
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic insights into a NOx storage-reduction (NSR) catalyst by spatiotemporal operando X-ray absorption spectroscopy†
Yasutaka
Nagai
 *a,
Akihiko
Kato
a,
Masaoki
Iwasaki
a and
Keisuke
Kishita
b
*a,
Akihiko
Kato
a,
Masaoki
Iwasaki
a and
Keisuke
Kishita
b
aToyota Central R&D Labs., Inc., Nagakute, Aichi 480-1192, Japan. E-mail: e1062@mosk.tytlabs.co.jp
bToyota Motor Corporation, Toyota, Aichi 471-8572, Japan
First published on 14th February 2019
Abstract
Spatially resolved monitoring of the catalytically active state and online catalytic activity measurements were applied to track the fast transient phenomena occurring along the axial direction of a NSR catalyst bed. This methodology clarified the distribution of stored NOx as well as the cause of NOx spike emission.
Lean-burn combustion engines, using fuels such as gasoline and diesel, provide greater fuel economy compared to conventional stoichiometric gasoline engines. However, the three-way catalysts used for conventional gasoline engines cannot purify NOx in the oxygen-rich exhaust emitted from lean-burn engines. Therefore, new catalytic approaches are needed to treat the NOx emissions from diesel and gasoline lean-burn engines.1 This situation has encouraged the development of new catalyst technology that can reduce NOx in excess oxygen, i.e., a NOx storage-reduction (NSR) catalyst for gasoline lean-burn engines,2 also known as a lean NOx trap (LNT). The NSR catalyst has been used practically in Japan in 1994.3 However, vehicle emission standards have been gradually strengthened on a global scale, leading to a demand for further improvements in NSR performance. Therefore, studies of lean NOx catalysts remain a challenging research task.4,5
Basically, NSR catalysts are precious group metals (PGMs; Pt, Rh and Pd) together with supporting oxides such as Al2O3 and barium oxide/carbonate as a NOx storage material, that can promote catalytic oxidation and reduction processes (Fig. S1 in the ESI†). During the fuel-lean phase (oxygen excess), NOx is oxidized over the precious metal, and then stored as barium nitrate by reaction with the barium storage component. However, during the short fuel-rich phase (reductive atmosphere) of catalyst regeneration, the stored NOx is released and subsequently reduced to N2 over the precious metal. Depending on the driving conditions, a burst of NOx emission, often called an NOx spike or NOx puff, can be observed at the outlet of the catalyst bed during a rich period, because a portion of the stored NOx is not reduced to N2, leading to a NOx slip at the outlet. Several studies have been conducted on the NOx reduction mechanism, and significant progress has been made.6–14 However, the nature of NOx spike generation during the fast transient reaction remains very complicated, and requires further investigation.
To elucidate the NOx spike generation mechanism, understanding the spatially resolved reactions occurring in the catalyst bed is necessary, because chemical species and catalytic active states have drastic gradients along the axial direction of the catalyst bed. Several research groups have conducted spatially resolved analysis of the NSR process using infrared (IR)/Raman spectroscopy that provides surface and bulk information about the Ba species,6–9 spatially resolved capillary-inlet mass spectrometry (SpaciMS) to monitor the composition of the gas phase,10–13 and IR thermography to obtain temperature measurements.14 Spatial analysis of the PGM active site is vitally important to understand the NSR process in the entire catalyst bed from front to rear. However, no studies have ever been conducted to track the PGM active state in the axial direction of the NSR catalyst. This is because the oxidation–reduction rate of the PGM, which is related to the catalytically active state, is very fast under the lean/rich perturbations at temperatures of about 200 °C and above,15,16 therefore, fast operando analysis methodology with millisecond temporal resolution is required to track the PGM active state.
Therefore, a spatiotemporal operando X-ray absorption spectroscopy (XAS) method was applied to obtain spatially resolved monitoring of PGM in the axial direction as well as for the measurement of online outlet gas components to clarify the NOx storage and reduction processes, especially the NOx spike mechanism. A Toyota beamline (BL33XU) of SPring-8, combining a servo-motor-driven Si channel-cut monochromator with a tapered undulator, allowed rapid acquisition of high-quality data for quick scan XAS (QXAS),17 along with an operando setup simulating automotive engine exhaust (Fig. S2a†). This study presents the importance of operando spatiotemporally resolved QXAS analysis for heterogeneous catalytic processes, and reveals the nature of the NOx storage and reduction process.
In this study, a 0.5 wt% Rh/BaO (10 wt% as Ba)/γ-Al2O3 catalyst was prepared. A capillary tube with a pellet catalyst sample (66 mg, sieve fraction 75–150 μm, ca. 8 mm in catalyst bed length) was set in a specially designed operando cell (Fig. 1a and b). The NSR reaction was operated at 450 °C, and a lean stream consisting of 0.07% NO, 7% O2, and He balance was introduced into the cell for 300 or 60 s, followed by a rich stream of 3% H2 and He balance for 240 s. The gas flowing over the sample was quickly changed from a lean to a rich atmosphere using a gas-actuated switching valve (Fig. S2b†). Measurement of inlet/outlet NO concentration confirmed that the amount of stored NOx during the lean 60 s supply corresponded to 0.050 mmol per g of catalyst (33% of the saturated NOx storage amount) and that during the lean 300 s supply, the amount corresponded to 0.151 mmol per g of catalyst (100% NOx storage amount). The experiments of 300 s and 60 s of lean supply are referred to as 100% and 33% NOx storage, respectively. Under the NSR operation, online mass spectra of the outlet gas species were obtained every 150 ms, along with XAS measurements.
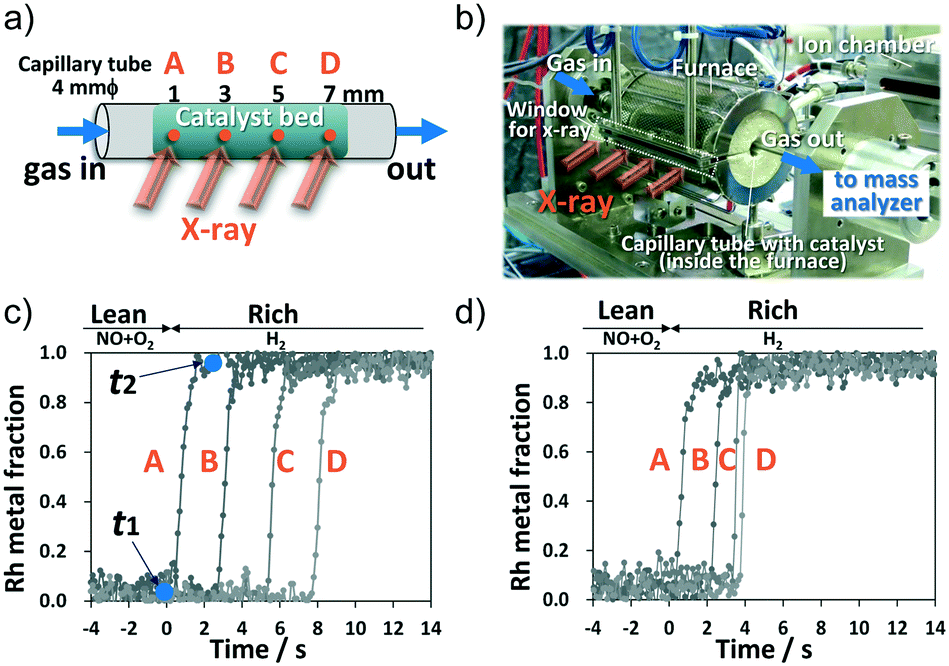 | ||
| Fig. 1 (a) Schematic illustration of the quartz capillary tube with the pellet catalyst. (b) The operando XAS cell for spatially-resolved analysis in the axial direction. Temporal dependence of the Rh metal fraction in the Rh/BaO/Al2O3 catalyst from lean to rich conditions for (c) 100% NOx storage and (d) 33% NOx storage. | ||
To track the catalytically active state, Rh K-edge X-ray absorption near-edge structure (XANES) spectra were collected every 100 ms at four positions: 1 (A), 3 (B), 5 (C), and 7 (D) millimeters from the bed front (Fig. 1a). High-quality data from the Rh K-edge XANES spectra were acquired using QXAS in transmission mode (Fig. S2c†), which enabled tracking of temporal changes in the Rh oxidation state as Rh metal fraction. Fig. 1c and d show the Rh metal fraction at each position (A–D) after switching from lean gas to rich gas at 450 °C. Under the lean conditions, all Rh species in the catalyst bed were Rh oxide (Rh2O3). After switching to the rich condition, for both 100% and 33% NOx storage, reduction in Rh occurred sequentially from the front A position to the rear D position. Thus, Rh at the downstream position (e.g., position B) was not reduced unless the reduction in Rh upstream (e.g., position A) was completed. The metallic Rh state is considered an active state for catalytic NO reduction18–20 because Rh metallic clusters are required for the NO dissociation step. This indicates that NO reduction with H2 over the Rh species occurred sequentially from upstream to downstream.
In Fig. 1c, “t1” and “t2” denote the onset time and the termination time, respectively, of Rh reduction, and the value obtained by subtracting t1 from t2 is defined as the “Rh reduction time” (calculation method in Fig. S3†). Fig. 2a presents the Rh reduction time at each axial position for 100% and 33% NOx storage along with that for 0% NOx storage. The 0% NOx storage conditions were for the experiment conducted under a lean stream of only 7% O2/He (without the NO gas component) for 300 s and then a rich stream of 3% H2/He for 240 s at 450 °C. Temporal dependencies of the Rh metal fraction at each position for 0% NOx storage are shown in Fig. S4.† In Fig. 2a, the NOx reduction time for 33% NOx storage at each axial position varied widely from 1.2 s at A to 0.2 s at D, whereas those of 100% and 0% NOx storage produced constant values (1.1–1.2 s for 100% storage and ∼0.2 s for 0% storage). These results indicate that the Rh reduction time correlates with the amount of stored NOx. As shown in Fig. S5,† the greater the amount of stored NOx, the longer the Rh reduction time because the stored NOx prolongs the reduction of Rh oxide. Thus, the Rh reduction time during the rich period was proportional to the amount of NOx stored in the Ba compounds in the lean period. Results shown in Fig. 2a clearly indicate that NOx is fully stored along the entire length of the catalyst bed after 300 s of lean supply (100% NOx storage), whereas for 33% NOx storage, NOx is stored only upstream at the A and B positions.
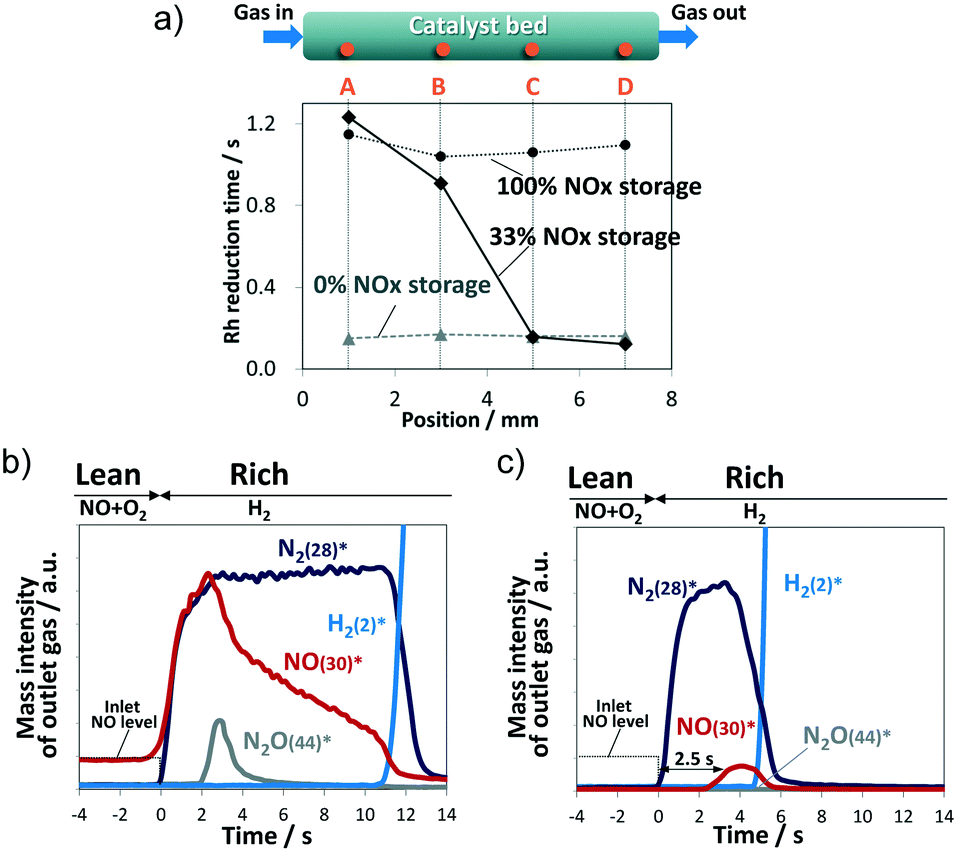 | ||
| Fig. 2 (a) Rh reduction time at each axial position. Evolution profiles of the outlet gases for (b) 100% NOx storage and (c) 33% NOx storage, *m/z is given in the parentheses. | ||
Fig. 2b shows the evolution profiles of outlet NO, N2, N2O, and H2 gases when the gas flow was switched from lean to rich conditions at 100% NOx storage. NH3 production was not observed within the measurement time in Fig. 2b. After switching to the rich period of H2/He flow, a spike in NO emission was observed along with N2 generation from the reaction of H2 with the stored NOx, indicating that the stored NOx decomposed during the rich period to gas phase NOx and was subsequently released to the outlet of the catalyst bed without being reduced to N2. Such NO emission lasted for about 11 s until unreacted H2 was evolved.
In addition to N2 as a NOx reduction product, a trace amount of N2O was confirmed at around 3 s. Kubiak et al. investigated mechanistic aspects in the formation of N2O over Pt/BaO/Al2O3 and Rh/BaO/Al2O3 by transient microreactor experiments and operando IR spectroscopy.21 They concluded that N2O formation involves the coupling of gaseous NO molecules with N-adspecies formed upon NO dissociation onto PGM sites. As for 100% NOx storage in our experiment, it is reasonable to consider that the concentration of gas phase NO became the highest at around 3 s, and trace N2O was generated accordingly.
The gas atmosphere along the axial direction inside the catalyst bed during the rich period was investigated. Since the reduction reaction rate of stored NOx is sufficiently faster than the gas flow velocity, NO reduction at the Rh metal sites proceeded sequentially from the upstream position with complete consumption of H2. Actually, at the elapsed time of 1 s (Fig. 1c), the Rh species at position A is reduced, but the Rh species in the region from position B to D remain as Rh oxide. Considering that the time for gas to flow through the catalyst bed was 60 ms for this experiment, the downstream region from B to D, after H2 was completely consumed at the upstream of position A, is basically under an inert gas atmosphere (He plus N2). A schematic for this process is shown in Fig. S6.†
Luo et al.11 and Choi et al.13 reported that the stored NOx during a lean period is released to the outlet of the catalyst bed as gaseous NOx during an inert gas purge at temperatures of 400 °C and above. In addition, Luo et al.11 reported that an oxidizing atmosphere (oxygen excess) stabilizes barium nitrates, whereas they become less stable under an inert gas atmosphere, resulting in the formation of a NOx puff. Urakawa et al.6 investigated the dynamic surface and bulk processes of Ba components during NSR reactions using spatiotemporal IR/Raman spectroscopy. Their results suggested that the surface Ba nitrite species decomposed during the rich period, releasing NO into the gas phase. According to their investigation, a portion of stored NOx in the region from B to D in this study became unstable under the inert gas atmosphere and was released to the outlet as a NOx spike emission.
All of these results led to the following conclusions about the NSR process for 100% NOx storage (Fig. 3a):
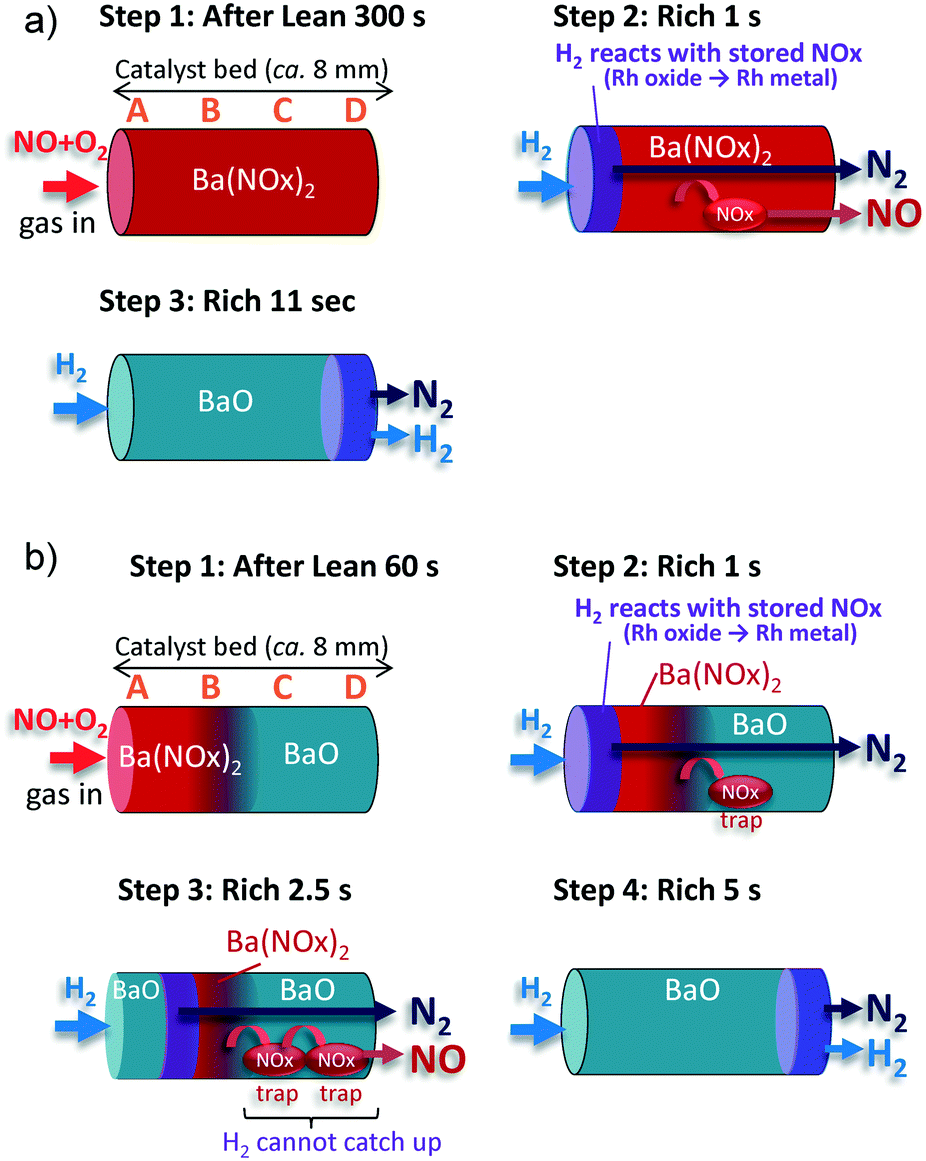 | ||
| Fig. 3 Schematic illustration of NOx storage and reduction behavior for (a) 100% NOx storage and (b) 33% NOx storage. | ||
Step 1: After the lean period of 300 s, NOx is fully stored along the entire length of the catalyst bed as barium nitrite or barium nitrate [Ba(NOx)2)].
Step 2: During the first second of the rich period, the stored NOx at the front position near A is reduced to N2 by H2 over the metallic Rh species, and N2 is released to the outside of the catalyst bed. At the same time, a portion of the stored NOx at the back position of B–D (e.g., the surface nitrites which are weakly adsorbed) is decomposed under the inert gas, and released into the gas phase. Therefore, NO spike emission and N2 generation begin to occur almost simultaneously. Besides, a trace amount of N2O is generated in the presence of gas phase NO at around 3 s.
Step 3: Step 2 continues for ∼11 s until H2 reaches the end of the catalyst bed. At this point, all of the stored NOx is reduced by H2, which is consistent with the termination of Rh reduction at position D (Fig. 1c).
Next, the 33% NOx storage case was examined by obtaining outlet gas profiles (Fig. 2c). The amount of N2 generation from 33% NOx storage (0.024 mmol g−1) was less than that for the 100% NOx storage case (0.059 mmol g−1), by a value that corresponds to the difference between the two NOx storage amounts (0.050 and 0.151 mmol g−1). H2 emission was observed at approximately 5 s into the rich period, which is consistent with the time when reduction of Rh species at position D was completed (Fig. 1d). Note the delay of 2.5 s for NO emission after N2 generation in the 33% NOx storage case. A similar delay was not observed for 100% NOx storage.
The delay of the NO spike was investigated (Fig. S7†). According to the Rh reduction behavior in Fig. 1d during the first second of the rich period, a H2 atmosphere exists upstream of position A; in contrast, the atmosphere downstream from B to D contains an inert gas. The NOx stored near position B under the inert gas is decomposed to the gas phase, resulting in desorbed NOx flowing downstream relatively slowly because the gas phase NOx is weakly trapped in the downstream C and D regions at vacant sites on the BaO surface. This slow travel via repetition of desorption/re-trapping results in a 2.5 s delay based on the onset time of N2 emission.
Incidentally, the ratio of the NOx spike emission amount to the amount of stored NOx for 33% NOx storage, calculated from the evolution profiles of the outlet gases (Fig. 2c), was 4%, which is significantly less than the 21% found for 100% NOx storage (Fig. 2b). This difference is interpreted as follows. Since no re-trap sites are available downstream in the 100% NOx storage case, the decomposed NOx exits unreduced from the catalyst bed. In contrast, for the 33% NOx storage case, some of the downstream re-trapped NOx species are reduced to N2 at the Rh sites by H2 coming from upstream, and the travelling NOx species that the H2 does not reach are released to the outlet.
Thus, the NSR process for 33% NOx storage is interpreted as follows (Fig. 3b):
Step 1: After the 60 s lean period, NOx is stored as Ba(NOx)2 upstream of the A and B positions. Downstream, from C to D, vacant sites (BaO) exist for NOx trapping.
Step 2: During the first second of the rich period, the NOx stored at the front position near A is reduced to N2 by H2 over the metallic Rh species, and released to the outside. At the same time, a portion of the NOx stored toward the back of B is decomposed under the inert gas and released into the gas phase. The decomposed NOx species are re-trapped downstream in the C and D regions.
Step 3: At 2.5 s into the rich period, the reduction front reaches position B. The decomposed NOx species travel farther downstream, repeating the desorption/re-trap process on the vacant sites. The traveling NOx is released to the outlet as an NO spike unless H2 gas reaches the traveling NOx.
Step 4: Step 3 continues until the H2 reaches the D position, and then H2 emission is observed at approximately 5 s into the rich period.
Conclusions
A spatiotemporal operando XAS technique was used to investigate the dynamic behavior of the NOx storage and reduction process of a Rh/BaO/Al2O3 catalyst. The Rh K-edges XAS in the axial direction of the catalyst bed were monitored every 100 ms, which was combined with online mass spectrometry. Using this methodology, the fast transient phenomena occurring inside the NSR catalyst were tracked and the NOx storage distribution during the lean period was clarified, along with the cause of the NOx spike emission.In this study, a simplified model gas containing NO + O2 and H2 was used, in order to gain mechanistic insights into the principal NSR process. However, an actual engine exhaust includes H2O, CO2, CO and so on. Using the spatiotemporal operando XAS methodology, we plan to investigate the effect of coexisting gases and reductants on the catalytic active site. Furthermore, combining SpaciMS with the current XAS set-up can give a deeper understanding of the overall mechanism occurring in the NSR convertor. We hope to establish an ultimate spatiotemporal operando system in the near future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Kazuhiko Dohmae, Toshitaka Tanabe, and Hideki Takagi (Toyota Central R&D) for their helpful discussions and excellent support.Notes and references
- A. Fritz and V. Pitchon, Appl. Catal., B, 1997, 13, 1 CrossRef CAS.
- N. Takahashi, H. Shinjoh, T. Iijima, T. Suzuki, K. Yamazaki, K. Yokota, H. Suzuki, N. Miyoshi, S. Matsumoto, T. Tanizawa, T. Tanaka, S. Tateishi and K. Kasahara, Catal. Today, 1996, 27, 63 CrossRef CAS.
- S. Matsumoto, Catal. Today, 1996, 29, 43 CrossRef CAS.
- T. C. Waltling, P. D. Bolton and D. Swallow, Chem. Eng. Sci., 2018, 178, 312 CrossRef.
- X. Mei, Q. Yan, P. Lu, J. Wang, Y. Cui, Y. Nie, A. Umar and Q. Wang, Sci. Rep., 2017, 7, 42862 CrossRef CAS PubMed.
- A. Urakawa, N. Maeda and A. Baiker, Angew. Chem., Int. Ed., 2008, 47, 9256 CrossRef CAS PubMed.
- N. Maeda, A. Urakawa and A. Baiker, Top. Catal., 2009, 52, 1746 CrossRef CAS.
- N. Maeda, A. Urakawa and A. Baiker, J. Phys. Chem. C, 2009, 113, 16724 CrossRef CAS.
- N. Maeda, A. Urakawa, R. Sharma and A. Baiker, Appl. Catal., B, 2011, 103, 154 CrossRef CAS.
- J. Choi, W. P. Partridge and C. S. Daw, Appl. Catal., A, 2005, 293, 24 CrossRef CAS.
- J. Y. Luo, M. Alharbi, M. Pang and W. S. Epling, Appl. Catal., B, 2011, 106, 664 CrossRef CAS.
- V. Easterling, Y. Ji, M. Crocker, M. Dearth and R. W. McCabe, Appl. Catal., B, 2012, 123–124, 339 CrossRef CAS.
- J. S. Choi, W. P. Partridge, J. A. Pihl, M. Y. Kim, P. Kočí and C. S. Daw, Catal. Today, 2012, 184, 20 CrossRef CAS.
- K. Aftab, J. Mandur, H. Budman, N. W. Currier, A. Yezerets and W. S. Epling, Catal. Lett., 2008, 125, 229 CrossRef CAS.
- K. Dohmae, Y. Nagai, T. Tanabe, A. Suzuki, Y. Inada and M. Nomura, Surf. Interface Anal., 2008, 40, 1751 CrossRef CAS.
- J. D. Grunwaldt, M. Beier, B. Kimmerle, A. Baiker, M. Nachtegaal, B. Grisesebock, D. Lutzenkrichen-Hecht, J. Stotzel and R. Frahm, Phys. Chem. Chem. Phys., 2009, 11, 8799 RSC.
- T. Nonaka, K. Dohmae, T. Araki, Y. Hayashi, Y. Hirose, T. Uruga, H. Yamazaki, T. Mochizuki, H. Tanida and S. Goto, Rev. Sci. Instrum., 2012, 83, 083112 CrossRef CAS PubMed.
- J. Evans and M. Tromp, J. Phys.: Condens. Matter, 2008, 20, 184020 CrossRef.
- A. J. Dent, J. Evans, S. G. Fiddy, B. Jyoti, M. A. Newton and M. Tromp, Faraday Discuss., 2008, 138, 287 RSC.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukada, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176 CrossRef CAS PubMed.
- L. Kubiak, R. Matarrese, L. Castoldi, L. Lietti, M. Daturi and P. Forzatti, Catalysts, 2016, 6, 36 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cy00176j |
| This journal is © The Royal Society of Chemistry 2019 |