
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow to tailor flexible silicone elastomers with mechanical integrity: a tutorial review
Piotr
Mazurek
 ,
Sindhu
Vudayagiri
and
Anne Ladegaard
Skov
*
,
Sindhu
Vudayagiri
and
Anne Ladegaard
Skov
*
Department of Chemical Engineering, Technical University of Denmark, Denmark. E-mail: al@kt.dtu.dk
First published on 11th February 2019
Abstract
The tutorial aims to equip the beginners in silicone research with the knowledge to formulate recipes and process elastomer networks, targeting specific properties related to soft applications such as stretchable electronics without compromising the mechanical integrity of the elastomer. In doing so, we hope to stimulate further research in the area of tailor-made soft silicone elastomers for novel applications and allow researchers to bypass the limitations imposed by the use of commercially available silicone elastomer formulations. Silicone elastomers are widely used due to the favourable properties, such as flexibility, durable dielectric insulation, barrier properties against environmental contaminants and stress-absorbing properties over a wide range of temperatures ≈ −100 °C to 250 °C. For research on flexible electronics and other emerging technologies, the most commonly utilised silicone elastomer formulation is Sylgard 184 which is easier to process than most other commercially available silicone elastomers, due to the fact that the premixes have low viscosity. Furthermore, curing is robust and not as sensitive to poisoning as other silicone elastomer formulations. However, Sylgard 184 is not suitable for all fields of research that require flexible and stretchable silicones. When much softer networks are needed, the Sylgard 184 premixes are either mixed in non-stoichiometric ratios, or they are blended with softer types of commercially available elastomers, which compromise the mechanical integrity of the elastomer. Therefore, it is advantageous for researchers to formulate their own custom-made silicone elastomers and not depend on premade formulations which often harbour a few unknown components.
 Piotr Mazurek | Piotr Mazurek is a scientist in Danish Polymer Centre at the Danish Technical University, DTU. He studied chemical engineering at West Pomeranian University of Technology (Szczecin, Poland). He received a PhD degree in polymer science from DTU in 2016 for the study on development of functional materials used in dielectric elastomer transducer (DET) technology. Currently he works on smart elastomeric silicone membranes and foams used in DET, biomedical drug delivery, and magneto-chromatic systems. |
 Sindhu Vudayagiri | Sindhu Vudayagiri is currently a PostDoc in the Danish Polymer Centre at the Danish Technical University, DTU. She has an MS (chemical engineering) from SUNY, Buffalo, NY. She received a PhD (chemical engineering) from DTU in 2014 and her research was about ‘Large scale processing of dielectric electroactive polymers.’ She has worked as a Post Doc at Copenhagen University (Centre for Ice and Climate) for evaluating the paleo elevations of the Greenland ice sheet from total air content of the ice cores. |
 Anne Ladegaard Skov | Anne Ladegaard Skov is a professor of polymer science and engineering specialising in silicone elastomers in the Danish Polymer Centre at Department of Chemical Engineering, DTU. She was a research fellow at Cambridge University, UK, before she took up a position as assistant professor at DTU. In 2018 she was promoted to full professor. She has worked with functionalisation and formulation of silicone elastomers with main focus on silicone elastomers utilised and optimised for dielectric elastomers and more recently for flexible electronics and drug delivery amongst others. |
Key learning points(1) Silicone elastomer formulations primarily consist of a silicone polymer, a crosslinker, a catalyst and reinforcing fillers. Multiple additives are generally added for ease of processing and to add extra functionality to the elastomer.(2) The structure and mechanical properties of the elastomeric network are a direct consequence of the ratio of the above components and the length of the polymer chain. (3) By mixing the network components in a non-stoichiometric ratio, a complex mixture of elastically active polymer chains and dangling substructures appears. The substructures have complex mechanical behaviours and may disguise the fact that a coherent elastomer may not have resulted from the reaction, simply because an elastomer prepared close to the gelation threshold may show elastic behaviour on short timescales but behave as a liquid over longer periods. (4) Most silicone elastomers are reinforced with fumed silica to improve ultimate tensile properties as well as tear strength. These particulate fillers introduce non-linearity to the mechanical properties. Such history and time dependant properties, explained in terms of the Payne and Mullins effect, must be considered when using these reinforced elastomers in various applications. (5) In addition to the precise formulation, appropriate processing techniques are required in order to get reliable and reproducible silicone elastomers. |
1 Introduction
Silicone elastomers, also dubbed as ‘silicone rubbers’, appear frequently in domestic products such as flexible baking moulds, bathroom sealants, skin adhesives, cosmetics and noise cancellation for car brakes, to mention but a few. Silicone elastomers are also extensively used in advanced technologies, in the form of stretchable electronics,1 drug delivery vehicles,2 super hydrophobic surfaces3 and implants.4 Essentially, such a wide range of application requires silicone elastomers with diverse properties. Detailed manuals and informative publications about the commercially available elastomer formulations with customized additives based on their end uses (high performance, pharmaceutical, microfluidic systems, soft stretchable devices, etc.) are available from industry and research groups.5–8 However, the amount of information available in the literature on how to formulate soft and stretchable silicone elastomer networks with electro-mechanical stability seems insignificant. Hence, the tutorial focuses on how to tailor-make soft silicone elastomers to meet different mechanical and electrical requirements that are stable upon repeated deformation cycles. First, a discussion on commercially available silicone elastomers is developed, following which the network structure is discussed briefly. Secondly, every component that goes into the elastomer formulation is explained and the formulation of reliable and durable silicone elastomers from polymeric and oligomeric starting materials are described. Subsequently, elastomer processing is discussed before proceeding to the mechanical properties of soft elastomers. Finally, a description of the peculiar mechanical properties that arise when elastomers are formulated with significant amounts of particulate fillers is elaborated.1.1 Commercial silicone elastomer formulations
Commercial formulations come in a great variety of grades, addressing the commercial demands of the existing technologies. Premade elastomer formulations possess superior mechanical properties and are usually relatively rigid/stiff. Most of the commercial preformulated elastomers are not ideal for application as stretchable electronics, where softness is a key performance indicator. Commercial formulations have the added disadvantage of harbouring materials which are intentionally kept unknown by the industry as a secret recipe. The presence of unknown components in the formulations may sometimes lead to unnecessary complications. It is essential for a researcher to know exactly what is being worked with. An elastomer primarily consists of a network of polymer chains, with the polymer being above its glass transition temperature and being amorphous, along with various additives and fillers. The infinite polymer network defines the elastomeric properties, whereas the additives and fillers provide enhanced tear strength, ultimate tensile properties and ease of processing to the elastomer. A covalent polymer network consists of polymer chains covalently connected either by a crosslinker (addition or condensation curing) or by inter-chain reactions (peroxide curing).In the context of elastomer formulations the general notion is that: “A silicone elastomer formulation consists of a polymer and a crosslinker, and if softer elastomers are desired, the two components are mixed in different ratios.” This notion expresses the simplicity with which silicone elastomers are usually perceived and signifies that silicone producers have indeed managed to prepare complex systems that are extremely easy to use. For mechanically reliable elastomers the formulations are to be used at or near to supplier-specified mixing ratios. Changing the mixing ratio may lead to loss of mechanical integrity and in the worst case result in critical elastomer compositions that are plastic or those that may start flowing over long time scales due to insufficient crosslinking. Such elastomeric networks cannot be regarded as reliable.
Silicone elastomers are typically crosslinked using hydrosilylation (usually Pt-catalysed addition), condensation (usually Sn-catalysed) and radical reaction. In Fig. 1, the two most commonly used curing chemistries are illustrated. Apart from applied chemistry, also temperature is frequently used as means to differentiate between various silicone elastomer types. Based on their curing temperatures, commercial formulations are named as room temperature vulcanising (RTV) and high temperature vulcanising (HTV), which are typically radical cured. A third type of silicone elastomer is named as liquid silicone rubber (LSR/LR), which is usually cured at high temperatures but with a distinction from HTV being that it is formulated to possess lower viscosity. Curing chemistry is not always implied in the naming but it is usually mentioned if the elastomer is based on condensation chemistry, because a certain level of humidity is required for this particular process. The focus here is on addition curing formulations (RTV and HTV), which result in reliable elastomers because the curing reactions are selective and efficient.
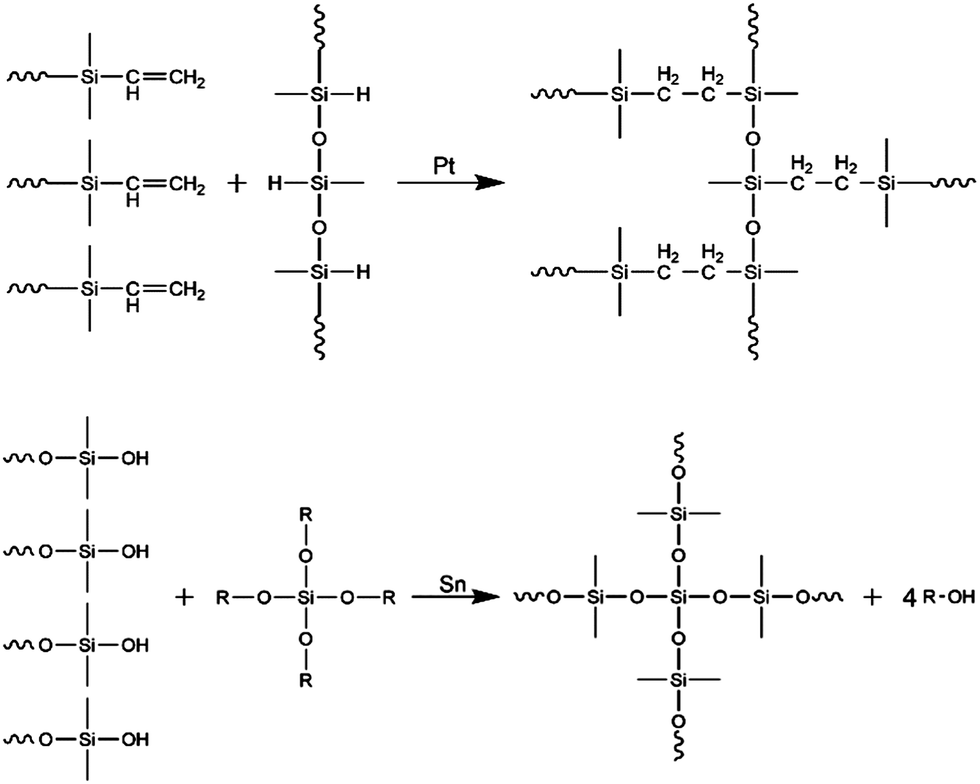 | ||
| Fig. 1 Crosslinking chemistries for silicone elastomers. The upper figure shows the Pt catalysed addition reaction (silylation) of telechelic three-functional polymers with a highly functional crosslinker with three remaining reaction sites. The lower figure shows the Sn-catalysed condensation reaction between four polymers and a 4-functional crosslinker, where volatiles (R–OH) result as a byproduct of the reaction. R indicates an aliphatic group, whereas ∼∼ indicates a connection to a larger ensemble of molecules (network or network sub-structure). Both types of polymers are shown as telechelic, which is the most commonly used polymer configuration. | ||
Typically, a commercially available elastomer kit consists of two premixes of an addition curing system named ‘A’ and ‘B’, where, most commonly, one of them is denoted as a crosslinker and the other a base or a polymer. This denotation, however, does not specify anything about the elastomer and the nomenclature is confusing and misleading for an untrained eye. The crosslinker premix contains a crosslinker (which in rare circumstances may also be polymeric) and some functional polymer (or potentially a silicone oil†), and the polymer premix contains functional polymer amongst other components (Table 1).
| Premix names | Contains at a minimum | May also contain | Precautions to be taken |
|---|---|---|---|
| BASE or POLYMER | Polymer + catalyst | Inhibitor, silica/reinforcing resin | Do not contact with sulphur or other catalyst poison |
| CROSSLINKER | Crosslinker + polymer | Reinforcing resin, silica | Flush container with inert gas after use to avoid hydrolysis |
In the crosslinker premix, the crosslinker and polymer are unreactive and stable, provided that there is no catalyst present and humidity is minimized. The hydride groups on the crosslinker are prone to hydrolysis over time in the presence of water (–Si–H → –Si–OH, Si–OH + Si–OH → –Si–O–Si–), and this reaction is one of the most important reactions in post-curing, i.e. further curing of the cured elastomer over time. The crosslinker premix contains both a crosslinker and a polymer in order to dilute the crosslinker prior to mixing the two A and B premixes. The required amount of crosslinker is slight, due to the combination of high functionality and low molecular weight. Dilution is necessary, since adding drops of pure crosslinker into the base premix may lead to immediate local crosslinking, which can be observed visually as macroscopic lumps forming in the mixture. Without dilution, the diffusion rate of the crosslinker into the base premix will be paltry and reaction will take place around the added drop only. This leads to inhomogeneous structures, and hinders overall macroscopic curing.
The base or polymer premix contains the polymer and a catalyst. Usually, silica particles or resins are added to this premix as well, and they are usually fumed silicas but may also be chemically surface-treated silicas for better compatibility with the silicone polymers. Silicone elastomers with no silica or resins have poor mechanical properties. The polymer premix has usually higher viscosity than the crosslinker premix and usually constitutes a significant larger amount than the crosslinker premix in the final formulation.
1.2 Silicone polymer properties
Silicone elastomers are used in a vast range of applications, due to their inherent flexibility, robustness and biocompatibility, just to mention three of the most favorable properties. Silicone polymers differ from almost all other polymers by having a polymer backbone of alternating units of silicon and oxygen (–Si–O–) rather than the traditional carbon-based backbone. Silicon is tetrahedral in nature, and the most common structure of silicone polymers is polydimethylsiloxane (PDMS), i.e. two side chains of methyl on each silicon. The –Si–O– backbone gives rise to properties different from other polymers, such as high inherent flexibility and softness, as well as high thermal stability amongst many other favorable properties.9 Silicone polymer and all other amorphous polymers above their glass transition temperatures increase significantly their elastic response when crosslinked into an elastomer, due to the covalent nature of the network. The network comprises in theory one molecule only, if ideally crosslinked. This will be discussed further in Section 2. Additionally, silicone polymers and silicone networks are almost entirely amorphous, and therefore there is no wear from microcrystalline domains within the elastomer.Despite silicone elastomers being inherent and excellent electrical insulators, and thereby fundamentally poor conductors, their use as flexible, electrically conducting elastomers has not been ruled out due to the ease of formulation with conductive fillers. Another important property is the integral stability and reliability of properly designed silicone elastomers. Their chemically inert nature enables them to be used in harsh and variable environments. They may swell in good solvents and undergo dimensional changes, resulting in changed mechanical properties, but this will usually take place in a reversible manner. Their operational temperature is from around −100 °C (glass transition temperature, Tg, being around −126 °C) and up to around 200–350 °C, depending on the more exact elastomer composition. Inertness to substances is also required for reliability over time, and silicone elastomers are more or less only prone to degradation by contact with strong bases or acids. Additionally, silicones possess favorable dielectric properties such as high dielectric breakdown strength and high resistivity. However, they suffer from low dielectric permittivity. Despite the later, they are among materials utilised the most frequently in imperceptible, stretchable electronics and lightweight dielectric electroactive transducers.
2 Network structure
In order to understand elastomers’ properties and to design them for specific uses, it is important to understand how reactants and curing affect the final properties. First, there is a focus on the curing chemistries utilised before proceeding to elastomer formulation, whereby additives and fillers are added by the producer to the network to provide enhanced properties.2.1 Ideal networks
An ideal elastomer network is based on irreversible covalent bonds such that elastic behaviour is not transient in the presence of large strains or increased temperatures. When the connections are covalent in nature, the elastomer is denoted as a ‘thermoset’. For thermosets, curing proceeds irreversibly and these elastomers cannot be reheated to allow for remoulding.From a theoretical point of view, the simplest possible polymer network is prepared from the reaction of a crosslinker with more than two reactive groups per polymer chain on average (functionality f > 2) and a polymer with a functionality of 2. The stoichiometry, r, of the reaction, which is a ratio between the two types of reactive groups, is defined as:
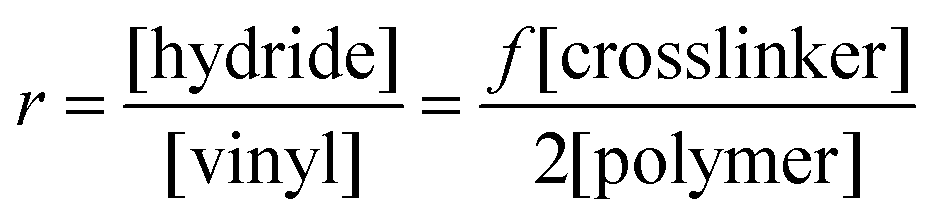 | (1) |
Furthermore, in the simplest case, intra-reactions do not take place and the network formation is based solely on the reaction between the functional groups of the crosslinker and the polymer. Side reactions are assumed to be non-existent. This is usually denoted as a ‘model system’, since network imperfections, such as elastically inactive loops, can be ignored under such ideal conditions. The system of telechelic vinyl-functionalized linear PDMS and an oligomeric (i.e. short) multifunctional hydride-functional siloxane crosslinker is regarded as a model system and is thus ideal for studying the dynamics of polymer networks.11 The reaction of a three-functional crosslinker (3f) is illustrated in Fig. 2.
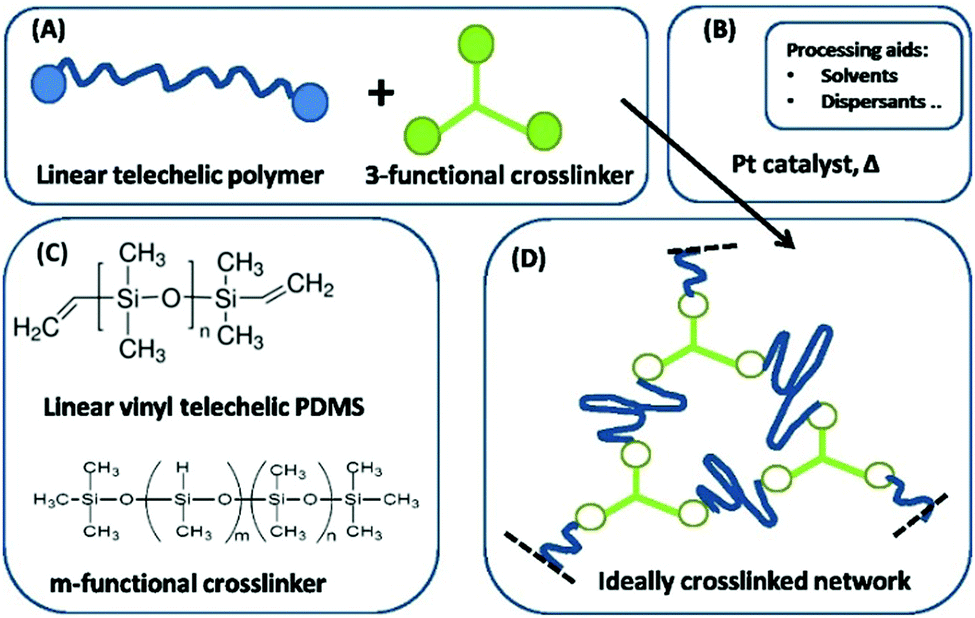 | ||
| Fig. 2 Illustration of a pathway to an ideal network by using a three-functional crosslinker. (A) Reactants. (B) Catalyst. (C) Structure of the utilised polymers. (D) The resulting ideal network consists of one single molecule. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
The involved crosslinking reaction, commonly referred to as ‘hydrosilylation’, between the hydride and the vinyl groups proceeds almost to completion, i.e. high conversion. Side reactions are limited, i.e. the reaction is very selective.11–13 Thereby, the final network consists of elastically active polymer chains connected at both ends to a crosslinker, as illustrated in Fig. 2. The elastic modulus, E, of this ideal network, with elastic chains acting as entropic springs, can then be calculated as:
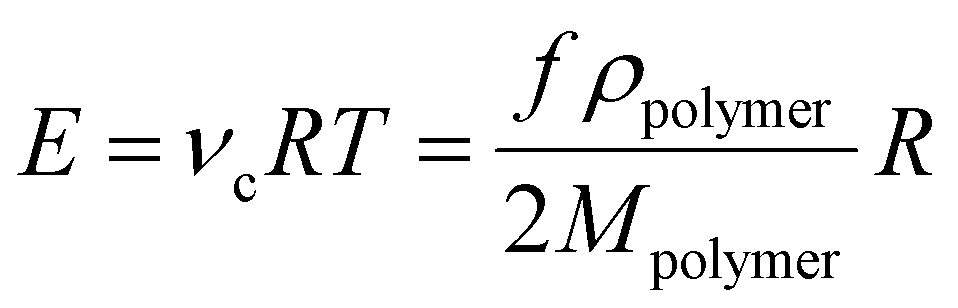 | (2) |
2.2 Imperfect networks and the dynamics thereof
While the network illustrated in Fig. 2D is ideal for visualisation, this is by far not the true structure of real networks, which consist of unreacted reactants due to steric hindrance or as a result of side reactions diminishing the targeted stoichiometric balance. Side reactions and/or stoichiometrically imbalanced reactants (i.e. r ≠ 1) result in dangling polymer chains and substructures as well as a sol fraction, i.e. a fraction not covalently connected to the polymer network,15 as illustrated in Fig. 3 (top figure). If the reactants are mixed in an off-stoichiometric ratio, a true network is not formed. This off-stoichiometric reaction leads, in extreme cases, to a viscoelastic liquid composition consisting of highly branched polymers, and this implies that the gelation threshold has not been exceeded. The gelation threshold (also often denoted the gel point) can be defined as a lower (too much polymer) and higher boundary (too much crosslinker) of r according to the Flory–Stockmayer theory:16,17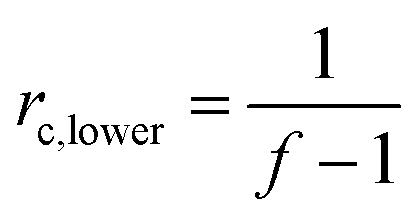 | (3) |
| rc,upper = f − 1 | (4) |
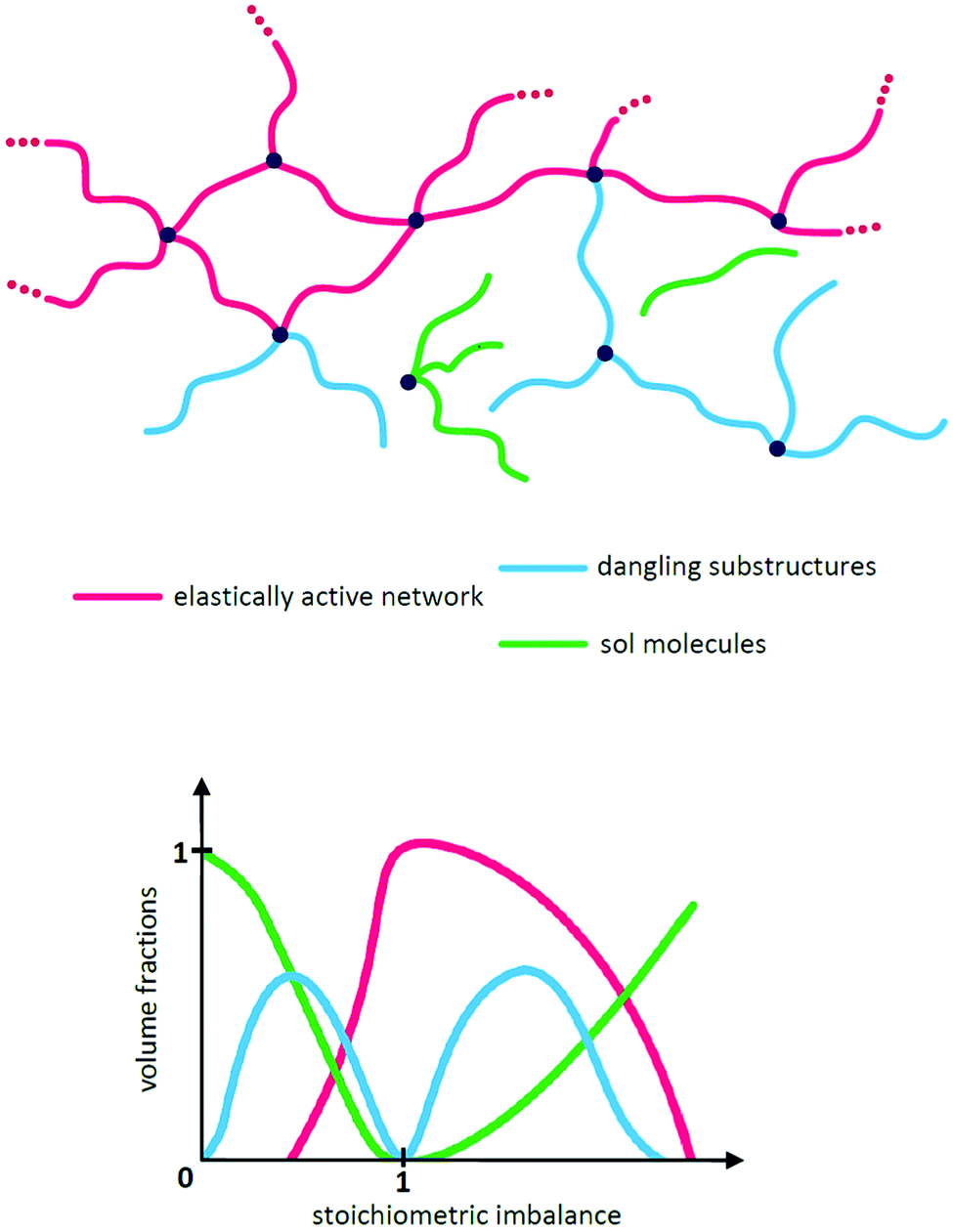 | ||
| Fig. 3 (top) Illustration of the three typical network structures, which are characteristic of every network structure when stoichiometry is not exactly 1. (bottom) Illustration of how the volume fractions of an ideal network structure vary with stoichiometry. At r = 1 there are elastically active chains only (ideally). The two gelation points (as defined in eqn (3) and (4)) are reached when the volume fractions of the elastically active network reach 0. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
An illustration of so-called ‘first order’ imperfections (dangling chains) is given in Fig. 4. Higher order dangling structures (i.e. with multiple sets of constituents in each highly branched structure) become structurally very complex and contribute with very slow dynamics.19 The most complex of these are encountered close to the two gel points, and the most common examples are illustrated in Fig. 5. The sol fraction gives rise mainly to viscous behaviour, but in an entangled state it may cause time-dependent elasticity, which results from the polymer chains not being able to release stress immediately during fast deformation, in which case they act elastically until all stress is released.20
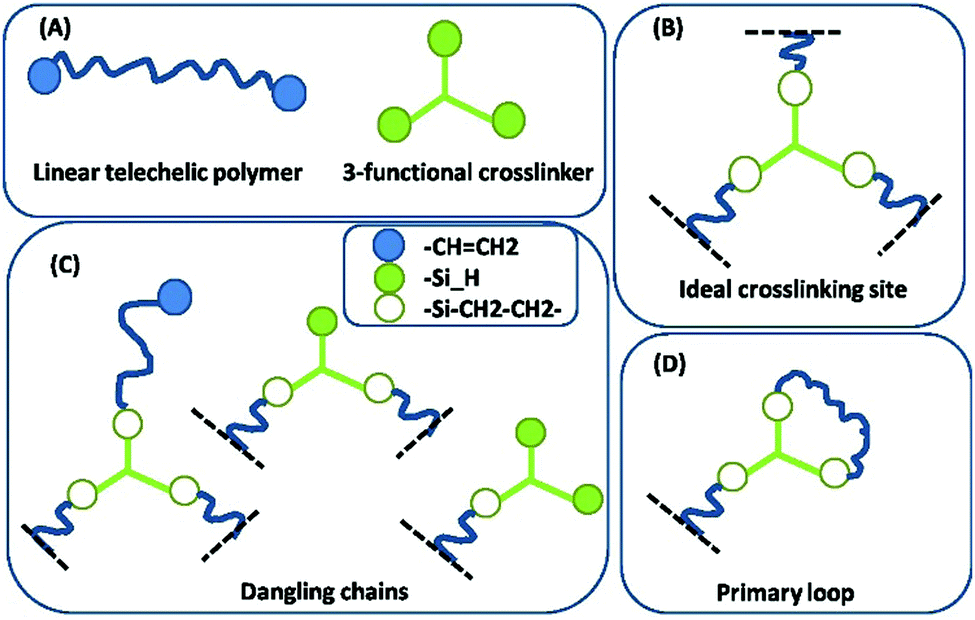 | ||
| Fig. 4 Some common first order imperfections in networks. (A) The two constituents in the network (reactants). (B) An ideal, elastic junction. The black dashed lines indicate connections to the infinite network. (C) Dangling chains of first order, i.e. dangling structures, which consist solely of one of each constituent or a set thereof. (D) A primary inelastic loop. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
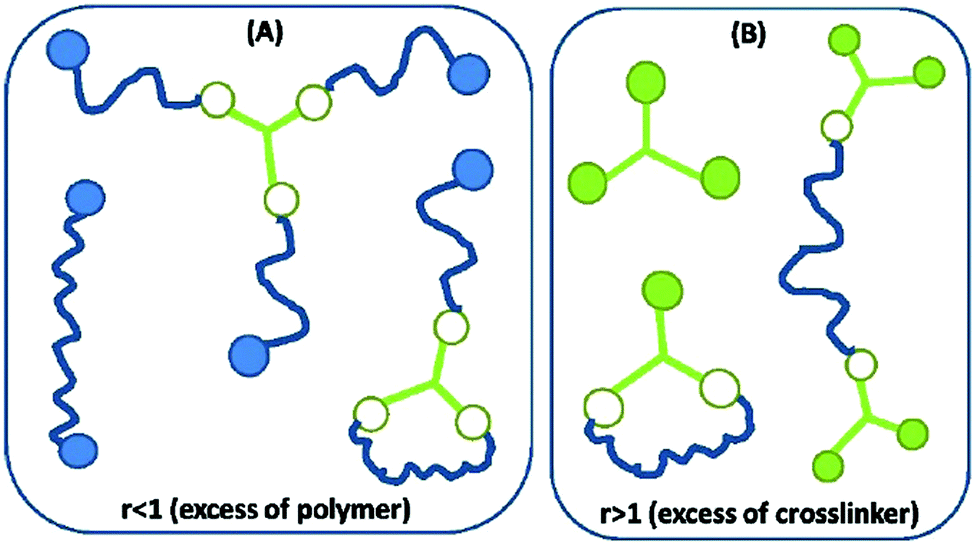 | ||
| Fig. 5 Illustration of the most common sol structures for the two scenarios of (A) excess of polymer and (B) excess crosslinker. Networks resulting from r < 1 possess by far the most complex dynamical behaviour, whereas those resulting from r > 1 may be prone to curing over time when the utilised crosslinker is hydride-functional. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
Most commercial formulations contain a fraction of non-functional or monofunctional polymers (telechelic polymers cannot be prepared with 100% yield) that will also increase the amount of imperfections in the network. Finally, loop formation, i.e. polymers reacting at both ends with the same crosslinker, may occur and lead to loss of the effective number of elastic strands (Fig. 4). All of these network imperfections decrease the effective crosslinking density (and thus also the elastic modulus) and introduce strongly time-dependent (transient) elasticity if the deformation is faster than the given relaxation time of the dangling substructure. Fast deformation results in the substructures acting elastically during the process but relax thereafter implying losses.
Mechanical properties of this complex ensemble of structures are modelled (rubber elasticity models) as a serial or a parallel combination of masses, springs and dashpots to account for behaviour of non-ideal elastomers. Losses are usually modelled as dashpots at slow deformations. The more sol fraction, the more dashpot behaviour. The mechanical behaviours of a mass, a spring and a dashpot are illustrated in Fig. 6.
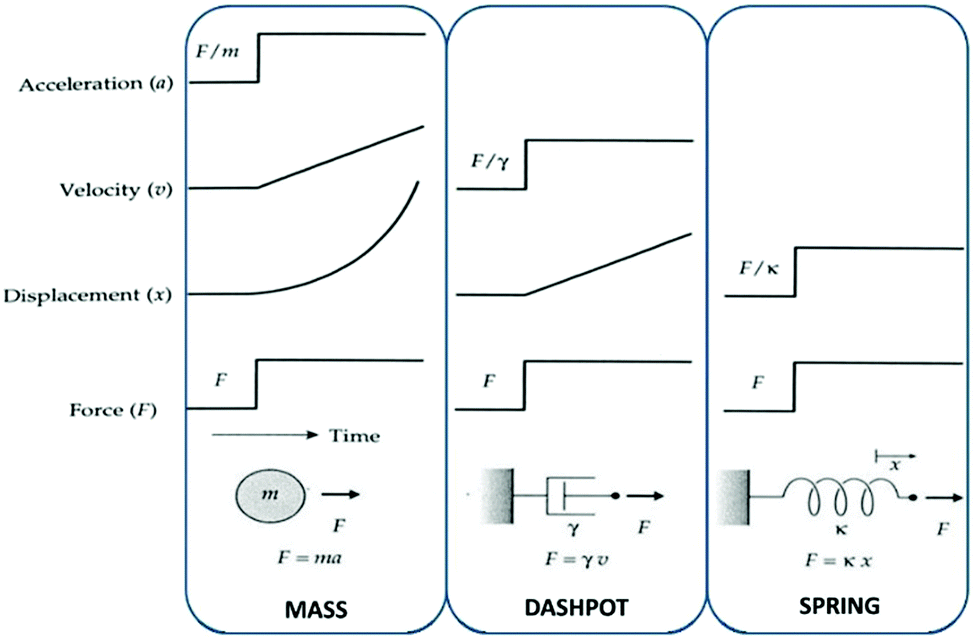 | ||
| Fig. 6 The mechanical behaviour of a mass, a dashpot and a spring. The mechanical properties of elastomers can be modelled as a combination of these three elements. In the ideal case, these mechanical properties arise solely from an entropic spring contribution. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
Models vary a lot in complexity, and for further knowledge regarding modelling rubber elasticity, the article by Arruda and Boyce,21 which contains a thorough description of the nomenclature and rudiments for describing large-strain deformations of elastomers/rubbers is recommended. For small strains (up to at least 10% for most silicone elastomers), the stress–strain relationship can be described by Hooke's law:
| σ = εE | (5) |
3 Formulation of tailor-made silicone elastomers
This section provides recommendations for the design of the network structure, to allow the reader to develop a qualified starting point for formulating the desired silicone elastomer. The raw materials required for making an addition curing elastomer network are (1) a telechelic vinyl terminated PDMS, (2) hydride-functional siloxane crosslinker, (3) Pt catalyst, (4) an inhibitor and (5) suitable fillers, resins or block copolymers. The following section throws light on how to choose each component, considering the properties of the end product.3.1 Designing the network structure
The first part of the formulation process is to decide which elastic modulus is being targeted. It is complicated to formulate reliable silicone elastomers with elastic moduli below 0.6 MPa, since this is the apparent elastic modulus of well-entangled silicone polymers (i.e. silicone polymers above approximately 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). The entanglements in crosslinked polymers behave as crosslinks with more dynamic behaviour, since they can slide in between each other and some of them can also disentangle. Those that can disentangle are commonly denoted as ‘trapped’, whereas ones that cannot do so are denoted as ‘locked’. They give rise to a constant contribution to elasticity when the polymer length is above the entanglement molecular weight. Above this length, the concentration of entanglement remains constant and thus a constant contribution to the modulus. However, recently, so-called ‘bottlebrush’ elastomers have been developed and they bypass this limitation, due to effectively changing the molecular entanglement weight.22,23 More details on entanglements and the characteristic properties of silicone polymers can be found in the article by Fetters et al.24 Alternatively, elastomers can be processed in the presence of a large amount of solvent, in order to dilute locked entanglements25 and thereby proceed below 0.6 MPa. However, this process is not always favorable, since the solvent may swell other components in the devices.
000 g mol−1). The entanglements in crosslinked polymers behave as crosslinks with more dynamic behaviour, since they can slide in between each other and some of them can also disentangle. Those that can disentangle are commonly denoted as ‘trapped’, whereas ones that cannot do so are denoted as ‘locked’. They give rise to a constant contribution to elasticity when the polymer length is above the entanglement molecular weight. Above this length, the concentration of entanglement remains constant and thus a constant contribution to the modulus. However, recently, so-called ‘bottlebrush’ elastomers have been developed and they bypass this limitation, due to effectively changing the molecular entanglement weight.22,23 More details on entanglements and the characteristic properties of silicone polymers can be found in the article by Fetters et al.24 Alternatively, elastomers can be processed in the presence of a large amount of solvent, in order to dilute locked entanglements25 and thereby proceed below 0.6 MPa. However, this process is not always favorable, since the solvent may swell other components in the devices.
It is relatively straight forward to formulate elastomers with the elastic moduli around 1 MPa and above. Silica fillers can be introduced in significant amounts, and so relatively high tear strength and electrical breakdown strength can also be achieved. Suggestions for formulations are given in Table 2. Explanations are provided in the following sections. For targeted elastic moduli below 1 MPa, the importance of tear strength must be evaluated. If tear strength is not a criterium then silica particles can be fully or partly omitted making easier to achieve lower elastic modulus. If tear strength is important, a low Young's modulus is difficult to achieve but it can be achieved by simultaneous entanglement dilution, reduction of fillers and decreased stoichiometry. Suggestions for formulations are given in Table 3, and explanations are given in the following sections.
| Traditional | Optical transparency | Low viscosity | |
|---|---|---|---|
| f = functionality of crosslinker, M = molecular weight of the silicone polymer in premixes (before crosslinking reaction), r = stoichiometry. | |||
| Network | r = 1.1–1.4 | r = 1.1–1.4 | r = 1.1–1.4 |
| f ≥ 8 | f ≥8 | f ≥8 | |
|
M ≈ 20000 to 70000 g mol−1 |
M ≈ 20000 to 70000 g mol−1 |
M < 20000 g mol−1 |
|
| Include | Silica | Resins | Resins/chain extender |
| Exclude | — | Silica | Silica |
| Adhesive | Moderately extensible | Highly extensible | |
|---|---|---|---|
| f = functionality of crosslinker, M = molecular weight of the silicone polymer in premixes (before crosslinking reaction), r = stoichiometry. | |||
| Network | ∼rc,lower < r < 1 | r < 1 | r < 1 |
|
M ≈ 70000 g mol−1 and upwards |
f ≥8 | f = 4–5 | |
|
M ≈ 50000 g mol−1 and upwards |
M ≈ 70000 g mol−1 and upwards |
||
| Include | Resins for better tack (if required) | Possibly resin (low loading) | Silica (low loading) |
| Exclude | Silica | Silica | — |
When optical transparency is desired, it is favorable to use resins rather than silica particles to reinforce the network as there is a noticeable difference in the refractive index between silica and silicone polymers. The formulation with resins is not directly included in this tutorial, but some details on them can be found in Section 5. If very high elongation (up to 5000%) is a key requirement, a novel elastomer system has been developed by Goff et al.26 and commercialized by Gelest as ExSil 100. The underlying principle here is the reaction of a silicone polymer with hydride-functionality at one end and vinyl at the other end. In this instance, in theory, only one big molecule may result from the inherent stoichiometric reaction.
000 g mol−1 and up to M = 60000 g mol−1 (possibly even higher) can be used (e.g. DMS-V31–35 from Gelest) without achieving overly viscous mixtures. The viscosity of silicone polymers increases in line with the molecular weight to a power of approximately 2.4 and in such cases special mixing and dispensing equipment is required. When the viscosity of premixes is to be kept low, then the relatively long polymer chains can be replaced with a shorter polymer and a so-called ‘chain extender’, which again is a telechelic, two-functional ‘crosslinker’. Consequently, in the systems under discussion, this would mean that a telechelic hydride PDMS such as the DMS-H series is mixed with telechelic vinyl PDMS, e.g. DMS-V21–22, both from Gelest, in the presence of the other components. Chain extenders come in a range of viscosities, from 2 to 10000 cSt, to accommodate easy mixing with the rest of the reactants. When a low viscosity silicone is mixed with a high viscosity one, phase separation may take place, and the two can be difficult to mix homogenously. This can be avoided by using a higher molecular weight chain extender, but of course in this case overall viscosity is also increased (which can then be compensated by using lower viscosity versions of the telechelic silicone polymer).
The crosslinker-to-chain extender ratio can be varied, but best results are achieved when the molar ratio is above 0.2, simply because the chain extender may introduce additional imperfections to the network compared to when a long telechelic polymer is used. The lower limit to the molecular weight of the polymer strands, whether resulting from chain extension or a single polymer, is that it should always be above approximately 20000 g mol−1; otherwise, the resulting elastomer may become brittle, simply because the crosslinking of the short chains introduces tension into the resulting elastomer.
3.2 Particulate fillers
Silicone networks with added particulate fillers are often denoted as ‘silicone composites’. We use this nomenclature in the following to distinguish silicone elastomers with particulate fillers from non-filled versions despite the fact that most silicone elastomers contain particulate fillers (i.e. silica). When formulating silicone composites, it is common to use the unit phr (parts per hundred rubber) and denote the concentration of filler as ‘loading’. The phr denotes the amount needed to add to 100 g of elastomer parts rather than recalculating it into weight per cent while working. Silicone polymers have a very low surface tension, thereby making them incompatible with most conventional fillers. Surface modification of fillers by thermal, chemical, electrochemical or coupling agent treatments are employed to achieve compatibility between silicone elastomer and the fillers.27 The necessary loading of filler in the elastomer is decided based on whether or not percolation is required. Percolation means that there is at least one macroscopic connection of particles through the entire macroscopic material. The percolation threshold, defined as exactly one connection, defines a strong change in properties. When a conductive filler is added to a silicone elastomer in low concentrations, the composite will behave in a non-conducting manner, since the particles (or particle agglomerates) are still isolated from one another that they cannot conduct electricity. When the filler concentration exceeds the percolation threshold, then the particles are no longer isolated in the elastomer matrix thus making the composite conductive.28 As filler concentrations increase further above the percolation threshold, the composites become more and more conductive. Hence, if a conductive composite is desired, the filler concentration should be at least as much as the percolation threshold and higher. Caution should be exercised while adding fillers as they significantly increase the elastic modulus and introduce complex behaviour as will be discussed in Section 6. Hence while loading conductive fillers, a balance between elasticity and conductivity should be considered.If permittivity-enhancing particulate fillers are added to non-conducting composites, then compositions approaching the percolation threshold should be avoided. Usually, percolation is determined in low electrical fields (e.g. by measurements in a dielectric spectrometer operating around 1 V) and the percolation threshold will not be the same at higher voltages, as discussed further below. Irrespective of the voltage dependency of the percolation threshold, it is important to know that mixtures close to percolation threshold may contain few domains above percolation threshold, if improperly mixed. So the properties of the composite become dependent on the efficiency of mixing. Therefore, it is always recommended to utilise very conservative loadings of conductive fillers. Filler concentrations below the percolation threshold by a factor of 10 should be used in composites in order to avoid undesired conductivity. Another issue to be concerned about when formulating elastomers with conductive fillers is that charges may build up on the particle surfaces, thereby leading to loss of efficiency.
As mentioned above, when particulate fillers – or other fillers for that matter (excluding silica, which is non-conducting) – are used in the formulation of non-conductive silicone elastomers, it is important to consider if the silicone composites show voltage-dependent conductivity. An example is shown in Fig. 7 for a glycerol-containing composite with around 50 wt% of glycerol (80 and 100 phr, respectively).‡ This composite still remains non-conducting, but this may not be a general conclusion.
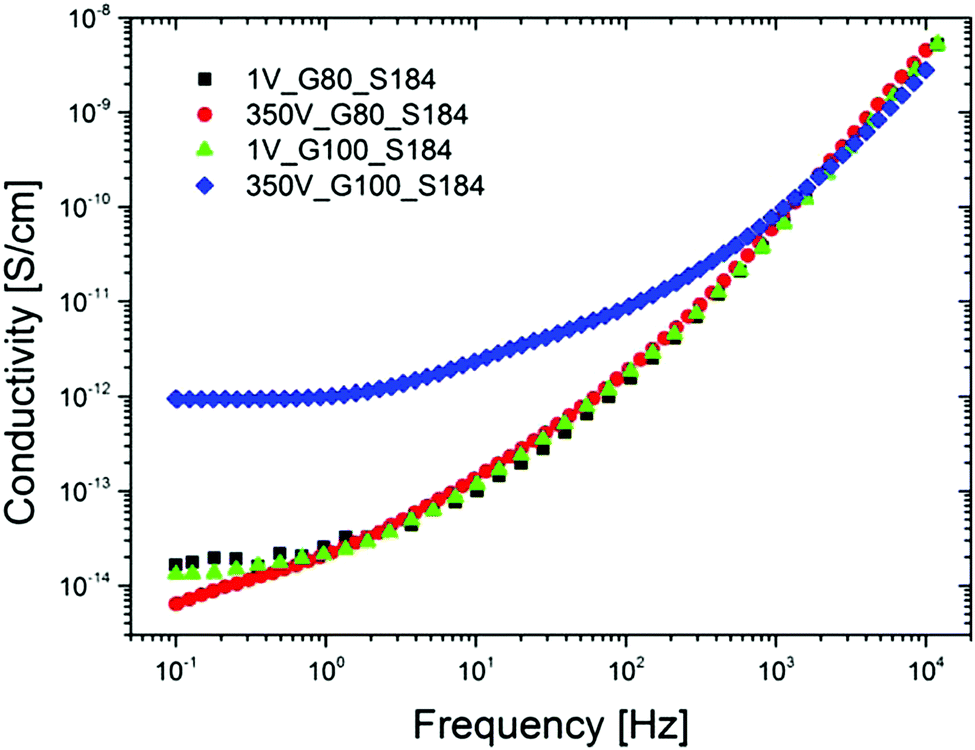 | ||
| Fig. 7 Illustration of the electrical field dependency of silicone elastomers with high loadings of high-permittivity substances (glycerol). Conductivity of various glycerol–Sylgard 184 composites with 80 phr and 100 phr loading, respectively, at 1 V and 350 V and at room temperature.29 Reproduced from ref. 29 with permission from the Wiley Periodicals, Inc., copyright 2016. | ||
While adding particulate fillers to an elastomer network, the stoichiometry of the network may need to be changed, since many fillers may consume some of hydrides of the crosslinker, due to the –OH groups present on the particles. Therefore, more crosslinker may be required, and so it is important to formulate the premixes such that the crosslinker does not react with the particles in the premix before the curing reaction. The exact amount of additional crosslinker required is difficult to quantify, since it depends on the amount and specific properties of the particles, albeit these may change significantly over time if stored in a humid environment.
3.3 Potential of elastomer design
Research within the area of dielectric elastomers has led to the development of many novel approaches to elastomer design. The breakthrough of grafting of high-permittivity moieties to the silicone backbone to attain better electrical sensitivity, e.g. for sensors and actuators was achieved in this context. The recent review by Madsen et al. gives an insight into the advancements of elastomer design in the field of dielectric elastomer transducers.30 Most of the approaches are based on Pt catalysed crosslinking and various clever pathways to circumvent Pt inhibition are proposed. Some research groups also utilise the inhibition to get softer networks, due to the low extent of the reaction. This method cannot be recommended if reliable elastomers are desired. Designing of network structure is crucial to achieve certain properties, and to make sure that the chemistries are efficient; otherwise, properties may not be static and network structures may be poor.Molecular grafting and the overall influence on electro-mechanical properties are still not fully understood. However, it is believed that there is a lot to be learned from the high-voltage industry, where so-called ‘voltage stabilizers’ are added to polyethylene in minute amounts, which increases the electrical breakdown strength by 50%.31 A similar but less significant effect has also been found in silicone elastomers, but identifying the ideal structure of the voltage stabilizer still remains elusive.32 Another interesting observation of the influence of grafted moieties is the change in breakdown patterns in the silicone elastomer films. In Fig. 8, the electrical breakdown zone of a silicone elastomer with a chloropropyl-functional grafted moiety is shown.33 This study demonstrated that there was significant boiling of the elastomer's degradation products during or after the electrical breakdown event. Thereby, a lot of energy was dissipated and the electrical breakdown events never led to tearing, as is otherwise common. This stresses that there are unexplored chemistry-related phenomena that may affect the physical and electrical properties of the silicone elastomers. Furthermore it is evidenced in the article by Yu and Skov and references within34 that the design of the elastomer structure not just determines elastic properties but also electrical properties (e.g. breakdown strength). This is a relatively unexplored area and holds great potential for developing soft silicone elastomers with excellent electrical reliability. For hard silicone elastomers, an increased elastic modulus is a good approach towards higher electrical breakdown strength, but for soft silicone elastomers, it becomes complicated due to electro-mechanical instability. For details on this instability, see the work of Suo and references within.35
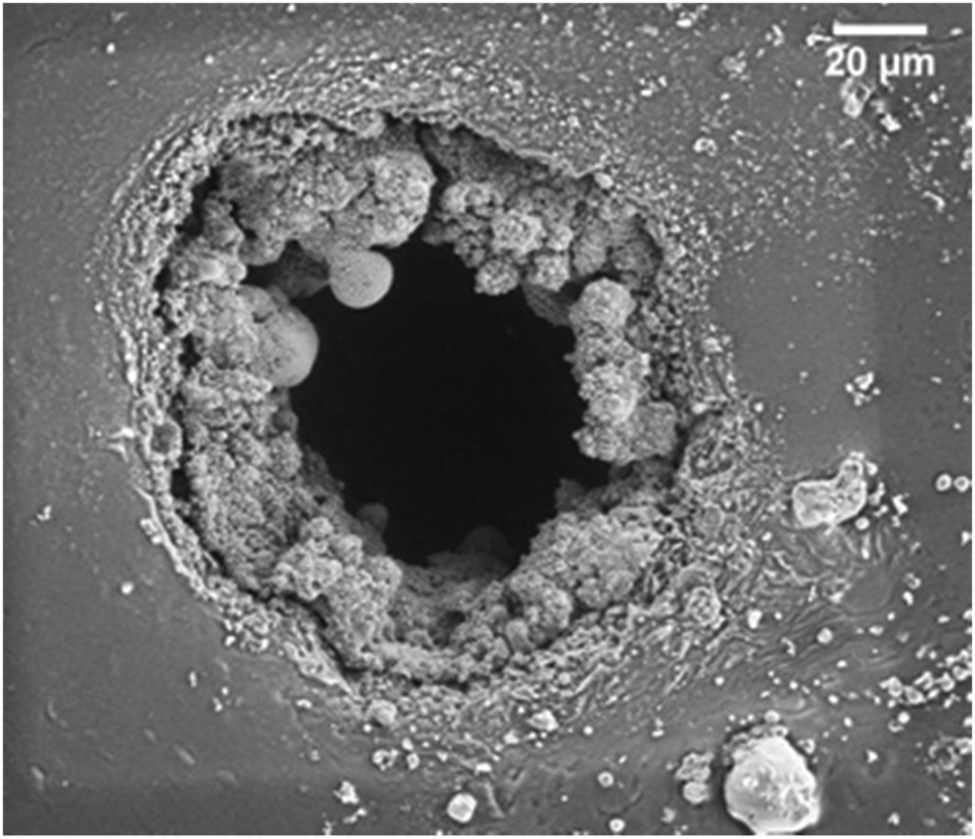 | ||
| Fig. 8 Breakdown zones of a chloropropyl-functional copolymer crosslinked into elastomers (details of elastomers in article).36 Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
4 Processing of silicone elastomers
The precision and perfection in processing determines the quality of the elastomer network. In this section, different scenarios and possible issues when preparing silicone elastomers will be discussed based on the example of an addition cure silicone composition with silica particles as the reinforcing agent.4.1 Homemade formulations
First step is to decide on the type of telechelic PDMS, hydride siloxane crosslinker and other desired fillers that will go into the formulation based on the end properties targeted. Secondly, the ratio/weight of these components needs to be calculated based on the stoichiometry that is required to obtain the target properties of the silicone network. Once the components are weighed accurately, it is recommended that they are mixed into a base called part A (silicone polymer + catalyst + filler + inhibitor) and a crosslinker portion called part B (crosslinker + silicone polymer) just like the commercial premixes. The base and crosslinker portions need not always be labelled as part A and part B, respectively. Mixing all the components together is not recommended as crosslinking will start before the components are well mixed. In part A, apart from the components listed above, other additives like resins and block copolymers can be mixed. Possible chain extenders should be mixed in part B with crosslinker. Silicone oil can be added in part A and part B if necessary. It is recommended that part A is mixed in steps using a speed mixer. The telechelic vinyl PDMS and catalyst should be mixed first, following which the filler should be added and mixed well. Lastly, the inhibitor should be added to part A and mixed well. Part A and part B can now be cold stored before use.4.2 Mixing
Properly mixing part A and part B is the first step in ensuring satisfactory silicone elastomer properties. A distinction between high- (solid-like), medium- (limited flow, e.g. liquid silicone rubber) and low viscosity (pourable compositions, e.g. Sylgard 184 from Dow Corning) silicone elastomers is proposed in this manuscript to make it easier to discuss various mixing scenarios.Addition (hydrosilylation) cure silicone elastomers usually consist of a higher viscosity part (A) and a part (B) with lower or similar viscosity. Blending both parts can be achieved by various methods. Very high viscosity silicones are mixed using, for example, roll mills, while low viscosity silicones are mixed using, for instance, mechanical stirrers (equipped with different stirring paddles), high-power mixers (e.g. Silverson L5M), dual asymmetric centrifuge mixers (often referred to as ‘speed-mixers’) and even by hand (see examples in Fig. 9). Processing high viscosity silicones is usually performed on sophisticated production lines designed specifically for this purpose. These high viscosity silicones are usually applied in the bulk productions where they are extruded (e.g. tubes, custom profiles), injection moulded, compression moulded, or calendared.
 | ||
| Fig. 9 Laboratory-scale mixers. From the left: roll mill with two rolls and two speed mixers. Optimised mixing is required for reliable elastomers, and for most silicone elastomer formulations high shear mixing is strictly required. | ||
Mixing medium viscosity silicones is usually challenging without applying specialized equipment. Various aids facilitating the mixing can be applied, such as the addition of compatible volatile solvents (e.g. OS-10 from Dow Corning) or flow control agents (e.g. Silicone Thinner from Smooth-On, Inc.). Lowering the viscosity of a silicone premix makes it substantially easier to disperse additional fillers in the formulation, e.g. titanium dioxide added to dielectric transducer films to improve dielectric properties. However, formulations prepared with additional solvents typically require an extra solvent removal step. Volatile solvents must be chosen with consideration for the curing temperature. A very rapid evaporation of a solvent during the crosslinking process results in the formation of air voids in the elastomer structure, while too slow evaporation will not eliminate the solvent from the elastomer and will thereby compromise its final mechanical properties. Moreover, it has to be remembered that the addition of non-volatile flow control agents permanently change the ultimate mechanical properties of the elastomer and might lead to unwanted side effects (e.g. sol fraction leaching from elastomer).
Proper mixing of fillers is obtained by applying high-shear forces to the mixture, which will break filler agglomerates and evenly distribute the particles. Homogenization can be facilitated further by applying ultrasound or microwaves, although with the two latter techniques it is important to ensure that the processes do not destroy the polymers and fillers. The process of incorporating additional filler results in a significant viscosity increase of the composition. It is sometimes beneficial to premix a composition by hand (to help ‘wet’ the surface to speed incorporation) to distribute the filler (or other high viscosity additive) before the main mixing process. Moreover mixing of highly viscous compositions often leads to overheating, which might trigger premature crosslinking. This problem can be overcome by cooling the premix in refrigerator before mixing. Silicone premixes do not freeze, due to the low glass transition, but only moderate cooling is recommended, since water from the air may condense on the cold surface of the silicone premix leading to hydrolysis.
Most of the mixing methods lead to the introduction of air bubbles to the premix compositions which must be eliminated for product reliability. A few air bubbles weaken the elastomer mechanically and electrically. Due to the low electrical breakdown strength of air, immediate failure of the elastomer is inevitable when it is used as dielectric elastomer transducer. The conventional means of removing air bubbles involves placing a container with a premixed silicone composition in a vacuum chamber/oven. The low pressure causes the air bubbles to migrate out of the liquid. Intuitively, higher vacuum or longer degassing times have to be applied to higher viscosity compositions. Moreover, it is beneficial to degas the silicone parts before curing because of the high solubility of gases in silicones. It should be considered that a vacuum might eliminate solvents from the composition, which might make subsequent processing steps more complicated. Degassing step is not required when using speed mixers, as the dual centrifuge rotation not only provides high shear forces, facilitating excellent mixing, but also efficiently removes gases from the mixture, thus making it the most efficient mixing tool for silicone formulations of low to medium viscosity. Speed mixers are efficient, since within a few minutes most silicone premixes are ideally mixed and ready to cast.
4.3 Casting of thin elastomeric films
Preparing thin silicone films starts with selecting an appropriate support/carrier web. The support has to be inert and maintain its physical properties and dimensions at the temperature of silicone elastomer curing. The adhesion strength between the silicone elastomer and the support must be lower than the cohesion strength of the film, enabling a smooth release of the elastomer film from the support after curing. Typically, polytetrafluoroethylene (PTFE)-coated polymeric films are used, since the PTFE layer is believed to minimize the adhesion of silicone to the support. In practice, a variety of materials can be used as support, e.g. polyester, polypropylene or poly(methyl methacrylate) (PMMA) sheet. It is advised to attach the carrier web to an additional rigid support, which will eliminate issues related to carrier web deflection (e.g. during transportation), which results in uneven films etc. The carrier web must be cleaned before the formulation can be cast on it. | ||
| Fig. 10 Casting of thin silicone elastomer films, using a commercial film applicator and knife. The bar across the plate can be set to move at different speeds. The knife can be varied in length and gap. Some knifes have built-in chambers for the liquid to be coated. | ||
4.4 Preparing bulk silicone samples
Silicone-based elements with a thickness above 1 mm are considered as bulk samples for the purposes of this tutorial. A multitude of moulds can be applied to manufacture bulk silicone elastomer samples. Flat elements (e.g. 5 mm thick squared pads) are typically prepared by casting a degassed premix composition on a horizontal mould consisting of a rigid bottom support fitted with a frame of desired thickness that defines element dimensions. A proper contact between the support and the frame has to be ensured. Silicone premix is carefully transferred to the mould and spread evenly after eliminating air bubbles by needle or degassing. Sophisticated elements, e.g. microfluidic devices, are manufactured by casting the premix onto a prefabricated mirror-image mould (prepared using photolithography or micro-milling technique). Sylgard 184 is the most frequently reported commercial silicone used for preparing elements with small feature size as this elastomer formulation has excellent flowability, low viscosity, and possesses the ability to reflect even submicrometer size element details.37,38Another method to produce bulk silicone elastomer products involves preparing a vertical mould consisting of two rigid side supports separated with a spacer defining sample thickness. The mould should be fitted with an inlet on top and outlet on the bottom. A silicone premix is poured or injected through the inlet and the composition flows by means of gravity and thereby fills the mould. The silicone excess is collected via the outlet. A horizontal design can also be applied, however, in this case pressure has to be applied to fill the mould with a silicone premix.
4.5 Curing of silicone elastomers
It is always advisable to cure commercial silicone elastomers at temperatures provided by the manufacturer, although curing at other temperatures is also possible. Different curing temperatures often result in elastomers with different mechanical properties.39,40 Typically, the higher the temperature, the higher the elastic modulus. High temperatures initially increase the diffusivity and mobility of polymer chains within the premix, and subsequently lock in entanglements rapidly without allowing for disentangling. Moreover, high temperatures evaporate volatiles such as solvent and additives more efficiently.Pot life is the property of silicones (and other premixes for thermosets) that defines the time from the moment of mixing the silicone premixes up to the point where viscosity is doubled. Pot life is used to define the time allowed for processing the premix and can be extended by applying curing inhibitors (e.g. 1,3-divinyltetramethyldisiloxane from Gelest), which hinder the hydrosilylation reaction before the inhibitor is either evaporated off or degraded.41 Different types of inhibitors deactivate at different temperatures, which allows for precise control over the curing reaction. Contrary to pot life, curing time and required curing temperature can be shortened by adding extra Pt catalyst to the system. The Pt catalyst should be premixed with a silicone base to ensure uniform distribution throughout the composition, as discussed earlier. Some studies recommend post-curing silicone elastomers for several hours at high temperatures, e.g. 200 °C, and for some medical devices this is a requirement,42 because in this way the maximum reactive group conversion is achieved, and all volatiles are released from the system. The curing and post curing at high temperatures increases the elastic modulus of the elastomer and this is not always favorable for stretchable electronics where softness is sought. It has been demonstrated that post-curing various types of silicone elastomers leads to highly improved electrical breakdown strengths which is advantageous for high-voltage applications.43
4.6 Cleaning and storage of silicone samples
Silicone elastomers tend to accumulate electrostatic charges on the surface and thereby attract impurities such as dust. They can be cleaned using a strong stream of air or by washing with water or water with soap. In addition, various solvents can be used to wipe silicone surfaces. However it has to be remembered that most solvents penetrate into the silicone network, and it is therefore advisable to choose appropriate solvents, e.g. according to the study presented by Lee and co-workers.44Although silicones retain excellent mechanical properties over long periods, it is recommended to store silicone elastomer samples at room temperature and with limited exposure to light.
4.7 Processing aids
Although silicone elastomers do not adhere to most of the materials used as a support, problems with demoulding the crosslinked silicone elements is encountered occasionally. In such cases, coating of the carrier web or the mould with a release agent is advised, e.g. Universal Mold Release from Smooth-On, Inc. To promote adhesion between silicone elastomers and other components, adhesion promoters/primers (e.g. Primer G 790 from Wacker Chemie AG) can be applied. The primer ensures good adhesion between a polymer or a non-polymer substrate and the silicone elastomer. Silicone plasticizers (e.g. Plasticizer 100 from Wacker Chemie AG) are added to silicone compositions in order to tune the ultimate mechanical properties of the elastomers as well as to improve the processability of the premix composition. Sometimes, simple non-reactive silicone-based (in rare cases non-silicone-based) oligomers and polymers are used in this regard.5 Mechanical properties of elastomers
A thorough understanding of mechanical properties of silicone elastomers is a prerequisite to direct their usage and to report mechanical data properly for the research community to benefit from one's experiments. For elastomers utilised in stretchable electronics, softness – as mentioned earlier – is key to most applications, but for such materials measurements become more complex than if measuring rigid elastomers. Therefore, it is always important to use a tensile testing machine with the right sensitivity for the materials under investigation. This section serves as an inspiration on how to evaluate mechanical data critically, since a common fault in the literature is to report the ultimate properties with multiple significant numbers relying on one single measurement.For ideal, soft elastomers, there are three primary mechanical properties of interest, namely (1) elastic modulus, (2) either ultimate stress or strain and (3) tear strength. Of course, for non-ideal elastomers the relationship between stress and strain become non-linear and dependent on deformation rate. These phenomena are discussed in more detail in Section 6.
5.1 Stress–strain properties
Silicone elastomers possess viscoelastic properties, i.e. they possess both elasticity and viscous dissipation, with the main contribution being elasticity. In terms of mechanical properties, their elastic moduli are usually very low (of the order of a few MPa as a maximum) and they are very extensible, with ultimate strains usually far above 300%.The ideal polymeric network is illustrated by means of a grid, to show the three-dimensional structure of the network with all reactive groups participating in covalent crosslinks and yielding ideal inter-connections between crosslinkers. Between the crosslinking points the polymer chains are illustrated as elastic strands, visualised as springs connected via the crosslinker. The spring changes its entropy and is thus denoted as an entropic spring. The spring constant of the series of interconnected entropic springs is then the elastic modulus, and thus it is proportional to crosslinking density. This is the basis for the derivation for Hooke's law (eqn (5)).
From the assumption of incompressibility (Poisson's ratio ν = 1/2) of the elastomer, following relation coupling, the elastic modulus (E) and the shear modulus (G) are derived:
| E = 3G | (6) |
5.2 Ultimate properties
The maximum strain and stress are often referred to as ‘ultimate’ properties. In general, for silicone elastomers, a softer elastomer results in larger ultimate strain and lower ultimate stress.In contrast to elastic moduli, the two ultimate measures are not entirely material parameters but rely strongly on the number of imperfections. A poorly cured elastomer may result in a significant higher maximum strain and lower maximum stress than the ideally cured version, while a poorly processed silicone elastomer where, for example, air was introduced into the composition during the coating process, may result in both lower ultimate stress and strain. Therefore, variations in ultimate property values are expected, and when determining these measures the assessment must be based on at least three to five measurements, to provide any valuable information.
In Fig. 11 the ultimate properties of 8 samples of identical LSR elastomers are shown. The elastomer is difficult to process, due to very high viscosity, so it is diluted with significant amounts of solvent. Evaporating the solvent may cause defects in the network if curing is not occurring ideally, and as can be observed from the measurements herein, one out of eight elastomer films had significant amounts of defects leading to lower ultimate properties. The seven other films show similar properties to each other in the sense that ultimate strains are within a 600–700% range and ultimate stresses within 10–13 MPa. Due to the processing influence, the results appear somewhat dispersed, however, such discrepancies are considered acceptable by the silicone research community. Additives alter the elastomer matrix in various degrees and while enhancing one property, they may compromise other properties. For every such scenario, the optimum amount of additive must be arrived at by analysing the effect of the additive on the various mechanical properties.
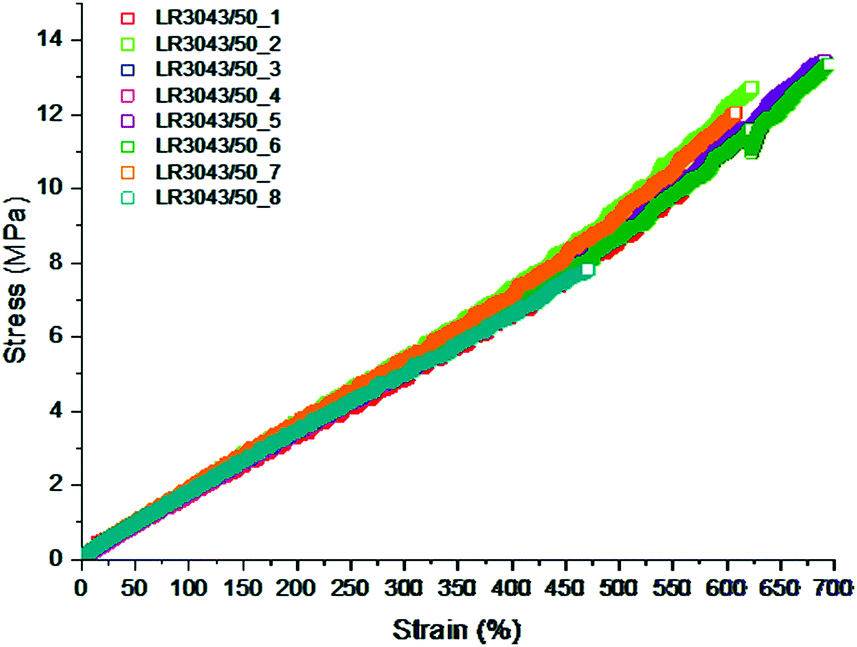 | ||
| Fig. 11 Ultimate properties of Wacker Chemie LR3043/50 determined from eight stress–strain experiments. The maximum extension falls between 600 and 700% with a single outlier at 450% as well as the ultimate stress ranges from 10 to 13 MPa with the same single outlier at 8 MPa. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
5.3 Tear strength
Tear strength (or tear resistance) is a measure of how well a material can withstand an imposed, sharp incision under mechanical load. The value becomes crucial in processing, especially when thin films are processed, as well as in applications where the silicone elastomer is in close contact with sharp objects. Also, tearing is a particular problem for dielectric elastomers after electrical breakdowns, as illustrated in Fig. 12, since this trauma introduces stress gradients, and thereby tearing propagates from the microscopic pinhole. A recent review by Park et al. deals with improving the tear strength of commercial silicone elastomers, and it is recommended for further information about the tear properties of various commercial elastomers mixed in different ratios.8 | ||
| Fig. 12 A commonly encountered result of electrical breakdown in silicone elastomers is a macroscopic mechanical failure, whereby the thin elastomer film is completely torn apart. The picture shows dielectric breakdown in a film which was under small load only. It leads to a microscopic pinhole, which then may propagate mechanical failure. The presence of enhanced local stresses at the cracks around the breakdown zone, which is solidified to some extent, leads to tear propagation and usually also complete film failure. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
The tear strength of silicone elastomers is commonly improved by adding fumed silica, albeit this increases the elastic modulus significantly, so the trade-off for a more tear-resistant elastomer is stiffness. There are no alternatives to silica if the tear strength of the elastomer is not to be compromised to some extent. For consistent tear strength, interactions between fillers and polymer network need to be strong i.e. of hydrogen-bonding strength. It is more or less only silica fillers and various types of silicone resins that have suitable strong interactions with the silicone polymer network. Resin-reinforced silicone elastomers usually possess lower tear strength than silica-reinforced elastomers. Some resins facilitate good tear resistance properties while having less impact on the elastic modulus. A silicone resin is usually an irregular three-dimensional network with principally tri- or tetra-functional units of chemical composition RnSiXmOp of molecular weights from 1000 to 10000 g mol−1, where R is methyl or phenyl, and X is hydrogen (H), hydroxyl (OH) or alkoxy group (OR). The resin TEOS (tetraethyl orthosilicate) is one of the cleaner versions, whereas the traditionally used MDTQ, DT and MQ resins contain significant amounts of salt. M stands for Me3SiO, D for Me2SiO2, T for MeSiO3 and Q for SiO4. The drawback with resin-based elastomers is the high content of salts, due to the reaction path necessary to produce them. Even in highly purified resins, some impurities remain, and this may be observed as a change in electrical properties when high DC voltage is applied for extended periods. Similar to resin-reinforced elastomers, bimodal elastomers can be prepared to possess very good tear resistance despite possessing low elastic modulus.46 These two methods will not be discussed in detail here, though, due to most resins being rather impure (and thus more conducting) along with the complexity of formulation for both types of elastomer.47
6 Non-linearity of reinforced silicones
For very soft elastomers, shear experiments are easier to perform than extensional experiments, and it is therefore convenient to determine the elastic modulus from oscillatory shear with a given angular frequency (ω). This method, though, must be used with precaution when dealing with filled elastomers. From shear experiments, one obtains the elastic contribution from the material (opposing force), which is denoted as the storage modulus, G′(ω), as well as the loss modulus, G′′(ω), which is the energy dissipated. However, the correlation in the case of filled elastomers should be specified strictly to:| E = 3G0′ | (7) |
1. Mullins effect: the filled elastomers remember the maximum extension that they have experienced. The higher the maximum extension a sample has experienced, the lower the elasticity, see Fig. 13.
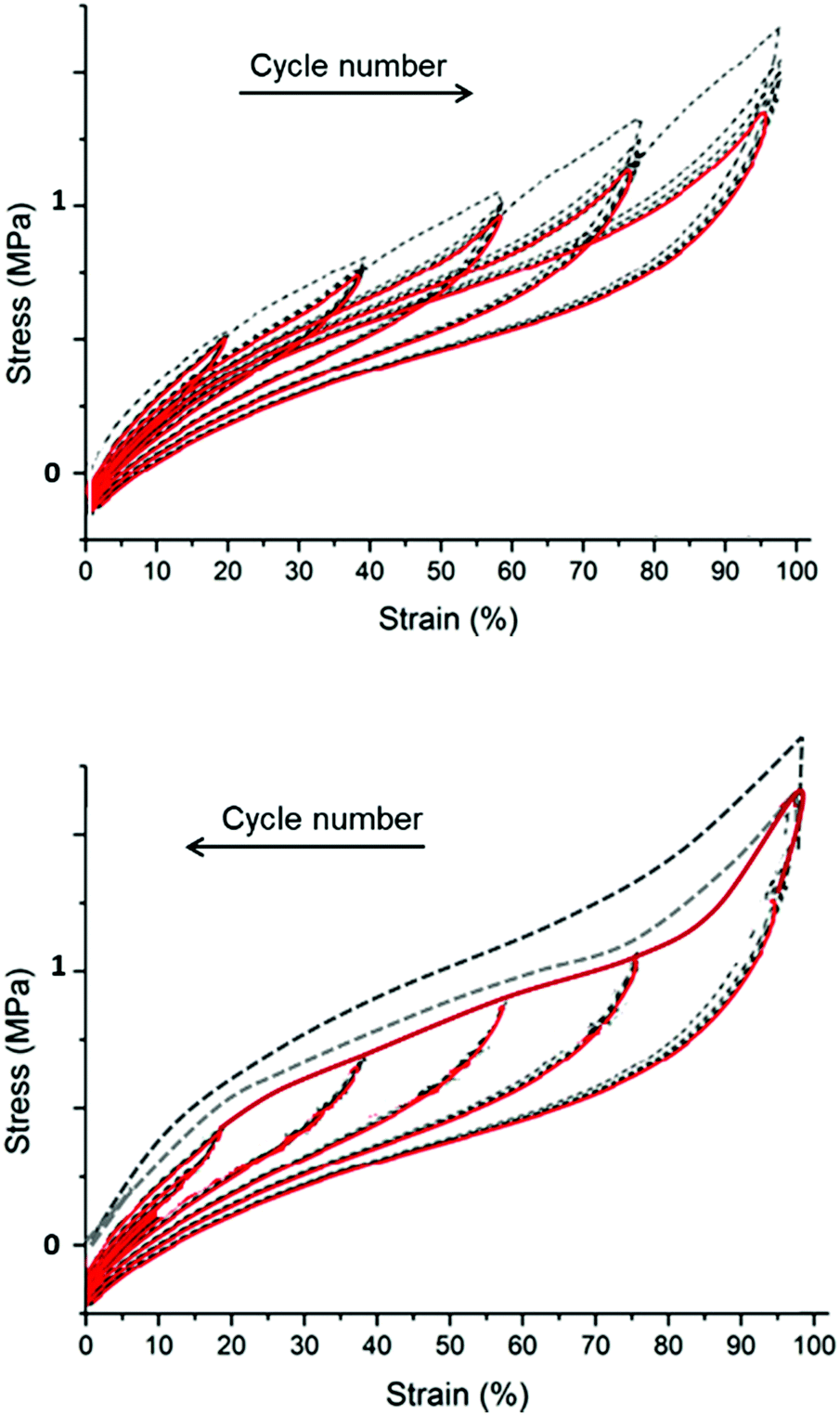 | ||
| Fig. 13 The Mullins effect on mechanical deformation cycles. The top figure shows repeated cycling to a given deformation followed by repeated cycling to a higher strain, whereas the lower figure shows repeated cycling but with decreasing strains utilised. The dotted curves are the initial cycle, whereas the red curves are the subsequent four cycles. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
2. Payne effect: the filled elastomer has two plateaus in the shear moduli. At small deformation rates, the particles act in an elastic manner and contribute significantly to elasticity, see Fig. 14.
 | ||
| Fig. 14 Shear rheology experiment displaying the Payne effect. Shear modulus decreases with increasing strain, eventually reaching the second plateau. The first plateau is usually below 1% for silicone elastomers, and the second plateau usually proceeds up to around 10%. Unfilled elastomers and most other materials have one plateau only. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
3. Permanent set: the filled elastomer does not return to the original zero-stress at no strain after it has been held at a given strain for an extended period of time. Rather, it extends a few per cent due to reorganizing the elastomer and potentially also post-curing in the extended state, see Fig. 15.
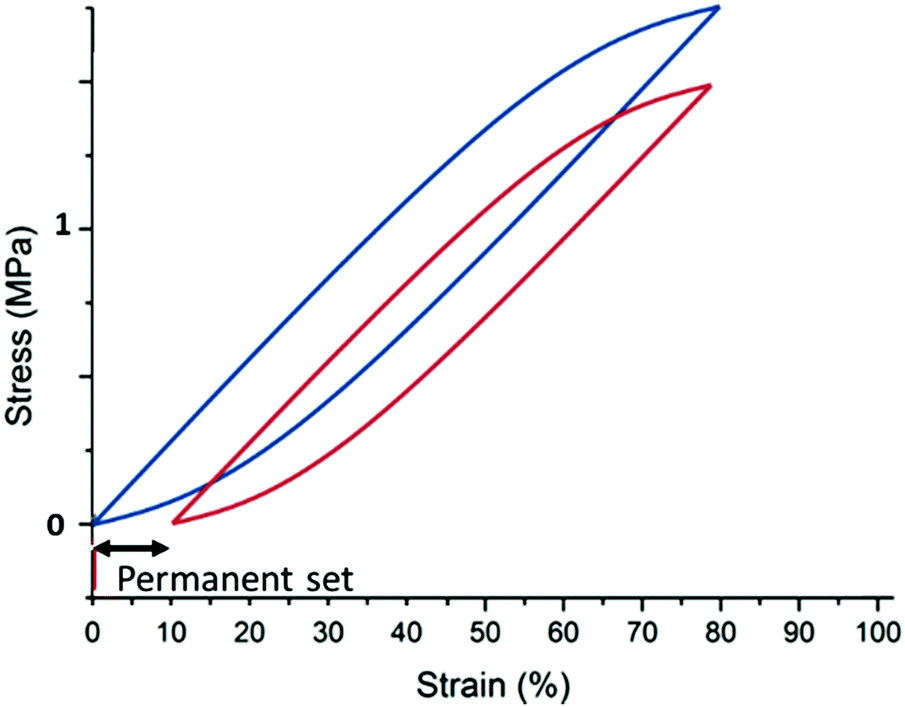 | ||
| Fig. 15 Illustration of a permanent set. The sample is initially cycled to 75% strain (blue curve) and then taken to a given strain (usually higher than 50% strain) and rested for some time (usually a day or more). The sample is then cycled again to 75% strain (red curve), but the zero stress state is now at 10% strain. This resting introduces the reorganization of the elastomer structure such that when released from the resting position, there will be an opposing force with a zero-stress condition at the rested strain. Reproduced from ref. 14 with permission from the Technical University of Denmark (DTU), copyright 2017. | ||
The Mullins effect is illustrated in Fig. 13. In the top figure, all cycles in steady-state conditions have different elastic moduli. The first deformation cycle indicates a stronger material than indicated by subsequent cycles, simply because the first time the elastomer experiences a strain greater than what it originally has experienced, it softens. When cyclic deformations are performed with decreasing strains, the elastomer has identical elastic moduli in all cycles, since the elastomer experiences the maximum strain in the initial cycle.
The Payne effect is illustrated in Fig. 14, which shows how a filled elastomer possesses two linear viscoelastic (LVE) regimes (i.e. where the modulus does not depend on the applied strain rate). For non-filled elastomers, this phenomenon is not encountered and the LVE is identified much more easily.
The permanent set of filled elastomers is illustrated in Fig. 15. It is a result of reorganizing or curing the elastomer in static stretched conditions, and it is evident that upon stretching for an extended time, the elastomer may elongate by a few per cent and in critical situations more than 10%. For the latter situation, the elastomer is so plastic in nature that it will most likely continue elongating in other situations. The reason for the elongation upon stretching is that hindered reaction sites may become accessible for crosslinking reactions, or complicated substructures may relax in an irreversible manner. Therefore, part of the elastomer gains a new zero-stress condition, which opposes the retractive force when it is released. The permanent set – despite its name – is not always permanent, and the elastomer may return to its original zero-stress strain within some days/weeks.
Expressed in terms of the stress response in the small deformation regime (also often denoted linear viscoelastic regime), this means that the stress has three contributions, namely:
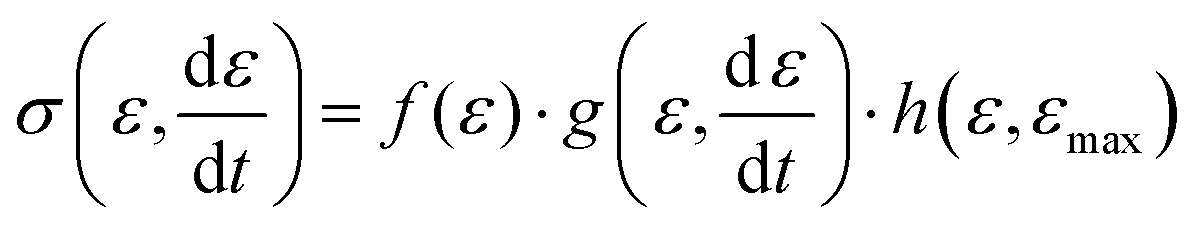 | (8) |
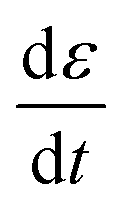 is the applied strain rate. f(ε) is the simple linear and strain rate-independent response of the sample when all the non-linear effects are ignored, i.e. εE.
is the applied strain rate. f(ε) is the simple linear and strain rate-independent response of the sample when all the non-linear effects are ignored, i.e. εE. 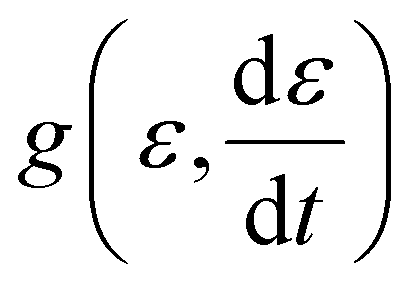 indicates that stress depends on both the strain and the strain rate (Payne effect), which is also common for other materials but not in the small strain limit. The function h(ε,εmax) can be regarded as softening from the Mullins effect (sample history effect).
indicates that stress depends on both the strain and the strain rate (Payne effect), which is also common for other materials but not in the small strain limit. The function h(ε,εmax) can be regarded as softening from the Mullins effect (sample history effect).
Practical precautions with respect to the three non-linear phenomena
For reproducibility and reliability, the elastomer should always be stretched significantly beyond what it has experienced during processing, thereby ensuring that the elastomer films have experienced the same maximum strain. In practice, elastomers with strong Payne effects in dynamic applications operated at high speeds may possess a certain strain rate at which the elastomer softens strongly. This needs to be addressed in the design, either by utilising this strong non-linearity favorably or by eliminating it. For determining the elastic properties of elastomers, it is important to ensure that the obtained LVE diagram corresponds to the utilised frequency in the application. The permanent set must be evaluated and be considered in design of products. Since permanent set strongly increases with particle loading, low filler concentrations should be used if possible. More detailed explanations of the three phenomena can be found in the ref. 48–50 and references therein.7 Conclusions and future outlooks
Silicone elastomers are widely investigated within the silicone industry, but this knowledge is not easily accessible for academia and thus the knowledge about elastomer formulating remains relatively unexplored in this sphere. This can be attributed to the dependency on the commercial silicone elastomers, but with many new silicone-based technologies emerging, e.g. stretchable electronics, commercial elastomers are experiencing a bit of a shortfall. It is essential and advantageous for researchers to formulate their own silicone elastomer recipes for ongoing research in order not to be constrained by commercial formulations. The correct formulation of silicone elastomers allows for a wide range of mechanical properties while maintaining mechanical integrity. Within this tutorial review, we concentrated on how to design soft, robust silicone elastomers by means of ensuring that a suitable network structure exists. A suitable network structure can only be achieved when the stoichiometry is altered in a controlled way and the catalyst concentration is maintained at a suitable level, to allow for efficient and fast curing. This is not always the case when commercial elastomer premixes are mixed in different ratios resulting in unreliable networks.The formulation strategy explained in this tutorial is only a starting point and can be expanded easily to incorporate silicone copolymers of various types and to include additives and fillers. The properties of silicone elastomers can be varied drastically by designing the network and additives suitably. The number of choices for elastomer constituents is very high and allows for a wide range of unexplored possibilities for self-formulated silicone elastomers.
Abbreviations
| D | Me2SiO2 |
| DC | Direct current |
| HTV | High temperature vulcanising |
| LR/LSR | Liquid (silicone) rubber |
| LVE | Linear viscoelastic |
| M | Me3SiO |
| Me | Methyl (–CH3) |
| PDMS | Polydimethylsiloxane |
| phr | Per hundred rubber |
| PMMA | Poly(methyl methacrylate) |
| PTFE | Polytetrafluoroethylene |
| Q | SiO4 |
| RTV | Room temperature vulcanising |
| T | MeSiO3 |
| TEOS | Tetraethyl orthosilica |
| Wt% | Weight per cent |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Independent Research Council Denmark. Frederikke Bahrt Madsen is acknowledged for commenting on the manuscript.Notes and references
- M. G. Mohammed and R. Kramer, All-Printed Flexible and Stretchable Electronics, Adv. Mater., 2017, 29, 1604965, DOI:10.1002/adma.201604965.
- P. Mazurek, M. A. Brook and A. L. Skov, Glycerol–Silicone Elastomers as Active Matrices with Controllable Release Profiles, Langmuir, 2018, 34, 11559–11566, DOI:10.1021/acs.langmuir.8b02039.
- H. Zhou, H. Wang, H. Niu, A. Gestos, X. Wang and T. Lin, Fluoroalkyl silane modified silicone rubber/nanoparticle composite: a super durable, robust superhydrophobic fabric coating, Adv. Mater., 2012, 24(18), 2409–2412, DOI:10.1002/adma.201200184.
- C. T. K. Khoo, Silicone synovitis: the current role of silicone elastomer implants in joint reconstruction, J. Hand. Surg. Eur. Vol., 1993, 18(6), 679–686, DOI:10.1016/0266-7681(93)90222-2.
- Concise Encyclopedia of High Performance Silicones, ed. A. Tiwari and M. D. Soucek, Scrivener Publishing, 2014 DOI:10.1002/9781118938478.
- K. Mojsiewicz-Pieńkowska, Review of Current Pharmaceutical Applications of Polysiloxanes (Silicones), Handb. Polym. Pharm. Technol., 2015, 2, 363–381, DOI:10.1002/9781119041412.ch13.
- M. P. Wolf, G. B. Salieb-Beugelaar and P. Hunziker, PDMS with designer functionalities—Properties, modifications strategies, and applications, Prog. Polym. Sci., 2018, 83, 97–134, DOI:10.1016/j.progpolymsci.2018.06.001.
- S. Park, K. Mondal and R. M. Treadway, et al., Silicones for Stretchable and Durable Soft Devices: Beyond Sylgard-184, ACS Appl. Mater. Interfaces, 2018, 10(13), 11261–11268, DOI:10.1021/acsami.7b18394.
- J. C. Carpenter, J. A. Cella and S. B. Dorn, Study of the degradation of polydimethylsiloxanes on soil, Environ. Sci. Technol., 1995, 29(4), 864–868, DOI:10.1021/es00004a005.
- A. S. Fawcett and M. A. Brook, Thermoplastic silicone elastomers through self-association of pendant coumarin groups, Macromolecules, 2014, 47(5), 1656–1663, DOI:10.1021/ma402361z.
- H. H. Winter and F. Chambon, Analysis of Linear Viscoelasticity of a Crosslinking Polymer at the Gel Point, J. Rheol., 1986, 30(2), 367–382, DOI:10.1122/1.549853.
- M. A. Villar, M. A. Bibbo and E. M. Valles, Influence of Pendant Chains on Mechanical Properties of Model Poly(dimethylsiloxane) Networks. 1. Analysis of the Molecular Structure of the Network, Macromolecules, 1996, 29(11), 4072–4080, DOI:10.1021/ma9506593.
- M. A. Villar and E. M. Vallés, Influence of pendant chains on mechanical properties of model poly(dimethylsiloxane) networks. 2. Viscoelastic properties, Macromolecules, 1996, 29(11), 4081–4089, DOI:10.1021/ma9506602.
- A. L. Skov, Silicone-based Dielectric Elastomers, 2017, https://findit.dtu.dk/en/catalog/2353448628 Search PubMed.
- S. M. G. Frankær, M. K. Jensen, A. G. Bejenariu and A. L. Skov, Investigation of the properties of fully reacted unstoichiometric polydimethylsiloxane networks and their extracted network fractions, Rheol. Acta, 2012, 51(6), 559–567, DOI:10.1007/s00397-012-0624-z.
- P. J. Flory, Molecular Size Distribution in Three Dimensional Polymers. I. Gelation, J. Am. Chem. Soc., 1941, 63(11), 3083–3090, DOI:10.1021/ja01856a061.
- W. H. Stockmayer, Theory of Molecular Size Distribution and Gel Formation in Branched Polymers II. General Cross Linking, J. Chem. Phys., 1944, 12(4), 125–131, DOI:10.1063/1.1723922.
- S. Sun, M. Li and A. Liu, A review on mechanical properties of pressure sensitive adhesives, Int. J. Adhes. Adhes., 2013, 41, 98–106, DOI:10.1016/j.ijadhadh.2012.10.011.
- A. L. Larsen, K. Hansen and P. Sommer-Larsen, et al., Elastic properties of nonstoichiometric reacted PDMS networks, Macromolecules, 2003, 36(26), 10063–10070, DOI:10.1021/ma034355p.
- A. L. Skov and L. Yu, Optimization Techniques for Improving the Performance of Silicone-Based Dielectric Elastomers, Adv. Eng. Mater., 2017, 20, 1700762, DOI:10.1002/adem.201700762.
- E. M. Arruda and M. C. Boyce, A three-dimensional constitutive model for the large stretch behavior of rubber elastic materials, J. Mech. Phys. Solids, 1993, 41(2), 389–412, DOI:10.1016/0022-5096(93)90013-6.
- J. Paturej, S. S. Sheiko, S. Panyukov and M. Rubinstein, Molecular structure of Bottlebrush polymers in melts, Sci. Adv., 2016, 2(11), 1601478, DOI:10.1126/sciadv.1601478.
- L. H. Cai, T. E. Kodger, R. E. Guerra, A. F. Pegoraro, M. Rubinstein and D. A. Weitz, Soft Poly(dimethylsiloxane) Elastomers from Architecture-Driven Entanglement Free Design, Adv. Mater., 2015, 27(35), 5132–5140, DOI:10.1002/adma.201502771.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Connection between Polymer Molecular Weight, Density, Chain Dimensions, and Melt Viscoelastic Properties, Macromolecules, 1994, 27(17), 4639–4647, DOI:10.1021/ma00095a001.
- A. L. Larsen, P. Sommer-Larsen and O. Hassager, Dynamic dilution effects in polymeric networks, e-Polymers, 2006, 42, 1–11, DOI:10.1515/epoly.2006.6.1.555.
- J. Goff, S. Sulaiman, B. Arkles and J. P. Lewicki, Soft Materials with Recoverable Shape Factors from Extreme Distortion States, Adv. Mater., 2016, 28(12), 2393–2398, DOI:10.1002/adma.201503320.
- S. Rezaei and B. Pourabbas, In situ generation of the surface-modified silica nanoparticles within the silicone matrix, Polym. Compos., 2015, 36(7), 1365–1370, DOI:10.1002/pc.23044.
- T. Q. Trung and N. Lee, Flexible and Stretchable Physical Sensor Integrated Platforms for Wearable Human-Activity Monitoring and Personal Healthcare, Adv. Mater., 2016, 28(22), 4338–4372, DOI:10.1002/adma.201504244.
- P. Mazurek, L. Yu, R. Gerhard, W. Wirges and A. L. Skov, Glycerol as high-permittivity liquid filler in dielectric silicone elastomers, J. Appl. Polym. Sci., 2016, 133, 1–8, DOI:10.1002/app.44153.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, The current state of silicone-based dielectric elastomer transducers, Macromol. Rapid Commun., 2016, 37(5), 378–413, DOI:10.1002/marc.201500576.
- Y. Yamano and H. Endoh, Increase in breakdown strength of PE film by additives of azocompounds, IEEE Trans. Dielectr. Electr. Insul., 1998, 5(2), 270–275, DOI:10.1109/94.671957.
- A. H. A. Razak and A. L. Skov, Silicone elastomers with covalently incorporated aromatic voltage stabilisers, RSC Adv., 2017, 7(1), 468–477, 10.1039/c6ra25878f.
- L. Yu, F. B. Madsen and A. L. Skov, Degradation patterns of silicone-based dielectric elastomers in electrical fields, Int. J. Smart Nano Mater., 2017, 1, 1–16, DOI:10.1080/19475411.2017.1376358.
- L. Yu and A. L. Skov, Molecular Strategies for Improved Dielectric Elastomer Electrical Breakdown Strengths, Macromol. Rapid Commun., 2018, 1800383, DOI:10.1002/marc.201800383.
- Z. Suo, Theory of dielectric elastomers, Acta Mech. Solida Sin., 2010, 23(6), 549–578, DOI:10.1016/S0894-9166(11)60004-9.
- F. B. Madsen, L. Yu, A. E. Daugaard, S. Hvilsted and A. L. Skov, A new sof dielectric silicone elastomer matrix with high mechanical integrity and low losses, RSC Adv., 2015, 5, 10254–10259, 10.1039/C4RA13511C.
- S. Park, H. Kim and M. Vosgueritchian, et al., Stretchable energy-harvesting tactile electronic skin capable of differentiating multiple mechanical stimuli modes, Adv. Mater., 2014, 26(43), 7324–7332, DOI:10.1002/adma.201402574.
- J. J. Wang, J. W. Tsai and Y. C. Su, Piezoelectric rubber films for highly sensitive impact measurement, J. Micromech. Microeng., 2013, 23(7), 075009, DOI:10.1088/0960-1317/23/7/075009.
- I. D. Johnston, D. K. McCluskey, C. K. L. Tan and M. C. Tracey, Mechanical characterization of bulk Sylgard 184 for microfluidics and microengineering, J. Micromech. Microeng., 2014, 24, 035017, DOI:10.1088/0960-1317/24/3/035017.
- F. Schneider, T. Fellner, J. Wilde and U. Wallrabe, Mechanical properties of silicones for MEMS, J. Micromech. Microeng., 2008, 18, 065008, DOI:10.1088/0960-1317/18/6/065008.
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley & Sons, New York, 2000 DOI:10.1021/ja004838y.
- M. A. Brook, H. U. Saier, J. Schnabel, K. Town and M. Maloney, Pretreatment of liquid silicone rubbers to remove volatile siloxanes, Ind. Eng. Chem. Res., 2007, 46(25), 8796–8805, DOI:10.1021/ie061666q.
- S. Zakaria, F. B. Madsen and A. L. Skov, Post Curing as an Effective Means of Ensuring the Long-term Reliability of PDMS Thin Films for Dielectric Elastomer Applications, Polym.-Plast. Technol. Eng., 2017, 56(1), 83–95, DOI:10.1080/03602559.2016.1211689.
- J. N. Lee, C. Park and G. M. Whitesides, Solvent Compatibility of Poly(dimethylsiloxane)-Based Microfluidic Devices, Anal. Chem., 2003, 75(23), 6544–6554, DOI:10.1021/ac0346712.
- T. K. Kim, J. K. Kim and O. C. Jeong, Measurement of nonlinear mechanical properties of PDMS elastomer, Microelectron. Eng., 2011, 88(8), 1982–1985, DOI:10.1016/j.mee.2010.12.108.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, Visualisation and characterisation of heterogeneous bimodal PDMS networks, RSC Adv., 2014, 4(14), 6939–6945, 10.1039/c3ra47522k.
- A. G. Bejenariu, L. Yu and A. L. Skov, Low moduli elastomers with low viscous dissipation, Soft Matter, 2012, 8(14), 3917–3923, 10.1039/c2sm25134e.
- L. Mullins, Softening of Rubber by Deformation, Rubber Chem. Technol., 1969, 42(1), 339–362, DOI:10.5254/1.3539210.
- A. R. Payne, The dynamic properties of carbon black-loaded natural rubber vulcanizates. Part I, J. Appl. Polym. Sci., 1962, 6(19), 57–63, DOI:10.1002/app.1962.070061906.
- L. R. G. Treloar, Crystallisation phenomena in raw rubber, Trans. Faraday Soc., 1941, 37, 84–97, 10.1039/tf9413700084.
Footnotes |
| † The use of oil as a crosslinking diluent is possible and plausible. To our knowledge, however, it is not applied in any commercial formulation. |
| ‡ The filler shown here is not particulate, but data on the voltage dependency of conductivity are scarce due to the cost of the utilised instrument. Particulate fillers will behave in a similar way, since charges can build up on the surface of the particles. |
| This journal is © The Royal Society of Chemistry 2019 |