
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimising hydrogen production via solar acetic acid photoreforming on Cu/TiO2†
Mikel
Imizcoz
 and
Alberto V.
Puga
*
and
Alberto V.
Puga
*
Instituto de Tecnología Química, Universitat Politècnica de València-Consejo Superior de Investigaciones Científicas, Avenida de los Naranjos, s/n, 46022 Valencia, Spain. E-mail: apuga@itq.upv.es
First published on 20th February 2019
Abstract
Valorisation of aqueous acetic acid—a model bio-oil component—by solar photocatalysis on Cu/TiO2 is efficient, yet decarboxylation tends to dominate over photoreforming due to copper co-catalyst oxidation. Selectivities to hydrogen were greatly enhanced by in situ photoreduction, ensuring the removal of passivation layers to unmask active Cu(0) surfaces, in a reversible fashion.
The utilisation of biomass for the production of hydrogen represents a key alternative to current commercial processes, relying chiefly on fossil carbon resources, i.e. natural gas and crude oil.1 A range of biomass-derived oxygenates (e.g. ethanol or glycerol) can be employed as convenient and readily transportable hydrogen carriers through reforming processes.2 Moreover, complete reforming results in the generation of H2/CO2 mixtures, which might be used as precursors for the subsequent production of liquid fuels.3 Thermocatalytic reforming of oxygenates is energy intensive, often requiring high temperatures which lead to degradation or side reactions.2,4 The photocatalytic alternative—photoreforming—proceeds under light irradiation at ambient temperatures, thus minimising degradation events.5
Carboxylic acids are relevant components of processed biomass streams (especially pyrolysis bio-oils), yet their valorisation is challenging.6 The most prominent example, acetic acid, has been frequently employed as a model bio-oil substrate for reforming, which takes place at elevated temperatures with unavoidable deactivation, chiefly due to coke deposition.3,7 For these reasons, the photocatalytic production of hydrogen from aqueous acetic acid represents an interesting option as a cleaner technology in this context.5 This process is however not devoid of selectivity issues, owing to the tendency of acetic acid to undergo decarboxylation into CH4 and CO2 (eqn (1)), which is a clearly more thermodynamically favourable reaction as compared to reforming (eqn (2)).
| CH3COOH(l) → CH4(g) + CO2(g) ΔG0 = −55.3 kJ mol−1 | (1) |
| CH3COOH(l) + 2H2O(l) → 4H2(g) + 2CO2(g) ΔG0 = 75.4 kJ mol−1 | (2) |
Photocatalytic decarboxylation of acetic acid occurs rapidly and preferentially over photoreforming on aqueous Pt/TiO2 suspensions under UV-vis irradiation.8–10 Selectivity to H2 is enhanced in increasingly basic media, although this is accompanied by decreases in overall activity.9 Higher H2/CH4 ratios can be also achieved by decreasing the initial acetic acid concentrations, yet at the expense of lower productivities.10 Complete photoreforming of acetic acid on Pt/TiO2 was also favoured under long-term inert gas flow operation.11
The prevalent use of platinum as the co-catalyst for acetic acid photoreforming is based on its known activity for photocatalytic hydrogen evolution.12 A recent comparative study on a range of metals (Pt, Rh, Au, Ag, Ir and Ru) demonstrated that indeed platinum outperformed the other contenders in terms of both activity and selectivity.10 Notwithstanding, it would prove certainly appealing to utilise more abundant and less costly metals to accomplish this task in the context of a sustainable energy scenario.13,14 Iron15 or copper16 have been studied as co-catalysts for the photoreforming of acetic acid under UV-rich irradiation, the latter resulting in superior activities and selectivities. Copper co-catalysts have been in fact used for H2 evolution in other photoreforming processes employing alcohols or polyols, and the dynamic evolution of its oxidation state under operation found to be a crucial aspect affecting performance.17–19 It is thus interesting to focus on copper as a promising inexpensive metal co-catalyst for the photoreforming of carboxylic acids. In this work, we have delved into the capability and mode of action of such Cu co-catalysts to optimise H2 production.
A preliminary screening comparing solar acetic acid photoreforming activity and selectivity on M(1%)/TiO2 was performed (M = none, Pt, Au, Ag or Cu; 1% metal vs. TiO2; Fig. 1, and Table S3† for a complete set of data on product yields). The materials were prepared by a photodeposition method (see ESI†), whereby co-catalyst particles are deposited in their metallic states. Materials based on Au or Ag produced low amounts of H2 and slightly higher amounts of CH4 (and CO2), showing little or no difference as compared to bare TiO2. Platinum induced higher activity, and whilst decarboxylation prevailed, the production of H2 was noticeable (33.5 μmol, Fig. 1), in line with previous reports (see Table S4† for a comparison with literature data).9,10 The use of copper was somewhat surprising since it resulted in greatly enhanced overall activities as compared to its group 10 noble counterparts (Au and Ag). Production of CH4 was even slightly higher on Cu(1%)/TiO2 than on the platinum analogue, albeit the amount of H2 produced was immeasurable. The performance of copper as a co-catalyst thus appeared to be remarkable in terms of activity, although its potential use for the photocatalytic production of H2 still remained a challenge.
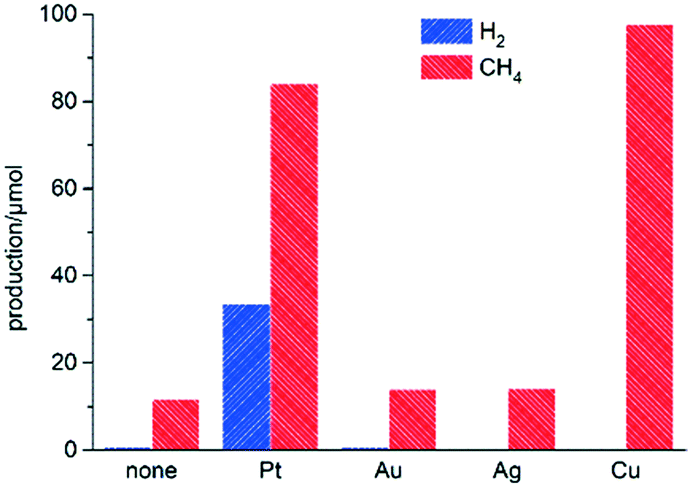 | ||
Fig. 1 Amounts of H2 (blue bars) and CH4 (red bars) formed upon simulated sunlight irradiation of aqueous acetic acid using M(1%)/TiO2 (M = none, Pt, Au, Ag or Cu; 1% w/w metal vs. TiO2) photocatalysts. Reaction conditions: CH3COOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 25 mL), suspended M/TiO2 (25 mg), simulated sunlight irradiation (AM1.5G, 100 mW cm−2), t = 2 h. :1 v/v, 25 mL), suspended M/TiO2 (25 mg), simulated sunlight irradiation (AM1.5G, 100 mW cm−2), t = 2 h. | ||
At that stage, it was hypothesised that the reason for the negligible H2 evolution on Cu(1%)/TiO2 might be surface oxidation (passivation) of the initially metallic copper particles under ambient air and/or in the reaction medium, and the consequent absence of presumably active Cu(0) surfaces. This reasoning was inspired by several investigations reporting dynamic evolution of the oxidation state of copper during alcohol photoreforming.14,17–21 In order to explore the possible relationship of both oxidation state and morphology with performance, we checked the speciation of copper in Cu(1%)/TiO2 by various characterisation techniques. No information could be drawn from X-ray diffraction (XRD, Fig. S1†), presumably due to low loading and small nanoparticle sizes. High-resolution transmission electron microscopy (HRTEM) did not clearly show the morphology of copper domains, and scanning transmission electron microscopy (STEM) barely allowed the observation of Cu nanoparticles (<6 nm, Fig. S2†). Diffuse reflectance UV-vis spectroscopy (DRUV) of Cu(1%)/TiO2 revealed a weak broad signal (>550 nm, and extending beyond 800 nm, Fig. S3†) indicative of Cu(II) species,19,21 in support of the existence of oxidised layers suggested above. Since no evidence for Cu(0) species were found, it could be inferred that the small Cu particles in Cu(1%)/TiO2 might have become totally oxidised to CuO.
Assuming that photo-generated electrons are transferred to Cu nanoparticles, eventually reducing the aforementioned oxidised domains, and that the resulting clean metallic surfaces would provide active sites for H2 evolution, selectivity was monitored for longer periods of irradiation on Cu(1%)/TiO2 (Fig. 2). Indeed, negligible amounts of H2 were detected for the first 5 h, whereas after that point hydrogen evolution became noticeable; in parallel, methane production declined after the same irradiation time. This indicated that active sites for active H2 formation (photoreforming, eqn (2)) were formed upon irradiation after an induction period, whereas decarboxylation sites disappeared concomitantly.
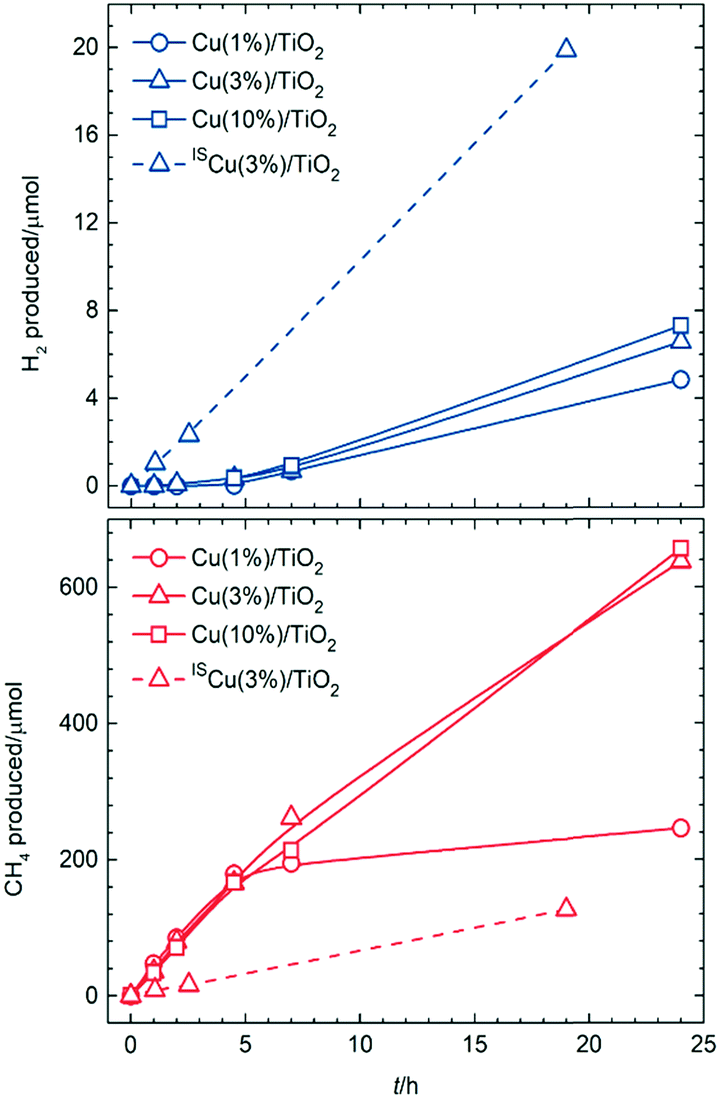 | ||
| Fig. 2 Amounts of H2 and CH4 formed upon simulated sunlight irradiation of aqueous acetic acid on conventional (Cu(X%)/TiO2) and in situ (ISCu(X%)/TiO2) photocatalysts as a function of increasing irradiation times. Reaction conditions as in Fig. 1. | ||
Similar induction periods were observed on analogue photocatalysts having higher amounts of copper (Fig. 2 and Table S5†). Long-term irradiations resulted in enhanced H2 production upon increasing Cu content, yet only marginally. On the other hand, decarboxylation activities were maintained for higher Cu loadings. This was related to the above proposed structure–activity relationship based on the passivation phenomenon taking place for all materials whereby CuO would cover the surface at initial stages, exclusively promoting CH4 (and CO2) formation, and would then become reduced to Cu(0) after sufficient irradiation time, giving rise to H2 evolution sites. The reason for extended decarboxylation at higher Cu loadings was believed to be related to the also higher amount of oxide layers in absolute terms, which would coexist with the nascent Cu(0) surfaces, and would require longer times for total reduction.
In order to ascertain this, HRTEM and STEM characterisation was undertaken for Cu(3%)/TiO2 (Fig. 3a and b). The presence of amorphous CuO particles of around 5 nm and larger were apparent in HRTEM micrographs (Fig. 3a), as opposed to the elusive observation of smaller particles in Cu(1%)/TiO2 (Fig. S2†). A more informative morphological examination by STEM, supported by energy-dispersive X-ray (EDX) spectroscopy, showed that a large number of small Cu nanoparticles (<6 nm), along with some bigger ones (up to 30 nm) were present (Fig. 3b and Fig. S4†). Based on XRD results, Cu(0) was present in this material even after exposure to air (Fig. S1†). A similar situation was found for Cu(10%)/TiO2 (Fig. S5†). Conversely, X-ray photoelectron spectroscopy (XPS) data clearly revealed that the outermost layers of copper particles in Cu(X%)/TiO2 (X = 3 or 10) were exclusively composed by Cu(II) (Fig. S6†). Taking these evidences together, the passivation hypothesis set out above becomes more solidly supported. As shown in Fig. 3, the Cu/TiO2 photocatalysts would consist of copper particles of a range of sizes, depending on co-catalyst loading. The smaller particles are expected to be totally oxidised to CuO in contact with ambient air, whereas larger particles are likely formed by a Cu(0) core surrounded by a passivation layer. Since Cu(0) nanoparticles in Cu(10%)/TiO2 were up to ca. 30 nm in diameter based on XRD signals for the largest, detectable, crystallites (Fig. S1†), as compared to the maximum ca. 40 nm observed by STEM (Fig. S5†), it can be concluded that the surface passivation layers extended to below 10 nm in thickness. This is also consistent with XPS (Fig. S6†), which suggested that Cu(II) layers were at least 5 nm deep.
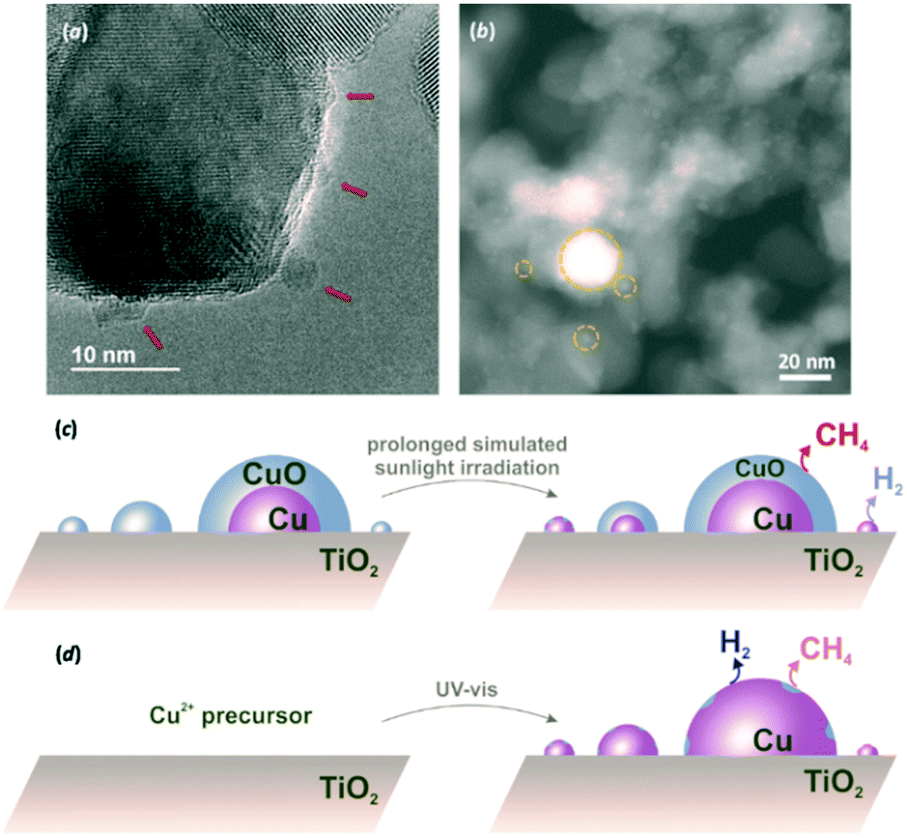 | ||
| Fig. 3 Cu(3%)/TiO2 HRTEM micrographs revealing the presence of amorphous CuO nanoparticles on TiO2 (examples marked by arrows, a), and STEM micrographs allowing the observation of copper domains of variable sizes spanning from the barely nanometric scale to tens of nanometers (examples inside dotted circles, b). Thus, conventional Cu(3%)/TiO2 contains a large number of small CuO nanoparticles along with larger particles formed by a metallic core and an passivation layer, as deduced from XPS and XRD data (c), yet copper reduction under extended solar irradiation progressively transforms the ubiquitous oxidic decarboxylation sites to Cu(0) hydrogen evolution surfaces. The photocatalyst prepared in situ consists of essentially reduced copper domains causing enhanced H2 production, although some minor surface oxidation may take place in the reaction medium (d). | ||
Upon solar irradiation, photogenerated electrons from the conduction band of TiO2 are capable of migrating to CuO nanoparticles and reduce them.14,18,19 For smaller particles, this process can proceed to completion, although the more extensive CuO crusts on larger particles might be more resistant to photoreduction (Fig. 3c). This is why similar induction periods were observed for different copper loadings. The fraction of smaller nanoparticles, present regardless of the copper amount, would take similar lengths of time to become reduced and thus, give rise to active sites for H2 evolution. In line with this, H2 production only improved marginally with increasing Cu loading (Fig. 2a), since larger particles thereby deposited, and contributing to a high proportion of the total copper surfaces (Fig. S4f and S5f†) are more prone to remain passivated. Conversely, oxide layers on such larger particles still resisting total photoreduction would have been responsible for the maintained CH4 production for Cu(3%)/TiO2 and Cu(10%)/TiO2, as opposed to Cu(1%)/TiO2 (see Fig. 2b).
Taking into account the model described in Fig. 3, the strategy envisioned to maximise surface copper reduction, and thus enhance H2 selectivities, was to photodeposit the metal in situ in the reaction suspension under UV-vis irradiation prior to solar photoreforming. Avoiding air contact in this way was expected to prevent passivation. A material prepared in this manner (ISCu(3%)/TiO2, IS: in situ, see ESI†) did not show any induction period for H2 evolution (Fig. 2a, and Table S5†). Also remarkably, lower decarboxylation activity at long irradiation times was observed relative to conventional Cu(3%)/TiO2 (Fig. 2b, and Table S5†). Copper nanoparticles in the ISCu(3%)/TiO2 material would expose reduced surfaces more readily and in consequence favour H2 production (Fig. 3d). Moreover, limited availability of active decarboxylation sites is presumably due to the lesser extent of CuO surface layers, which might still be formed due to slight re-oxidation in the aqueous medium. The morphology and composition of the copper co-catalyst nanoparticles, dominated by Cu(0) surfaces was the reason for the significantly enhanced photoreforming selectivity (the H2/CH4 ratio increased by two orders of magnitude, see Fig. 2 and Table S5†), comparable to UV-irradiated Cu/TiO2 systems or to state-of-the-art Pt/TiO2 materials (Table S4†). The photonic efficiency for ISCu(3%)/TiO2 (10.8% at 350 nm) is comparable to those of similar photocatalytic processes (see Table S6†) yet slightly lower than observed for hydroxyl-containing substrates, as expected due to the higher propensity of the latter to undergo hole-induced oxidation.5
The marked selectivity switch—from decarboxylation to photoreforming—observed on Cu/TiO2 by ensuring reduction of the co-catalyst surfaces, is an assertive proof that the use of earth-abundant and inexpensive copper can be optimised for photocatalytic H2 production by controlling its oxidation state. The tendency of Cu to become progressively reduced under solar irradiation had been previously demonstrated for alcohols and polyols.17–20 The use of acetic acid, a more challenging substrate in terms of selectivity, is herein shown to serve as a sensitive tool to track the oxidation state of copper in parallel to H2 evolution efficiency. The colour of the suspensions also indicated the changes in copper oxidation states. During photodeposition of Cu(3%)/TiO2, bright purple-pink colours were apparent, consistent with the surface plasmon resonance (SPR) of Cu(0) nanoparticles,19,20 yet after isolation, the colour of the solid materials faded into pale blue; re-suspending in CH3COOH/H2O resulted in off-white suspensions, indicative of extensive copper oxidation (Fig. S7†). We aimed at checking whether this unwanted re-oxidation could be readily reversed. To do so, a redox cycling experiment was performed (Fig. 4 and Table S7, see ESI† for details). By starting with conventional Cu(3%)/TiO2, essentially selective to CH4 due to passivation (Fig. 4, #ox1), it was shown that a simple photodeposition step under UV-vis irradiation caused a colour change to purple-pink, characteristic of Cu(0), and to a remarkable increase in H2 production; the evolution of CH4 also decreased significantly (Fig. 4, #red1). To unambiguously prove the reversibility of these photocatalyst transformations, a deliberate air oxidation procedure was carried out. The suspension discoloured (Fig. 4, #ox2), indicating the formation of passivated CuO surfaces, again suppressing H2 production and enhancing that of CH4. Finally, another photoreduction cycle (Fig. 4, #red2) restored the H2 evolution sites on the clean metallic copper surfaces formed anew. The second oxidation (#ox2) and reduction (#red2) cycles reveal that the production of CH4 decreased, whereas that of H2 increased slightly, probably due to a memory effect whereby part of the reduced copper species became resistant to oxidation upon prolonged exposure to irradiation. The scheme shown in Fig. 4 illustrates the morphological and compositional changes in copper co-catalysts governing the selectivity of the solar photocatalytic transformation of aqueous acetic acid on Cu/TiO2. Such a reversible system is expected to allow for improved H2 production through appropriate operational control.
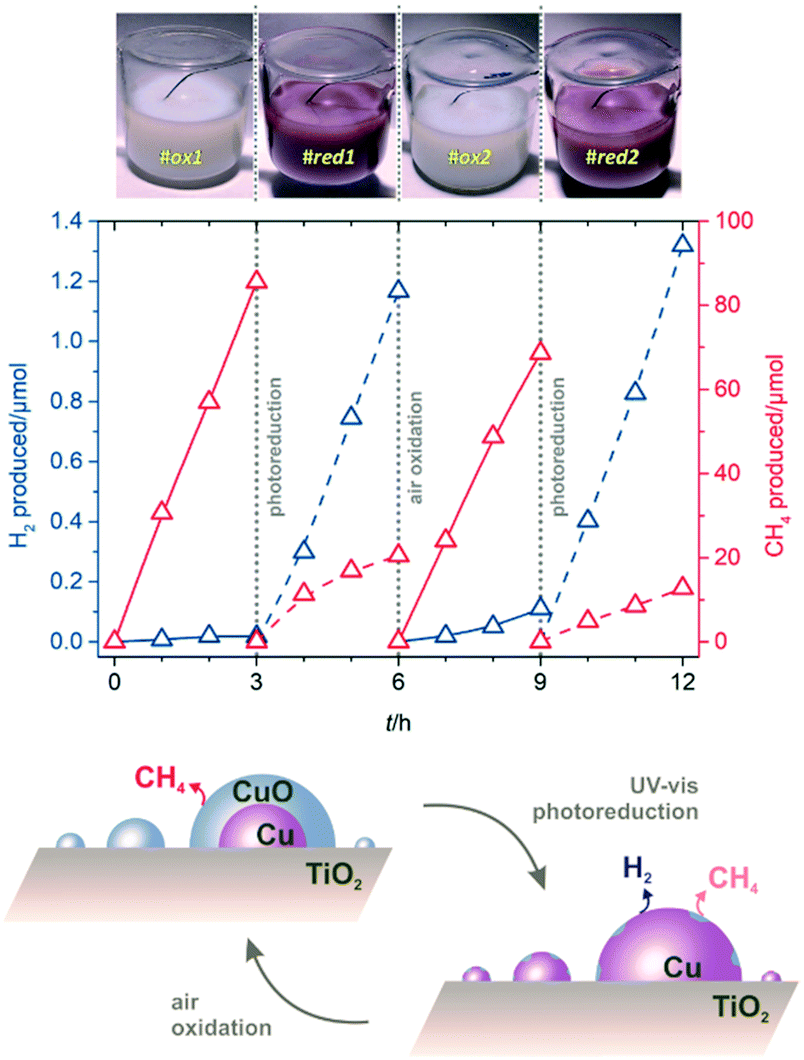 | ||
| Fig. 4 Cyclic redox behaviour during simulated sunlight irradiation of aqueous acetic acid on Cu(3%)/TiO2. The top pictures illustrate the aspect of the suspensions during the successive cycles, reversibly changing from white to purple-pink, indicative of oxidised copper due to air exposure, and photoreduction to Cu(0), respectively. Middle graph: production profiles for H2 and CH4; reaction conditions as in Fig. 1. The bottom scheme illustrates the reversible selectivity switching ascribed to oxidised and reduced copper sites, preferentially responsible for decarboxylation or reforming pathways, respectively. | ||
Conclusions
Acetic acid is a challenging substrate for photocatalytic H2 production, given the more thermodynamically favourable decarboxylation suppressing H2 and switching selectivity to CH4 as the main energy-containing product. In this work, we delved into the possibility of using earth-abundant Cu as the co-catalyst on TiO2 for this process using acetic acid as a model substrate for more recalcitrant acid-containing biomass-derived feedstocks. Direct use of conventional Cu/TiO2 photocataysts results in remarkably high decarboxylation activities due to severe passivation of copper nanoparticles. Although this might be an interesting valorisation solution per se, photoreduction is an efficient method to readily and reversibly force the exposure of clean Cu(0) surfaces active for H2 evolution. Via direct photodeposition of copper in aqueous acetic acid suspensions of TiO2, high H2 selectivities—on a par with platinum co-catalyst contenders—were achieved.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Financial support from the Spanish Government (Ministry of Science, Innovation and Universities) through its “Severo Ochoa” excellence programme (SEV 2016-0683) is acknowledged. A. V. P. thanks the Spanish Government (Agencia Estatal de Investigación) and the European Union (European Regional Development Fund) for a grant for young researchers (CTQ2015-74138-JIN, AEI/FEDER/UE). M. I. thanks the Spanish Catalysis Society (SECAT) for a Masters Grant. The authors also thank the Microscopy Service of UPV for kind help on TEM measurements.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed; R. M. Navarro, M. C. Sánchez-Sánchez, M. C. Alvarez-Galvan, F. del Valle and J. L. G. Fierro, Energy Environ. Sci., 2009, 2, 35–54 RSC; R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef PubMed.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS.
- D. Li, X. Y. Li and J. L. Gong, Chem. Rev., 2016, 116, 11529–11653 CrossRef CAS PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS PubMed; R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef PubMed.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS; K. C. Christoforidis and P. Fornasiero, ChemCatChem, 2017, 9, 1523–1544 CrossRef; L. I. Granone, F. Sieland, N. Zheng, R. Dillert and D. W. Bahnemann, Green Chem., 2018, 20, 1169–1192 RSC; M. Yasuda, T. Matsumoto and T. Yamashita, Renewable Sustainable Energy Rev., 2018, 81, 1627–1635 CrossRef; T. H. Jeon, M. S. Koo, H. Kim and W. Choi, ACS Catal., 2018, 8, 11542–11563 CrossRef.
- D. Mohan, C. U. Pittman, Jr. and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS; J. P. Diebold, A Review of the Chemical and Physical Mechanisms of the Storage Stability of Fast Pyrolysis Bio-oils, National Renewable Energy Laboratory, 2000 Search PubMed.
- D. Wang, S. Czernik, D. Montané, M. Mann and E. Chornet, Ind. Eng. Chem. Res., 1997, 36, 1507–1518 CrossRef CAS; L. Garcia, R. French, S. Czernik and E. Chornet, Appl. Catal., A, 2000, 201, 225–239 CrossRef; C. Rioche, S. Kulkarni, F. C. Meunier, J. P. Breen and R. Burch, Appl. Catal., B, 2005, 61, 130–139 CrossRef.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 5985–5992 CrossRef CAS; H. Yoneyama, Y. Takao, H. Tamura and A. J. Bard, J. Phys. Chem., 1983, 87, 1417–1422 CrossRef; J. Abrahams, R. S. Davidson and C. L. Morrison, J. Photochem., 1985, 29, 353–361 CrossRef; S. Hamid, R. Dillert, J. Schneider and D. W. Bahnemann, Catal. Sci. Technol., 2018, 8, 5886–5899 RSC.
- T. Sakata, T. Kawai and K. Hashimoto, J. Phys. Chem., 1984, 88, 2344–2350 CrossRef CAS; S. Hamid, I. Ivanova, T. H. Jeon, R. Dillert, W. Y. Choi and D. W. Bahnemann, J. Catal., 2017, 349, 128–135 CrossRef.
- S. Hamid, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2018, 122, 12792–12809 CrossRef CAS.
- A. Patsoura, D. I. Kondarides and X. E. Verykios, Catal. Today, 2007, 124, 94–102 CrossRef CAS.
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed; A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- P. Fornasiero and K. C. Christoforidis, ChemCatChem, 2018, 11, 368–382 Search PubMed.
- S. Mozia, A. Heciak and A. W. Morawski, Catal. Today, 2011, 161, 189–195 CrossRef CAS; S. Mozia, A. Heciak and A. W. Morawski, J. Photochem. Photobiol., A, 2010, 216, 275–282 CrossRef.
- A. Heciak, A. W. Morawski, B. Grzmil and S. Mozia, Appl. Catal., A, 2013, 140, 108–114 CrossRef.
- T. Montini, V. Gombac, L. Sordelli, J. J. Delgado, X. Chen, G. Adami and P. Fornasiero, ChemCatChem, 2011, 3, 574–577 CrossRef CAS; V. Gombac, L. Sordelli, T. Montini, J. J. Delgado, A. Adamski, G. Adami, M. Cargnello, S. Bernal and P. Fornasiero, J. Phys. Chem. A, 2010, 114, 3916–3925 CrossRef PubMed.
- Z. Wang, K. Teramura, T. Shishido and T. Tanaka, Top. Catal., 2014, 57, 975–983 CrossRef CAS.
- V. Polliotto, S. Livraghi, A. Krukowska, M. V. Dozzi, A. Zaleska-Medynska, E. Selli and E. Giamello, ACS Appl. Mater. Interfaces, 2018, 10, 27745–27756 CrossRef CAS PubMed.
- J. Nie, A. O. T. Patrocinio, S. Hamid, F. Sieland, J. Sann, S. Xia, D. W. Bahnemann and J. Schneider, Phys. Chem. Chem. Phys., 2018, 20, 5264–5273 RSC.
- A. Petala, E. Ioannidou, A. Georgaka, K. Bourikas and D. I. Kondarides, Appl. Catal., A, 2015, 178, 201–209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: synthetic and characterisation details, photocatalytic data. See DOI: 10.1039/c8cy02349b |
| This journal is © The Royal Society of Chemistry 2019 |