
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective synthesis of 4-hydroxyisophorone and 4-ketoisophorone by fungal peroxygenases†
Carmen
Aranda
a,
Martí
Municoy
b,
Víctor
Guallar
bc,
Jan
Kiebist
d,
Katrin
Scheibner
d,
René
Ullrich
e,
José C.
del Río
a,
Martin
Hofrichter
e,
Angel T.
Martínez
 *f and
Ana
Gutiérrez
*a
*f and
Ana
Gutiérrez
*a
aInstituto de Recursos Naturales y Agrobiología de Sevilla, CSIC, Reina Mercedes 10, E-41012 Seville, Spain. E-mail: anagu@irnase.csic.es
bBarcelona Supercomputing Center, Jordi Girona 31, E-08034, Barcelona, Spain
cICREA Passeig Lluís Companys 23, E-08010, Barcelona, Spain
dJenaBios GmbH, Löbstedter Str. 80, 07749 Jena, Germany
eTU Dresden, Department of Bio- and Environmental Sciences, Markt 23, 02763 Zittau, Germany
fCentro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, E-28040 Madrid, Spain. E-mail: atmartinez@cib.csic.es
First published on 26th February 2019
Abstract
The recently discovered unspecific peroxygenases (UPOs) from the ascomycetes Chaetomium globosum and Humicola insolens were capable of selectively hydroxylating isophorone to 4-hydroxyisophorone (4HIP) and 4-ketoisophorone (4KIP), which are substrates of interest for the pharmaceutical and flavor-and-fragrance sectors. The model UPO from the basidiomycete Agrocybe aegerita was less regioselective, forming 7-hydroxyisophorone (and 7-formylisophorone) in addition to 4HIP. However, it was the most stereoselective UPO yielding the S-enantiomer of 4HIP with 88% ee. Moreover, using H. insolens UPO full kinetic resolution of racemic HIP was obtained within only 15 min, with >75% recovery of the R-enantiomer. Surprisingly, the UPOs from two other basidiomycetes, Marasmius rotula and Coprinopsis cinerea, failed to transform isophorone. The different UPO selectivities were rationalized by computational simulations, in which isophorone and 4HIP were diffused into the enzymes using the adaptive PELE software, and the distances from heme-bound oxygen in H2O2-activated enzyme to different substrate atoms, and the corresponding binding energies were analyzed. Interestingly, for process upscaling, full conversion of 10 mM isophorone was achieved with H. insolens UPO within nine hours, with total turnover numbers up to 5500. These biocatalysts, which only require H2O2 for activation, may represent a novel, simple and environmentally-friendly route for the production of isophorone derivatives.
Introduction
Isophorone derivatives, such as 4-hydroxyisophorone (4HIP) and 4-ketoisophorone (4KIP), are of interest as flavour-and-fragrance additives,1 and as intermediates in the synthesis of pharmaceuticals, vitamins and natural pigments.2,3 A variety of chemical methods is available for the production of 4HIP and 4KIP. Thus, both derivatives have been synthesized from β-isophorone,1,4 which – on its part – can be obtained by isomerization of isophorone (also known as α-isophorone). The rearrangement of β-isophorone to the α-isomer, however, is a main drawback of this process. The direct oxidation of isophorone to 4KIP with molecular oxygen (O2) appeared to be the solution, using copper(II) acetylacetone or molybdenum-based systems as catalysts,5 but required toxic heavy metals and led to the formation of undesired side products. Moreover, a direct chemical oxidation process of isophorone to 4HIP is not available and this compound is usually synthesized by reduction of 4KIP,6,7 which can be a rather expensive starting material.Alternatively, some biological processes for the synthesis of 4HIP and 4KIP have been described, often using cytochrome P450 monooxygenases (P450s). Among them, the microbial biotransformation of isophorone was described for fungi – like Aspergillus niger, Alternaria alternata and Neurospora crassa –8,9 with 4HIP and 7-hydroxyisophorone (7HIP) as main metabolites. More recently, a process using recombinant Escherichia coli transformed with the P450-BM3 gene (together with the gene of NADPH-regenerating glucose dehydrogenase) allowed the scaled-up selective production of 4HIP at kilogram scale.10 On the other hand, 4KIP has been produced either in an one-pot two-step enzymatic process or as a cascade process employing cells co-expressing P450-WAL and Cm-ADH10 dehydrogenase.11 However, isolated P450s generally suffer the disadvantage of rather higher instability and the frequent need of auxiliary enzymes/domains and expensive cofactors.
Unspecific peroxygenases (UPOs, EC.1.11.2.1) are novel and appealing biocatalysts for organic synthesis, since their ‘simplicity’ (only H2O2 is required for activation) and stability (as secreted enzymes) circumvent major disadvantages of P450s while catalyzing the same kind of oxyfunctionalization reactions.12 The first enzyme of this class was discovered in 2004 in the basidiomycete Agrocybe aegerita13 and since then, new peroxygenases came out from Coprinellus radians,14Marasmius rotula,15 and more recently from Chaetomium globosum.16 Their widespread occurrence in the fungal kingdom has been demonstrated by the analysis of basidiomycete, ascomycete and other fungal genomes and revealed over one-thousand putative peroxygenase genes.17 This allowed the production of recombinant enzymes, like those of Coprinopsis cinerea18 or Humicola insolens,16 which are heterologously expressed by Novozymes A/S (Bagsvaerd, Denmark) in the mold Aspergillus oryzae.
The spectrum of reactions catalyzed by these enzymes is steadily increasing and includes oxygenations of both aromatic19,20 and aliphatic compounds,21–24 fatty acids epoxidation25 and chain-shortening,26 and also reactions of rather complex and bulky substrates like steroids,27,28 and secosteroids29,30 that are subject to epoxidation, side-chain hydroxylation or side-chain removal.
In the present work, the hydroxylation of isophorone by several UPOs with different selectivities is presented for the first time, to be included in the portfolio of reactions catalyzed by these novel and exciting enzymes.12,17
Materials and methods
Enzymes
AaeUPO (isoform II, 46 kDa), the first UPO described in 2004, is a wild-type (i.e. non-recombinant) peroxygenase from cultures of A. aegerita TM-A1, grown in soybean-peptone medium, which was purified as described by Ullrich and Hofrichter.31MroUPO is another wild-type peroxygenase (32 kDa) from cultures of M. rotula DSM-25031 (German Collection of Microorganisms and Cell Cultures, Braunschweig), which was purified as described by Gröbe et al.15CglUPO (36 kDa) is a third wild-type peroxygenase from cultures of C. globosum DSM-62110, which was purified as recently described by Kiebist et al.16 The recombinant enzymes rCciUPO (44 kDa) and rHinUPO were provided by Novozymes A/S. rCciUPO corresponds to the protein model 7249 from the sequenced C. cinerea genome available at the JGI (http://genome.jgi.doe.gov/Copci1) used in several studies.22,27,30 The rHinUPO sequence has been more recently reported32 and used for oxyfunctionalizations that are not catalyzed by other UPOs.16 Both UPOs were expressed by Novozymes in Aspergillus oryzae (patent WO/2008/119780). All UPO proteins were purified by fast protein liquid chromatography (FPLC) using a combination of size exclusion chromatography (SEC) and ion exchange chromatography on different anion and cation exchangers. Purification was confirmed by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and UV-visible spectroscopy following the characteristic heme-maximum around 420 nm (Soret band of resting-state heme-thiolate proteins). Enzyme concentration was estimated according to the characteristic UV-vis band of the reduced UPO-complex with carbon monoxide.33Model compounds
3,5,5-Trimethyl-2-cyclohexen-1-one (isophorone) from Sigma Aldrich (97% purity) was tested as substrate of the above UPOs. 3,5,5-Trimethyl-2-cyclohexen-1,4-dione (4-ketoisophorone, 4KIP) also from Sigma Aldrich, and chemically-synthesized 4-hydroxy-3,5,5-trimethyl-2-cyclohexen-1-one (4-hydroxyisophorone, 4HIP) by 4KIP reduction, were used as standards in gas chromatography-mass spectrometry (GC-MS) analyses. 4HIP, obtained as a racemic mixture by chemical reduction of 4KIP,7 was used as substrate in enzymatic reactions together with isophorone.Enzyme reactions
Reactions (1 mL volume) with isophorone (0.1 mM) were performed at 30 °C, in 50 mM phosphate buffer, pH 7. The enzyme concentrations ranged from 50 nM to 10 μM, using 2.5 to 5 mM H2O2. In control experiments, substrates were treated under the same conditions (including H2O2) but without enzyme. After 30 min reaction, products were extracted with methyl tert-butyl ether, which was evaporated under N2, and the products dissolved in chloroform to be analyzed by GC-MS. Reactions at higher isophorone concentration (10 mM) were performed with CglUPO and rHinUPO (2–5 μM). H2O2 was added with a syringe pump to give concentrations in the reaction mixture of 1 or 5 mM h−1 during 48 h or 12 h, respectively. Reactions for chiral analyses were carried out with 10 mM isophorone or 4HIP (racemic mixture) and 5 μM enzyme (CglUPO, rHinUPO and AaeUPO) for 60 min, with H2O2 manually added in small doses to a final concentration of 5–20 mM. Products were extracted with ethyl acetate and directly analyzed by GC-MS or dried and dissolved in the mobile phase to be analyzed by HPLC.Enzyme kinetics
Reactions were carried out with 6.25–6400 μM substrate and 100 nM enzyme. They were initiated adding 0.5 mM H2O2 and stopped by vigorous shaking in 5 mM sodium azide. Reaction times, the reaction velocity of which was linear, were previously selected: 5 min for AaeUPO, 3 min for CglUPO and 1 min for rHinUPO. All reactions were performed in triplicates. Product quantification was carried out by GC-MS using external standard curves, and kinetic parameters – turnover number (kcat), Michaelis constant (Km) and catalytic efficiency (kcat/Km) – were obtained by fitting the data to the Michaelis–Menten equation, or to the corresponding variation of this equation when substrate inhibition is occurring, using SigmaPlot (Systat Software Inc., San Jose, CA, USA).GC-MS
The analyses were performed in a Shimadzu GC-MS QP 2010 Ultra system, using a fused-silica DB-5HT capillary column (30 m × 0.25 mm internal diameter, 0.1 μm film thickness) from J&W Scientific. The oven was heated from 50 °C (1.5 min) to 90 °C (2 min) at 30 °C min−1, and then from 90 °C to 250 °C (15 min) at 8 °C min−1. The injection was performed at 250 °C and the transfer line was kept at 300 °C. Compounds were identified by comparing their mass spectra and retention times with those of available commercial or synthesized authentic standards, and by search in the NIST library.Chiral HPLC
Chiral analyses were performed with a Shimadzu LC-2030C 3D system equipped with a photo-diode array detector using a chiral column Chiralpak IG (5 μM particle size, 4.6 mm diameter × 150 mm, Daicel Chemical Industries Ltd.) equipped with a Chiralpak IG guard column (5 μM particle size, 4.0 mm diameter × 10 mm). The column was eluted in isocratic mode with 95% hexane and 5% isopropanol at 0.5 mL min−1 for 60 min, and the absorbance was monitored at 254 nm. Enantiomers were identified based on the elution order previously reported.34Molecular models
AaeUPO and MroUPO models were obtained from the chain-A of the 2YP1 and 5FUJ crystal structures, after removing the Cys227 dimerization disulfide bridge from the second structure. Homology models for CglUPO, rHinUPO and rCciUPO were obtained at the Swiss-Model server,35,36 with related crystal structures as templates. The heme cofactor and the Mg ion (along with its two coordinated water molecules) were then superimposed using 2YP1 and 5FUJ as templates, followed by an initial minimization to release steric clashes. An oxygen atom (iron-oxo) was finally added to all structures modeling heme compound I. All systems were prepared at pH 7 with the protein preparation wizard from Schrödinger.37 Heme charges were obtained from a quantum mechanics/molecular mechanics (QM/MM) minimization using QSite at the DFT M06-L(lacvp*)/OPLS level of theory. Based on previous experience in heme-bound systems,38 the charge of the Mg ion was set to 1.2. isophorone and 4HIP (S- and R-enantiomers) substrates were built with Maestro and optimized at the OPLS level of theory. In addition, two explicit water molecules were placed in the active site when exploring diffusion of the ligands. The presence of a water molecule has been highlighted in compound I activity and might be important when diffusing polar substrates such as 4HIP.39Ligand diffusion simulations
The new adaptive-PELE software40 was used to study ligand diffusion and binding on the different UPO structures. PELE uses a Monte Carlo (MC) procedure to describe the protein-ligand conformational dynamics. At each MC iteration, the algorithm performs: 1) ligand perturbation (translation and rotation); 2) protein perturbation following normal modes; 3) explicit water sampling; 4) side chain prediction; and 5) overall minimization. The final structure is then accepted or rejected based on a Metropolis criterion. The adaptive protocol improves PELE's sampling by running multiple short simulations (epochs) where initial conditions are selected through a reward function aiming at sampling non-visited areas. We also used a new version of PELE that allows for explicit water sampling, where water molecules are allowed to freely move (with 100 small translations and rotations) after the backbone sampling. Two sets of simulations were performed for each ligand and structure. In the initial one the ligand was placed on the protein surface, next to the entrance to the active site (at ∼16 Å from the heme's iron atom), and allowed to diffuse freely into the heme distal site. The structure with the best (ligand) binding energy from the initial simulation was then selected for a second local refinement run, where the ligand center of mass was constrained to move within 8 Å from the heme's iron. All simulations used 50 epochs of 16 MC PELE steps each with 128 computing cores. Interaction/binding energies (kcal mol−1) were derived as Eab − (Ea + Eb), where Eab is the total energy of the complex, Eb the energy of the ligand and Ea the energy of receptor (everything but the ligand), all of them obtained at the OPLS2005 level of theory with a surface GB implicit solvent model.Results and discussion
Regioselectivity in isophorone transformation by UPOs
In the present work, the ability of several UPOs to oxidize isophorone was analyzed, and different transformation patterns were observed (Fig. 1). The peroxygenases from the ascomycetes C. globosum (CglUPO) and H. insolens (rHinUPO), and the basidiomycete A. aegerita (AaeUPO) were found to transform the substrate, although with different regioselectivities (Table 1). In contrast, the enzymes from the basidiomycetes C. cinerea (rCciUPO) and M. rotula (MroUPO) were unable to convert the substrate, even at the highest (10 μM) enzyme doses tested.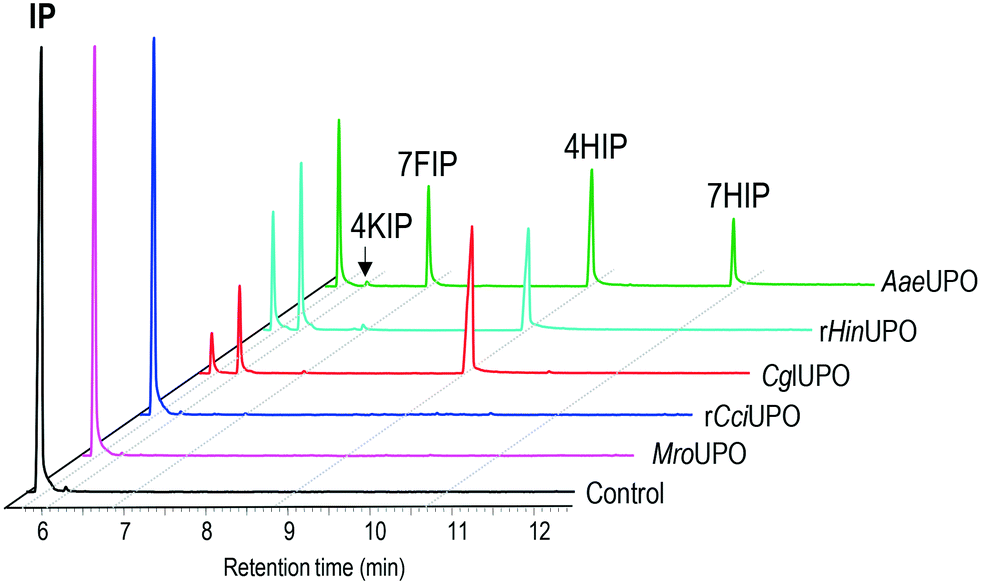 | ||
| Fig. 1 GC-MS analysis of 0.1 mM isophorone reactions (30 min) with 0.1 μM of MroUPO, rCciUPO, CglUPO, rHinUPO, AaeUPO and control without enzyme, showing the remaining substrate (IP, isophorone) and the hydroxylated (4HIP and 7HIP) and oxo (4KIP and 7FIP) derivatives. | ||
| Conversion | 4HIP | 7HIP | 4KIP | 7FIP | |
|---|---|---|---|---|---|
| a Reactions using 0.1 μM UPO. b Reactions using 0.25 μM UPO. c Reactions using 0.5 μM UPO. d Reactions using 2.5 mM H2O2. e Reactions using 5 mM H2O2; tr denotes traces. | |||||
| AaeUPOa,d | 72 | 46 | 25 | tr | 29 |
| AaeUPOb,d | 96 | 46 | 19 | tr | 35 |
| CglUPOa,d | 95 | 79 | — | 20 | 1 |
| CglUPOc,e | 100 | — | — | 100 | — |
| rHinUPOa,d | 84 | 53 | — | 45 | 2 |
| rHinUPOc,e | 100 | 3 | — | 97 | — |
CglUPO and rHinUPO selectively oxidized isophorone in position 4 (Scheme 1A) yielding mono-hydroxylated 4HIP and the keto-derivative 4KIP, which were identified by their retention times compared with authentic standards (Fig. S1†) and their mass spectra (Fig. S2†). Besides the 4HIP and 4KIP molecular ions at m/z 154 and 152, respectively, their mass spectra showed a shift from α-isophorone fragments at m/z 54 and 82 to m/z 70 and 98 in 4HIP and to m/z 68 and 96 in 4KIP, which corresponds to the insertion of a hydroxyl or keto group. Isophorone conversion by rHinUPO and CglUPO (84 and 95%, respectively) was already efficient at low enzyme dose (0.1 μM) and both enzymes showed the tendency to over-oxidize 4HIP to form 4KIP. Over-oxidation was more pronounced in the case of rHinUPO as shown by the ratio of 4HIP to 4KIP of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (compared to 4:1 in the case of CglUPO) (Table 1).
:1 (compared to 4:1 in the case of CglUPO) (Table 1).
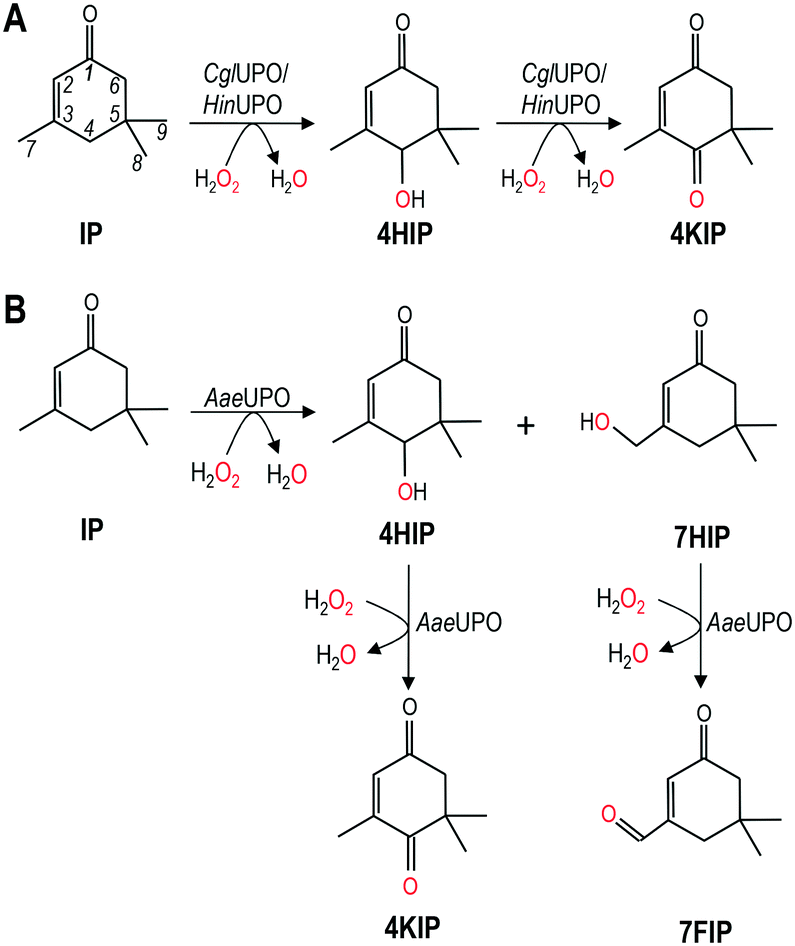 | ||
| Scheme 1 Isophorone (IP) hydroxylation catalyzed by CglUPO and rHinUPO (A) and AaeUPO (B), showing the hydroxylated 4HIP (4-hydroxyisophorone) and 7HIP (7-hydroxyisophorone) and the oxo 4KIP (4-ketoisophorone) and 7FIP (7-formylisophorone) derivatives. | ||
AaeUPO was found to be less regioselective in oxidizing isophorone, since – in addition to 4HIP – other mono-hydroxylated and keto-derivatives were formed (Scheme 1B). These side products were identified as 7HIP and 7-formylisophorone (7FIP) due to their mass spectra (Fig. S3†) that matched with those previously reported,34 with the singular mass fragment at m/z 125 in 7HIP, different from that at m/z 112 in 4HIP.
Reactions with low AaeUPO dose (0.1 μM) showed that hydroxylation took place in similar proportion at C4 (46%) and C7 (54%), but 7HIP was rapidly over-oxidized to form the corresponding aldehyde, and 7FIP and 4HIP were the major final products of the reaction (Table 1). Interestingly, 4HIP was barely further oxidized to 4KIP.
The regioselectivity observed in the hydroxylation of isophorone by some UPOs is similar to that reported for certain P450s. Among them, P450cam-RhFRed variants have been reported to yield 4HIP, 7HIP and isophorone oxide (2,3-epoxy-3,5,5-trimethyl-1-cyclohexanone) as major products, with 4HIP as the only product from one of the variants.11 4HIP was also the main product of reactions with CYP102A1 and CYP101A1, although minor amounts of the epoxide, 7HIP and further oxidation products were observed.34 P450s, unlike rHinUPO or CglUPO, seem to be unable to oxidize 4HIP into 4KIP and, therefore, two enzymes (a P450 and an alcohol dehydrogenase) are necessary to obtain 4KIP from isophorone.11
In view of the higher selectivity to form the products of interest (4HIP and 4KIP), CglUPO and rHinUPO were selected to perform reactions with higher (×100) substrate load (Fig. 2). These experiments revealed a faster substrate conversion by rHinUPO that completely transformed isophorone within 6 h, while CglUPO needed 12 h for 87% conversion. As expected, a higher proportion of 4KIP was observed in the rHinUPO reactions. A higher enzyme dose would be needed to complete conversion into 4KIP, as it was already found when lower substrate concentrations were tested (Table 1).
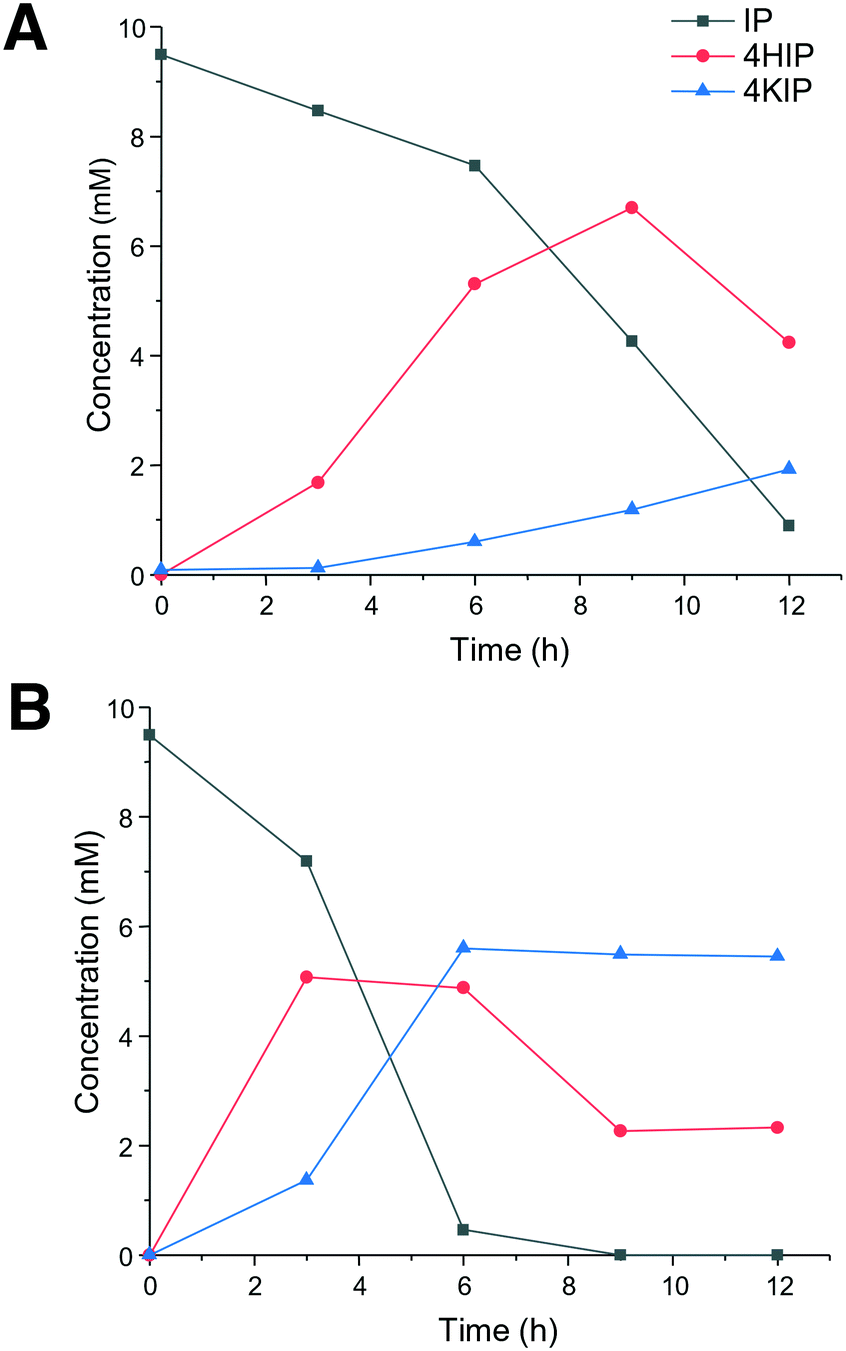 | ||
| Fig. 2 Time course of 10 mM isophorone (IP) reaction with 5 μM CglUPO (A) and rHinUPO (B) and H2O2 (added with a syringe pump to give 5 mM h−1 concentration), showing substrate and products (4HIP and 4KIP) concentrations. | ||
Kinetics of isophorone hydroxylation by UPOs
Despite the difficulties to determine initial enzymatic reaction rates by GC-MS, kinetic curves for isophorone hydroxylation by the three UPOs could be obtained (Fig. S4†) and reaction constants (kcat, Km and kcat/Km) were estimated (Table 2). There were differences in enzyme affinities, since the Km values were four- and two-fold higher for AaeUPO than for CglUPO and rHinUPO, revealing the higher isophorone affinity of the two latter enzymes. Moreover, rHinUPO displayed a ten-fold higher turnover number (kcat) compared to CglUPO, which resulted in five-fold higher catalytic efficiency, while the efficiencies of AaeUPO and CglUPO were similar.| k cat (s−1) | K m (μM) | k cat/Km (mM−1 s−1) | |
|---|---|---|---|
| a Data represent mean values of three replicates with standard deviations. | |||
| AaeUPO | 20.1 ± 1.1 | 1380 ± 200 | 14.6 ± 2.2 |
| CglUPO | 4.4 ± 0.4 | 309 ± 55 | 14.2 ± 2.8 |
| rHinUPO | 42.0 ± 9.8 | 633 ± 201 | 66.4 ± 26.1 |
The catalytic efficiency of these UPOs hydroxylating isophorone is in the range of previously reported for the hydroxylation of cyclohexane by other UPOs.41 On the other hand, the Km of CglUPO and rHinUPO for isophorone hydroxylation is similar to values (380–440 μM) reported for CYP102A1 variants when decoy molecules were used, and the turnover numbers of these variants (2.5–5.5 s−1) were similar as well to that reported herein for CglUPO (4.4 s−1).34
Higher total turnover numbers (TTNs) were attained with rHinUPO (2660) than with CglUPO (1820) under the same reaction conditions, although these values could be up to 5600 and 3600 when lower enzyme doses were used (Table 3). The TTNs are in the range of those reported for two-step (1567) or one-step (3421) isophorone oxidation by the combination of P450-WAL and Cm-ADH10.11
UPO enantioselectivity on isophorone and racemic 4HIP
Enantioselectivity in the synthesis of 4HIP by the three UPOs hydroxylating isophorone was determined by HPLC (Fig. S5†). The results of the chiral analysis showed that only the reaction with AaeUPO can be considered as stereoselective, with an enantiomeric excess (ee) of 88% S-4HIP (Table 4). The three UPOs preferentially formed the S-enantiomer, in contrast to P450s that rather formed the R-enantiomer.11,42The above values were estimated under reaction conditions where the product concentration was similar for all three enzymes (2.0–3.3 mM) and 4HIP over-oxidation was minimal, since it was observed that the ee of the hydroxylation product changed when 4KIP was formed. This was due to higher velocity in the conversion of the S-enantiomer compared to the R-enantiomer, as observed in rHinUPO reactions with racemic 4HIP as substrate (Fig. S6†). That way, a kinetic resolution of the racemate, with ee of 99–100% and 60–75% recovery, can be achieved with rHinUPO and CglUPO. In contrast, AaeUPO just slowly converted the racemic mixture of 4HIP, as it was also observed in the reaction of the enzyme with isophorone, where only traces of 4KIP were formed (Fig. 1), and no enantiomeric enrichment was produced.
Computational analyses: molecular modeling
Isophorone diffusion was done in two simulation steps using PELE software.40 For the initial simulation, after preparing all systems placing isophorone on the surface next to the entrance channel, the substrate was allowed to move freely within 16 Å of the heme iron. In all systems, the substrate moved easily into the active site. Structures in direct contact with the heme produced the best binding-energy poses, although they presented different substrate orientations. Therefore, after selecting the best binding-energy structure, we run a local refinement, where the ligand was forced to move within ∼8 Å of the heme iron. Fig. 3 shows an example of these two runs for CglUPO, where we display the binding-energy profile with respect to the C4-oxo distance for the initial (black) and the refinement (red) simulations.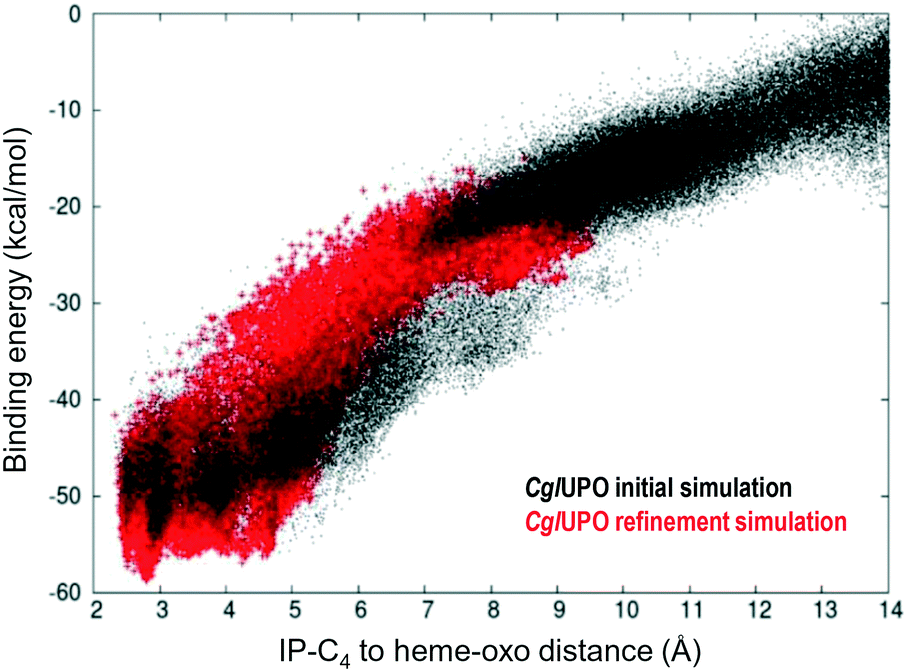 | ||
| Fig. 3 Example of the initial (black) and refinement (red) simulations on isophorone (IP) diffusion (C4-oxo distance being monitored with respect to binding/interaction energy) on CglUPO, using two-step adaptive PELE.35 | ||
Fig. 4A shows the binding energy along the C4-oxo distance for isophorone in the refinement runs for the five UPO structures (identified with different colors). Among them, three UPOs show distances lower than 3 Å: CglUPO (red), AaeUPO (green) and rHinUPO (cyan) (see Fig. S7† for the individual PELE plots). Such positioning will largely facilitate the hydrogen-atom abstraction by compound I, in agreement with the experimental results (Table 1). Interestingly, different isophorone reactive poses were detected for the different UPOs, as shown in Fig. 4B and C for CglUPO and AaeUPO, respectively. A closer examination of these structures showed shorter distances to the heme oxo for the pro-S (1.9 Å in both) than for the pro-R (3.5 and 2.6 Å, respectively, not shown) hydrogen atoms, which explained the preferential formation of the 4HIP S-enantiomer, as shown in Table 4.
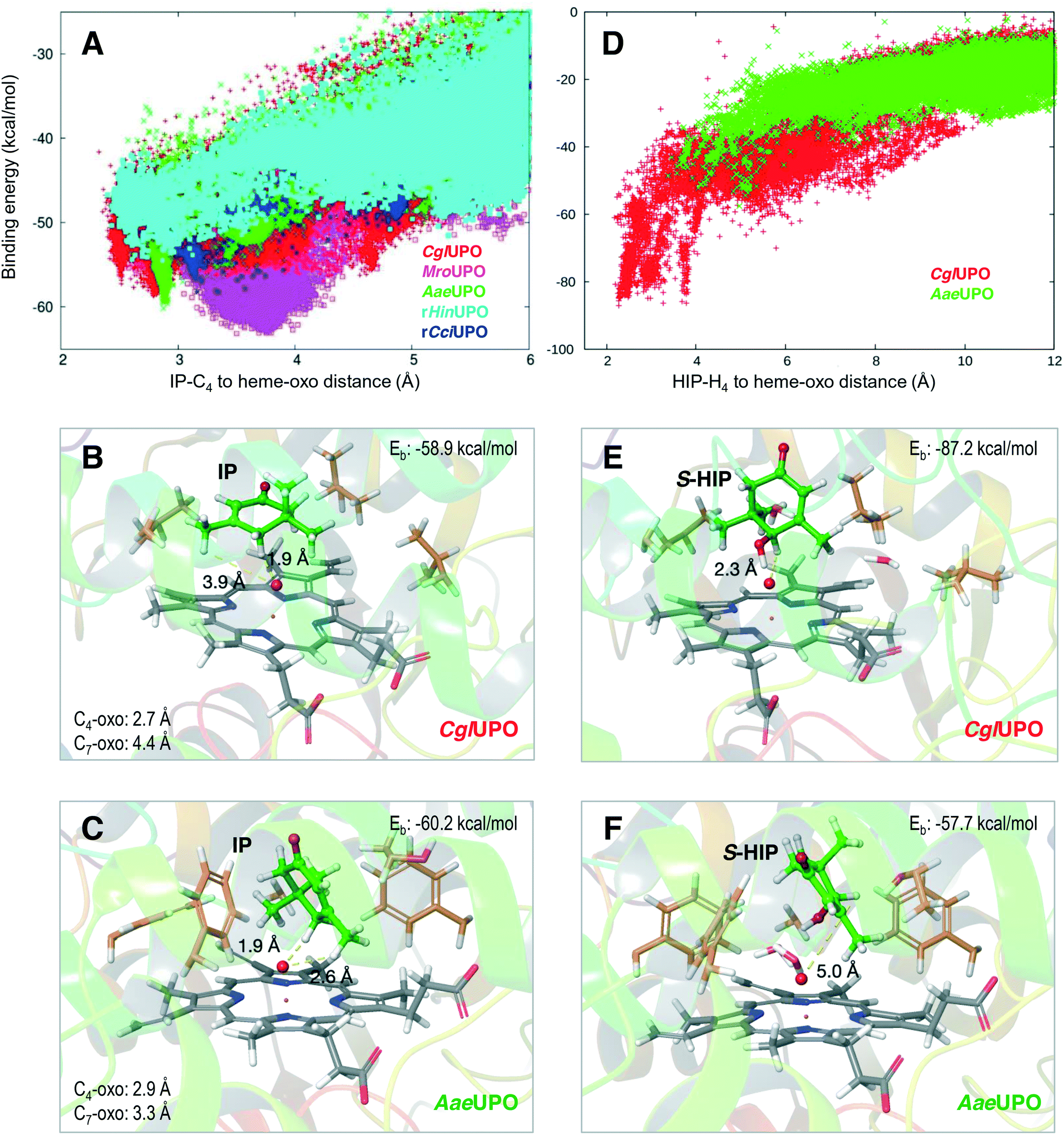 | ||
| Fig. 4 Isophorone (IP, left) and (S)-4-hydroxyisophorone (S-HIP, right) diffusion refinement on five UPOs, with adaptive PELE35 monitoring the distance between the substrate and the oxo atom (red sphere) of the H2O2-activated heme with respect to the binding energy (Eb). A) C4 distance vs. energy plot for IP diffusion in CglUPO (red), MroUPO (magenta), AaeUPO (green), rHinUPO (cyan) and rCciUPO (blue) (see Fig. S7† for individual PELE plots of the five UPO systems). B and C) IP at the two lowest binding-energy positions during the CglUPO and AaeUPO simulations (A), respectively. D) H4 distance vs. energy plot for S-HIP diffusion on CglUPO (red) and AaeUPO (green). E and F) S-HIP at the two lowest binding-energy positions during the CglUPO and AaeUPO simulations (D), respectively. | ||
The resulting S-4HIP was also diffused with PELE, and strong differences in the C4-oxo distances and energies were obtained, as illustrated in Fig. 4D for two of the systems, with CglUPO closely approaching the heme oxo (Fig. 4E) while no catalytic distances were attained by AaeUPO (Fig. 4F). The above results agree with isophorone oxidation to 4KIP by CglUPO, while 4HIP is the main product from AaeUPO (Table 1), as well as with the deracemization results of chiral 4HIP (Fig. S6†). On the other hand, although additional PELE calculations showed a similar C4-oxo distance for the R-enantiomer (data not shown), the slightly worse (5 kcal mol−1) binding energy of the different pose adopted (with respect to S-4HIP) is in agreement with the S preference experimentally observed (Fig. S6†).
Finally, the dual hydroxylation at the isophorone C4 and C7 positions by AaeUPO, compared with the selective oxidation at C4 by CglUPO and rHinUPO (Table 1) was also analyzed in the PELE simulations. The isophorone C7 position is not at a catalytically relevant distance in CglUPO (4.4 Å, Fig. 4B and S8A†), while the C7-oxo and C4-oxo distances for AaeUPO (3.3 and 2.9 Å, respectively, Fig. 4C and S8B†) are within reaction limits (the oxo to hydrogen distances are also shown in Fig. 4B and C). The above results explain the lack of C7 hydroxylation by CglUPO and the similar percentages of C7-derivatives (7HIP + 7FIP) and C4-derivatives (4HIP + 4KIP) by AaeUPO (Table 1).
Conclusions
We report a new enzymatic route for isophorone oxidation to form 4HIP and 4KIP, which are interesting products for the flavour-and-fragrance and pharmaceutical industries. The direct enzymatic oxidation of isophorone to 4KIP (with only one enzyme) is reported here for the first time for two fungal peroxygenases (CglUPO and rHinUPO). The above represents an advantage over the route with P450s, since the latter needs two enzymes (a P450 and an alcohol dehydrogenase) to obtain 4KIP from isophorone. However, process optimization of isophorone conversion by UPO is needed to attain the high-scale transformations reported for whole-cell P450 systems.43Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EnzOx2 (H2020-BBI-PPP-2015-2-1-720297) EU-project, the AGL2014-53730-R (BIORENZYMERY) and CTQ2016-79138-R projects of the Spanish MINECO (co-financed by FEDER) and the CSIC (201740E071) project. Novozymes (Bagsvaerd, Denmark) is acknowledged for providing samples of rCciUPO and rHinUPO.References
- S. Krill, K. Huthmacher and S. Perrin, US Pat., 6469215B1, 2002 Search PubMed.
- M. Eggersdorfer, D. Laudert, U. Létinois, T. McClymont, J. Medlock, T. Netscher and W. Bonrath, Angew. Chem., Int. Ed., 2012, 51, 12960–12990 CrossRef CAS PubMed.
- O. Isler, H. Lindlar, M. Montavon, R. Rüegg, G. Saucy and P. Zeller, Helv. Chim. Acta, 1956, 39, 2041–2053 CrossRef CAS.
- J. J. Becker, U. P. Hochstrasser and W. Skorianetz, US Pat., 3944620, 1972 Search PubMed.
- W. Zhong, L. Mao, Q. Xu, Z. Fu, G. Zou, Y. Li, D. Yin, H. Luo and S. R. Kirk, Appl. Catal., A, 2014, 486, 193–200 CrossRef CAS.
- M. Hennig, K. Püntener and M. Scalone, Tetrahedron: Asymmetry, 2000, 11, 1849–1858 CrossRef CAS.
- M. Ishihara, T. Tsuneya, H. Shiota, M. Shiga and K. Nakatsu, J. Org. Chem., 1986, 51, 491–495 CrossRef CAS.
- I. Kiran, O. Ozsen, T. Celik, S. Ilhan, B. Y. Gürsu and F. Demirci, Nat. Prod. Commun., 2013, 8, 59–61 CAS.
- Y. Mikami, Y. Fukunaga, M. Arita, Y. Obi and T. Kisaki, Agric. Biol. Chem., 1981, 45, 791–793 CAS.
- I. Kaluzna, T. Schmitges, H. Straatman, D. van Tegelen, M. Müller, M. Schürmann and D. Mink, Org. Process Res. Dev., 2016, 20, 814–819 CrossRef CAS.
- M. Tavanti, F. Parmeggiani, J. R. G. Castellanos, A. Mattevi and N. J. Turner, ChemCatChem, 2017, 9, 3338–3348 CrossRef CAS.
- Y. Wang, D. Lan, R. Durrani and F. Hollmann, Curr. Opin. Chem. Biol., 2017, 37, 1–9 CrossRef PubMed.
- R. Ullrich, J. Nuske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- D. H. Anh, R. Ullrich, D. Benndorf, A. Svatos, A. Muck and M. Hofrichter, Appl. Environ. Microbiol., 2007, 73, 5477–5485 CrossRef CAS PubMed.
- G. Gröbe, M. Ullrich, M. Pecyna, D. Kapturska, S. Friedrich, M. Hofrichter and K. Scheibner, AMB Express, 2011, 1, 31–42 CrossRef PubMed.
- J. Kiebist, K. U. Schmidtke, J. Zimmermann, H. Kellner, N. Jehmlich, R. Ullrich, D. Zänder, M. Hofrichter and K. Scheibner, ChemBioChem, 2017, 18, 563–569 CrossRef CAS PubMed.
- M. Hofrichter, H. Kellner, M. J. Pecyna and R. Ullrich, Adv. Exp. Med. Biol., 2015, 851, 341–368 CrossRef CAS PubMed.
- J. E. Stajich, S. K. Wilke, D. Ahren, C. H. Au, B. W. Birren, M. Borodovsky, C. Burns, B. Canbäck, L. A. Casselton, C. K. Cheng, J. X. Deng, F. S. Dietrich, D. C. Fargo, M. L. Farman, A. C. Gathman, J. Goldberg, R. Guigo, P. J. Hoegger, J. B. Hooker, A. Huggins, T. Y. James, T. Kamada, S. Kilaru, C. Kodira, U. Kües, D. Kupfert, H. S. Kwan, A. Lomsadze, W. X. Li, W. W. Lilly, L. J. Ma, A. J. Mackey, G. Manning, F. Martin, H. Muraguchi, D. O. Natvig, H. Palmerini, M. A. Ramesh, C. J. Rehmeyer, B. A. Roe, N. Shenoy, M. Stanke, V. Ter Hovhannisyan, A. Tunlid, R. Velagapudi, T. J. Vision, Q. D. Zeng, M. E. Zolan and P. J. Pukkila, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11889–11894 CrossRef CAS PubMed.
- C. Aranda, R. Ullrich, J. Kiebist, K. Scheibner, J. C. del Río, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Catal. Sci. Technol., 2018, 8, 2394–2401 RSC.
- M. Hofrichter and R. Ullrich, Curr. Opin. Chem. Biol., 2014, 19, 116–125 CrossRef CAS PubMed.
- A. Olmedo, C. Aranda, J. C. del Río, J. Kiebist, K. Scheibner, A. T. Martínez and A. Gutiérrez, Angew. Chem., Int. Ed., 2016, 55, 12248–12251 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, Biotechnol. Bioeng., 2013, 110, 2332 CrossRef PubMed.
- A. Gutiérrez, E. D. Babot, R. Ullrich, M. Hofrichter, A. T. Martínez and J. C. del Río, Arch. Biochem. Biophys., 2011, 514, 33–43 CrossRef PubMed.
- S. Peter, M. Kinne, X. Wang, R. Ulrich, G. Kayser, J. T. Groves and M. Hofrichter, Rev. Geophys., 2011, 278, 3667–3675 CAS.
- C. Aranda, A. Olmedo, J. Kiebist, K. Scheibner, J. C. del Río, A. T. Martínez and A. Gutiérrez, ChemCatChem, 2018, 10, 3964–3968 CrossRef CAS.
- A. Olmedo, J. C. del Río, J. Kiebist, K. Scheibner, A. T. Martínez and A. Gutiérrez, Chem. – Eur. J., 2017, 23, 16985–16989 CrossRef CAS PubMed.
- E. D. Babot, J. C. del Río, M. Cañellas, F. Sancho, F. Lucas, V. Guallar, L. Kalum, H. Lund, G. Gröbe, K. Scheibner, R. Ullrich, M. Hofrichter, A. T. Martínez and A. Gutiérrez, Appl. Environ. Microbiol., 2015, 81, 4130–4142 CrossRef CAS PubMed.
- R. Ullrich, M. Poraj-Kobielska, S. Scholze, C. Halbout, M. Sandvoss, M. J. Pecyna, K. Scheibner and M. Hofrichter, J. Inorg. Biochem., 2018, 183, 84–93 CrossRef CAS PubMed.
- F. Lucas, E. D. Babot, J. C. del Río, L. Kalum, R. Ullrich, M. Hofrichter, V. Guallar, A. T. Martínez and A. Gutiérrez, Catal. Sci. Technol., 2016, 6, 288–295 RSC.
- E. D. Babot, J. C. del Río, L. Kalum, A. T. Martínez and A. Gutiérrez, ChemCatChem, 2015, 7, 283–290 CrossRef CAS.
- R. Ullrich and M. Hofrichter, FEBS Lett., 2005, 579, 6247–6250 CrossRef CAS PubMed.
- H. Lund, L. Kalum, M. Hofrichter and S. Peter, US Pat., 9908860B2, 2018 Search PubMed.
- C. R. Otey, Methods Mol. Biol., 2003, 230, 137–139 CAS.
- S. Dezvarei, J. H. Z. Lee and S. G. Bell, Enzyme Microb. Technol., 2018, 111, 29–37 CrossRef CAS PubMed.
- M. Biasini, S. Bienert, A. Waterhouse, K. Arnold, G. Studer, T. Schmidt, F. Kiefer, T. G. Cassarino, M. Bertoni, L. Bordoli and T. Schwede, Nucleic Acids Res., 2014, 42, W252–W258 CrossRef CAS PubMed.
- N. Guex, M. C. Peitsch and T. Schwede, Electrophoresis, 2009, 30, S162–S173 CrossRef PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, J. Comput.-Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- V. Guallar and F. H. Wallrapp, Biophys. Chem., 2010, 149, 1–11 CrossRef CAS PubMed.
- A. Altun, V. Guallar, R. A. Friesner, S. Shaik and W. Thiel, J. Am. Chem. Soc., 2006, 128, 3924–3925 CrossRef CAS PubMed.
- D. Lecina, J. F. Gilabert and V. Guallar, Sci. Rep., 2017, 7, 8466 CrossRef PubMed.
- S. Peter, M. Kinne, R. Ullrich, G. Kayser and M. Hofrichter, Enzyme Microb. Technol., 2013, 52, 370–376 CrossRef CAS PubMed.
- I. Kaluzna, T. Schmitges, H. Straatman, D. van Tegelen, M. Müller, M. Schürmann and D. Mink, Org. Process Res. Dev., 2016, 20, 814–819 CrossRef CAS.
- I. Kaluzna, T. Schmitges, H. Straatman, D. van Tegelen, M. Müller, M. Schürmann and D. Mink, Org. Process Res. Dev., 2016, 20, 814–819 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cy02114g |
| This journal is © The Royal Society of Chemistry 2019 |