, S. Aravinthan, M. Bjrøgen, K.-P. Lillerud, S. Kolboe, I. M. Dahl and U. Olsbye, J. Catal., 2006, 241, 243–254 CrossRef CAS.
- K. X. Wang, H. F. Xu, W. S. Li, C. T. Au and X. Zhou, Appl. Catal., A, 2006, 304, 168–177 CrossRef CAS.
- Z. Liu, Z. Zhang, W. Xing, S. Komarneni, Z. Yan, X. Gao and X. Zhou, Nanoscale Res. Lett., 2014, 9, 1–12 CrossRef PubMed.
- M. Ibáñez, M. Gamero, J. Ruiz-Martínez, B. M. Weckhuysen, A. T. Aguayo, J. Bilbao and P. Castaño, Catal. Sci. Technol., 2016, 6, 296–306 RSC.
- E. McFarland, Science, 2012, 338, 340–342 CrossRef CAS PubMed.
- X.-P. Zhou, I. M. Lorkovic, G. D. Stucky, P. C. Ford, J. H. Sherman and P. Grosso, US Pat., 6464243, 2002 Search PubMed.
- D. C. Upham, Z. R. Snodgrass, M. T. Zavareh, T. B. McConnaughy, M. J. Gordon, H. Metiu and E. W. McFarland, Chem. Eng. Sci., 2017, 160, 245–253 CrossRef CAS.
- J.-S. Park, C. Chen, N. L. Wieder, J. M. Vohs and R. J. Gorte, Electrochim. Acta, 2011, 56, 1581–1584 CrossRef CAS.
- J. Pérez-Ramírez, C. Mondelli, T. Schmidt, O. F.-K. Schlüter, A. Wolf, L. Mleczko and T. Dreier, Energy Environ. Sci., 2011, 4, 4786–4799 RSC.
- M. Moser, I. Czekaj, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2014, 53, 8628–8633 CrossRef CAS PubMed.
- V. Paunović, R. Lin, M. Scharfe, A. P. Amrute, S. Mitchell, R. Hauert and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 9791–9795 CrossRef PubMed.
- S. G. Podkolzin, E. E. Stangland, M. E. Jones, E. Peringer and J. A. Lercher, J. Am. Chem. Soc., 2007, 129, 2569–2576 CrossRef CAS PubMed.
- V. Paunović, G. Zichittella, A. Bodi, P. Hemberger and J. Pérez-Ramírez, ACS Catal., 2019, 9, 1710–1725 CrossRef.
- I. M. Lorkovic, S. Sun, S. Gadewar, A. Breed, G. S. Macala, A. Sardar, S. E. Cross, J. H. Sherman, G. D. Stucky and P. C. Ford, J. Phys. Chem. A, 2006, 110, 8695–8700 CrossRef CAS PubMed.
- H. L.-H. Fong and R. D. Swain, US Pat., 2010/0234637A1, 2010 Search PubMed.
- K. Ding, A. R. Derk, A. Zhang, Z. Hu, P. Stoimenov, G. D. Stucky, H. Metiu and E. W. McFarland, ACS Catal., 2012, 2, 479–486 CrossRef CAS.
- E. W. Kilgren and E. Gorin, US Pat., 2498552, 1950 Search PubMed.
- G. A. Olah, R. Renner, P. Schilling and Y. K. Mo, J. Am. Chem. Soc., 1973, 95, 7686–7692 CrossRef CAS.
- P. Batamack, I. Bucsi, Á. Molnár and G. A. Olah, Catal. Lett., 1994, 25, 11–19 CrossRef CAS.
- V. Degirmenci, A. Yilmaz and D. Uner, Catal. Today, 2009, 142, 30–33 CrossRef CAS.
- H. Joo, D. Kim, K. S. Lim, Y. N. Choi and K. Na, Solid State Sci., 2018, 77, 74–80 CrossRef CAS.
- P. T. D. Batamack, T. Mathew and G. K. S. Prakash, J. Am. Chem. Soc., 2017, 139, 18078–18083 CrossRef CAS PubMed.
- S. K. Kaiser, R. Lin, S. Mitchell, E. Fako, F. Krumeich, R. Hauert, O. V. Safonova, V. A. Kondratenko, E. V. Kondratenko, S. M. Collins, P. A. Midgley, N. López and J. Pérez-Ramírez, Chem. Sci., 2019, 10, 359–369 RSC.
- M. Milina, S. Mitchell, P. Crivelli, D. Cooke and J. Pérez-Ramírez, Nat. Commun., 2014, 5, 3922 CrossRef.
- R. Van Grieken, J. L. Sotelo, J. M. Menéndez and J. A. Melero, Microporous Mesoporous Mater., 2000, 39, 135–147 CrossRef CAS.
- G. M. Lari, P. Y. Dapsens, D. Scholz, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2016, 18, 1249–1260 RSC.
- F. Kapteijn and J. A. Moulijn, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2008, ch. 9, pp. 2019–2045 Search PubMed.
- B. Kraushaar-Czarnetzki and S. P. Müller, in Synthesis of Solid Catalysts, ed. K. P. De Jong, Wiley-VCH, Weinheim, 2009, ch. 9, pp. 173–199 Search PubMed.
- R. W. van den Brink, P. Mulder, R. Louw, G. Sinquin, C. Petit and J.-P. Hindermann, J. Catal., 1998, 180, 153–160 CrossRef CAS.
- P. Payra and P. K. Dutta, in Handbook of Zeolite Science and Technology, Dekker, New York, 2003, ch. 1, pp. 1–24 Search PubMed.
- S. J. Kirkby and H. Frei, J. Phys. Chem. B, 1998, 102, 7106–7111 CrossRef CAS.
- L. R. Aramburo, S. Teketel, S. Svelle, S. R. Bare, B. Arstad, H. W. Zandbergen, U. Olsbye, F. M. F. de Groot and B. M. Weckhuysen, J. Catal., 2013, 307, 185–193 CrossRef CAS.
- J. Lv, Z. Hua, J. Zhou, Z. Liu, H. Guo and J. Shi, ChemCatChem, 2018, 10, 2278–2284 CrossRef CAS.
- B. L. Farrell, V. O. Igenegbai and S. Linic, ACS Catal., 2016, 6, 4340–4346 CrossRef CAS.
- N. N. Semenov, in Some Problems of Chemical Kinetics and Reactivity, Pergamon Press, London, 1958, ch. 6, pp. 183–208 Search PubMed.
- Y. J. Xu, J. Q. Li, Y. F. Zhang and W. K. Chen, J. Chem. Phys., 2004, 120, 8753–8760 CrossRef PubMed.
- H. L. Chen, S. P. Ju, H. T. Chen, D. G. Musaev and M. C. Lin, J. Phys. Chem. C, 2008, 112, 12342–12348 CrossRef CAS.
- A. J. Jones, S. I. Zones and E. Iglesia, J. Phys. Chem. C, 2014, 118, 17787–17800 CrossRef CAS.
- C. Baerlocher, W. M. Meier and D. H. Olson, Atlas of Zeolite Framework Types, Elsevier, Amsterdam, 2001 Search PubMed.
- R. W. Broach, in Zeolites in Industrial Separation and Catalysis, ed. S. Kulprathipanja, Wiley-VCH, Weinheim, 2010, ch. 2, pp. 27–59 Search PubMed.
- V. Paunović, P. Hemberger, A. Bodi, N. López and J. Pérez-Ramírez, Nat. Catal., 2018, 1, 363–370 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |