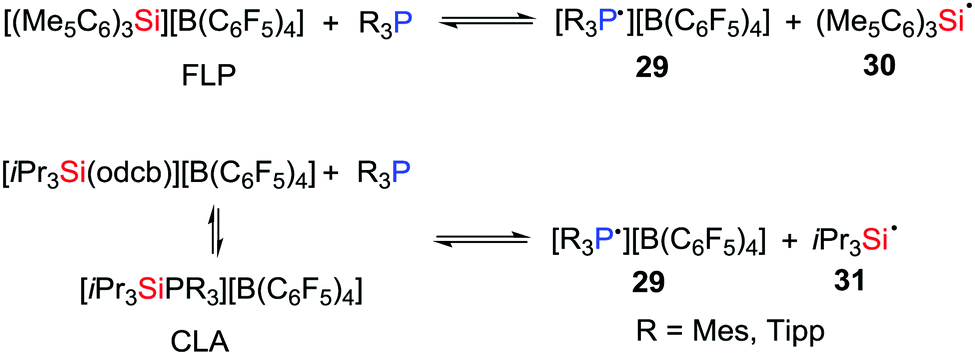DOI:
10.1039/C8CS00940F
(Tutorial Review)
Chem. Soc. Rev., 2019,
48, 3454-3463
Radicals derived from Lewis acid/base pairs
Received
27th November 2018
First published on 6th February 2019
Abstract
While conventional approaches to stabilizing main group radicals have involved the use of Lewis acids or bases, this tutorial review focuses on new avenues to main group radicals derived from combinations of donor and acceptor molecules. Reactions involving the use of both classical Lewis acid–base adducts and frustrated Lewis pair systems are discussed and the subsequent reactivity is considered.

Liu Leo Liu
| Liu Leo Liu is a post-doctoral fellow in Prof. Stephan's group at the University of Toronto. Prior to this, he received his PhD degree in 2016 from a joint program under the supervision of Prof. Yufen Zhao at Xiamen University (2011–2013, 2015–2016) and Prof. Guy Bertrand at University of California, San Diego (2013–2015). He was awarded the National Scholarship for outstanding PhD students in 2014 from the Chinese Ministry of Education. He has published over 50 peer-reviewed papers, and his research spans synthetic and computational chemistry with the focus of unusual main group compounds. |

Douglas W. Stephan
| Douglas W. Stephan began independent career at the University of Windsor (1982). In 2008 he moved to the University of Toronto as a Professor and Canada Research Chair. A highly successful researcher in inorganic chemistry/catalysis, he is best known for articulating the concept of “frustrated Lewis pairs”. Stephan has received a number of awards. He was named a Fellow of the Royal Society of Canada (2005), a Fellow of the Royal Society (2013, London), a Corresponding Member of North-Rhein-Westfaelia Academy (2014, Germany), and an Einstein Visiting Professor at the Technical University of Berlin (2016–2019) and made the Thomson Reuters Highly Cited List (2014–2018). |
Key learning points
1. The development of Lewis pair chemistry has contributed to advancements in generation of persistent or transient radicals.
2. Frustrated radical pairs have been shown to cleave chemical bonds in a homolytic fashion.
3. Single electron delivery to Lewis pairs leads to transient radical anions that split a variety of chemical bonds.
4. A variety of stable radicals have been achieved via reactions involving frustrated Lewis pairs or classical Lewis adducts.
|
Introduction
In 1923, Gilbert N. Lewis classified electron-pair donors and electron-pair acceptors as Lewis bases (LBs) and Lewis acids (LAs) respectively.1 Since then, classical Lewis acid–base adducts (CLAs) derived from the two electron donation from a Lewis base (LB) to a Lewis acid (LA) were thought to be thermodynamically stable and generally unreactive. This paradigm changed in 2006 with the discovery that a reversible activation of H2 could be mediated by p-(Mes2P)C6F4(B(C6F5)2) (Mes = mesityl).2 In this case, the Lewis basic phosphorus and acidic boron centres are sterically encumbered and this precludes the dative bond formation permitting the reaction with H2. Subsequently this observation was generalized to combinations of LAs and LBs with appropriate steric hindrance to deter dative bonding. Indeed, access to dissociative equilibria has proved sufficient to such frustrated Lewis pairs (FLPs) to react with a wide variety of substrates,3–6 as well as to effect metal-free hydrogenation catalysis.
Over the last decade the above development of FLP chemistry has been predicated on the notion that the chemistry results from two-electron processes in which the Lewis bases and acids act as two electron donors and acceptors respectively. However, a number of recent studies have either inferred or demonstrated the role of radical pathways in reactions involving Lewis acids and bases, both in FLP chemistry and beyond. At the same time, carbon-based radicals have an extended history dating from the landmark work of Gomberg more than one century ago.7 In contrast, main group radicals have received much less attention. The conventional approaches to stabilizing main group radicals has involved the use of LAs or LBs. For example, LA-complexation of a radical donor has been known for some time8 and the reactivity of such species has been exploited more recently.9,10 The complementary approach in which LBs are used to stabilize radicals has also been employed. For example, several recent studies have used singlet carbenes to stabilize main group radicals11–14 In this tutorial review, we focus on new avenues to main group radicals derived from combinations of donor and acceptor molecules. Reactions involving the use of both CLA and FLP systems are discussed and the subsequent reactivity of the derived radicals is considered.
Donor–acceptor induced homolysis
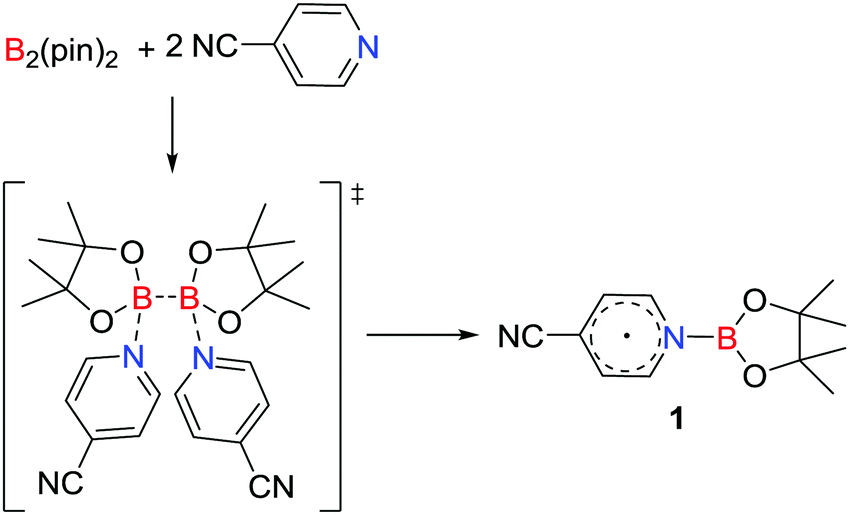
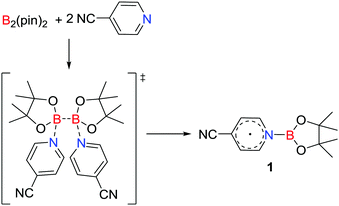
A unique approach to the generation of a main group radical involves base induced homolysis of an element–element bond. In 2016, Li, Zhu and co-workers15 described such a strategy involving the transient formation of a donor–acceptor adduct derived from the addition of two equivalents of 4-cyanopyridine to B2(pin)2. This prompted homolytic cleavage of the B–B bond affording the pyridine-ligated boryl radical [NCC5H4NBpin]˙ 1 (Scheme 1). The donor 4-cyanopyridine behaves as a one-electron acceptor, reminiscent of homolytic cleavage reactions of a base stabilized diborane (4).16
 |
| | Scheme 1 Synthesis of a pyridine-ligated neutral boryl radical 1via a homolytic cleavage of the B–B bond in B2(pin)2. | |
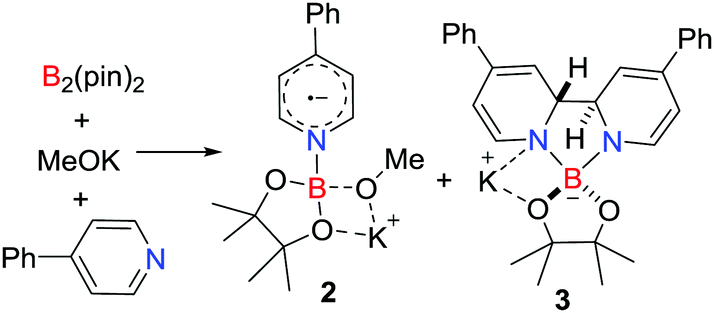
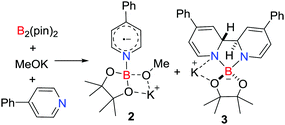
In extending this approach Jiao and Zhang showed that the combination of B2(pin)2, MeOK and 4-phenylpyridine afforded the novel pyridine-boryl species [PhC5H4NBpin(OMe)˙][K] 2 and [(PhC5H4N)2Bpin][K] 3 (Scheme 2).17 In the absence of MeOK, no radical was observed, inferring that the pyridine-boryl adduct reacts with MeOK to prompt the homolytic B–B bond cleavage to produce 2 and 3. These species behave as super electron donors, and can be used in catalytic radical borylation reactions and reductive cleavage reactions.17,18
 |
| | Scheme 2 Synthesis of 2 and 3. | |
Radical capture by FLPs
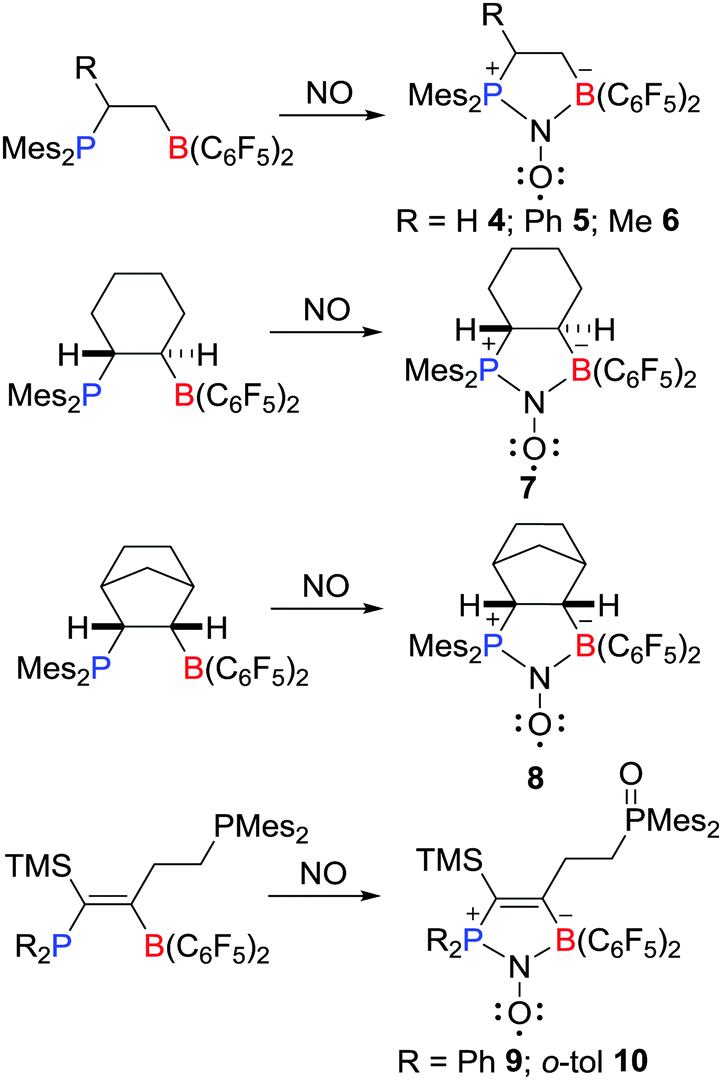
Recently, Erker, Grimme, Warren et al. prepared a novel type of aminoxyl radical 4via the reaction of an intramolecular FLP Mes2PCH2CH2B(C6F5)2 with NO (Scheme 3).19,20 Compound 4 was fully characterized including single crystal X-ray diffraction. The Mulliken spin densities of O and N atoms in 4 are 0.54 and 0.34 a.u., respectively. Shortly thereafter, Erker and co-workers reported analogous reactions leading to a variety of related aminoxyl radicals 5–10 (Scheme 3).20–22
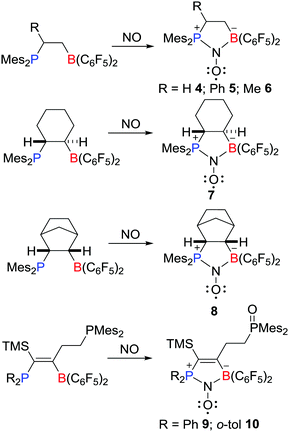 |
| | Scheme 3 Examples of aminoxyl radicals derived from reactions of FLPs with NO. | |
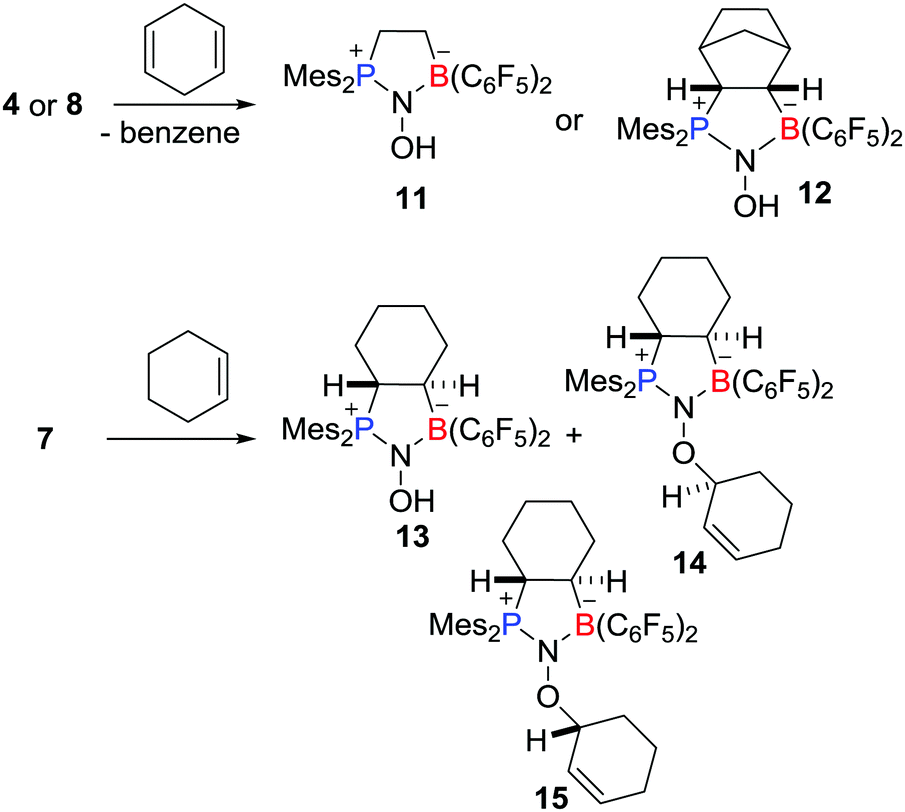
These aminoxyl radicals were shown to engage readily in H-atom abstraction reactions (Scheme 4). For example, compounds 4 and 8 rapidly reacted with 1,4-cyclohexadiene to afford the corresponding diamagnetic products 11 and 12, respectively, along with the formation of benzene. Compound 7 underwent abstraction of H-atoms with cyclohexene to give 13 and a pair of diastereoisomers of 14 and 15.
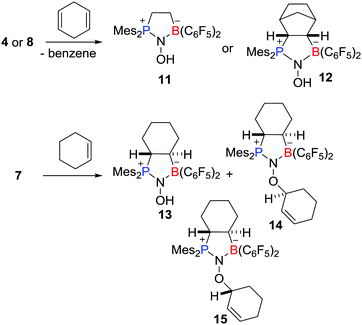 |
| | Scheme 4 Examples of hydrogen atom abstraction reactions of aminoxyl radicals. | |
Frustrated radical pairs
A triarylamine radical cation
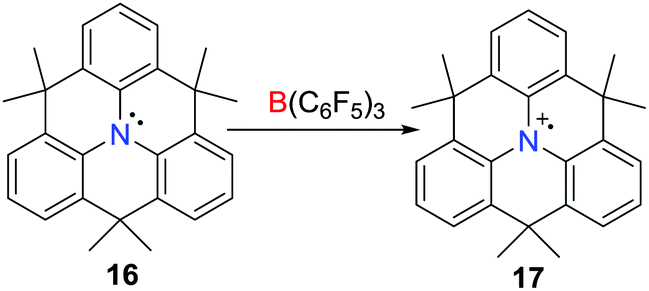
Although the one-electron oxidation of transition-metal complexes by B(C6F5)3 has been described,23,24 Wang et al. reported the first example of a one-electron oxidation of a triarylamine (C6H3CMe2)3N 16 by B(C6F5)3 in 2013 (Scheme 5),25 affording the deep blue triarylamine radical cation 17. Although the salt of the weakly coordinating alanate 17[Al(ORF)4] (ORF = OC(CF3)3) was isolated, the formation of the boron centred radical anion in the reaction with B(C6F5)3 was not confirmed as this species decomposes rapidly (vide infra).26
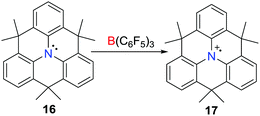 |
| | Scheme 5 Synthesis of 17via a one-electron oxidation by B(C6F5)3. | |
Phosphine/Lewis acid frustrated radical pairs (FRPs)
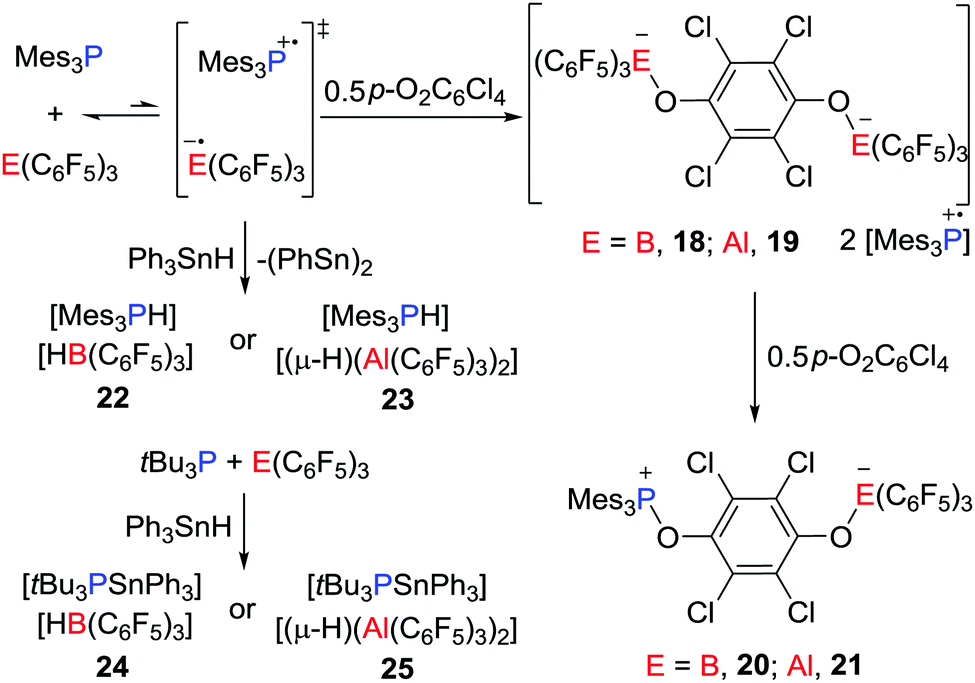
Stephan and co-workers have recently reported evidence for a mechanism for FLP reactivity that involves a radical pathway.27 Upon dissolving Mes3P and E(C6F5)3 (E = B or Al) in very dry toluene or chlorobenzene, the formation of a trace amount of the purple-colored [Mes3P˙]+ was observed. Although the EPR spectrum of the purple solution generated from Mes3P/B(C6F5)3 displayed a weak signal, the solution generated from the corresponding Al reaction revealed a room-temperature doublet EPR resonance with a g value of 2.0089 and a hyperfine coupling constant of 238 G. This confirmed the presence of [Mes3P˙]+, however, the presumed radical anion [˙E(C6F5)3]− was not observed as it was rapidly quenched by reactions with solvent.25,26 Nonetheless, these data support the notion of single-electron transfer (SET) equilibria between the FLP and FRP of the form [Mes3P˙][˙E(C6F5)3]. Generation of these FRPs in the presence of tetrachloro-1,4-benzoquinone (p-O2C6Cl4) prompts the formation of the radical salt [Mes3P˙]2[(C6F5)3EOC6Cl4OE(C6F5)3] (E = B, 18; Al, 19) (Scheme 6).
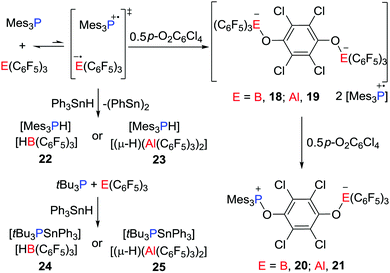 |
| | Scheme 6 Reactivity of R3P/E(C6F5)3 toward p-O2C6Cl4 and Ph3SnH. | |
These species react further with p-O2C6Cl4 to give the 1,6-addition products [Mes3POC6Cl4OE(C6F5)3] (E = B, 20; Al, 21) (Scheme 6). Alternatively, the FRPs homolytically split the Sn–H bond in Ph3SnH to give [Mes3PH][HB(C6F5)3] 22 or [Mes3PH][(μ-H)(Al(C6F5)3)2] 23, respectively (Scheme 6), along with (Ph3Sn)2. In sharp contrast, the combination of tBu3P/E(C6F5)3 shows no evidence of radical formation. Indeed, addition of Sn–H to these FLPs prompts heterolytic Sn–H cleavage affording [tBu3PSnPh3][HB(C6F5)3] 24 and [tBu3PSnPh3][(μ-H)(Al(C6F5)3)2] 25, respectively (Scheme 6). This contrasting behaviour demonstrates that FLPs can react either via consecutive one-electron steps or via a classic two electron process.
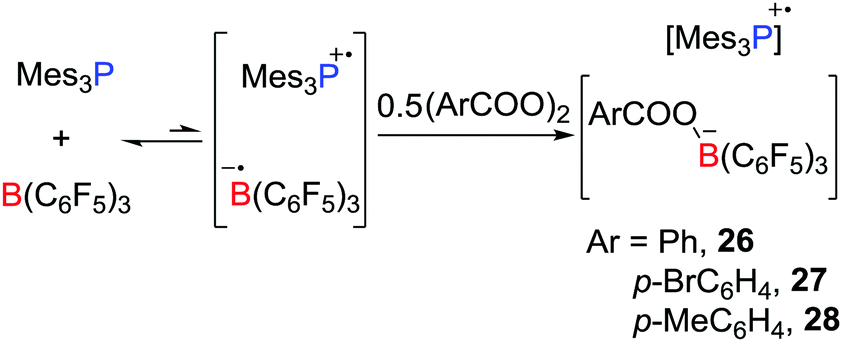
In a recent and related study, the reactivity of the Mes3P/B(C6F5)3 pair with benzyl peroxide derivatives was shown to proceed to efficiently split the O–O bond in a homolytic fashion, giving radical salts [Mes3P˙][PhCOOB(C6F5)3] 26, [Mes3P˙][p-BrC6H4COOB(C6F5)3] 27 and [Mes3P˙][p-MeC6H4COOB(C6F5)3] 28 (Scheme 7).28 Control experiments showed that the combination of B(C6F5)3 with the peroxides gave an adduct mixture. These reactions represent the first examples where FLPs react via a SET process affording the homolytic cleavage of a homodiatomic bond. It is also noteworthy that related phosphoniumyl radical cations [Tipp3P˙]+ (Tipp = 2,4,6-triisopropylphenyl) and [Tipp2MesP˙]+ were prepared and structurally characterized by Wang et al.29 employing the weakly coordinating anions such as [Al(ORF)4]− (ORF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OC(CF3)3) and [Al(ORMe)4]− (ORMeOC(CF3)2Me).
OC(CF3)3) and [Al(ORMe)4]− (ORMeOC(CF3)2Me).
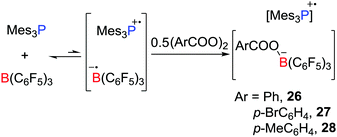 |
| | Scheme 7 Reactivity of Mes3P/B(C6F5)3 toward benzyl peroxide derivatives. | |
In 2018, Müller, Klare et al. demonstrated that a SET as a viable reaction pathway for silylium ion/phosphine Lewis pairs (Scheme 8). For example, in solution, both FLPs consisting of R3P/[(Me5C6)3Si][B(C6F5)4] (R = Tipp or Mes) and CLAs derived from the combination of R3P/[iPr3Si(odcb)][B(C6F5)4] (odcb = 1,2-dichlorobenzene) promote the establishment of a SET equilibrium mixture with the corresponding radical pairs 29–31, indicating that complete frustration of the Lewis pairs is not necessarily a prerequisite for such SET processes.
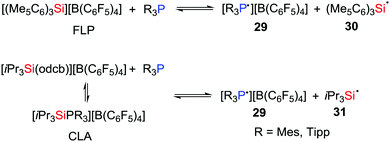 |
| | Scheme 8 One-electron oxidation of phosphines in silylium ion/phosphine Lewis pairs. | |
Single electron transfer in a Lewis acid–base reaction
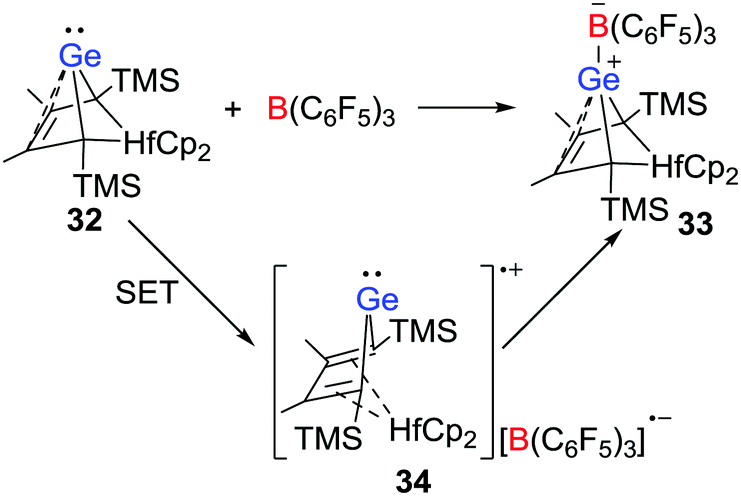
More recently, the group of Müller disclosed that a Lewis acid–base reaction between the nucleophilic hafnocene-based germylene 32 and B(C6F5)3 leading to a Ge-B CLA 33 is initiated by a SET process (Scheme 9).30 The radical salt 34 generated by a SET from the germylene to B(C6F5)3 was identified by EPR and UV-vis spectroscopy. In addition, the nature of the radical cation was confirmed via independent synthesis of the [B(C6F5)4]− salt via the reaction of 32 with [Ph3C][B(C6F5)4]. This work provided evidence for a SET in CLA formation reactions.
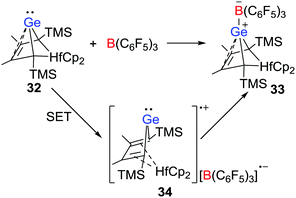 |
| | Scheme 9 Synthesis of 33via a radical complex 34. | |
Electron transfer in FLP-reactions
C–H activation by FRPs
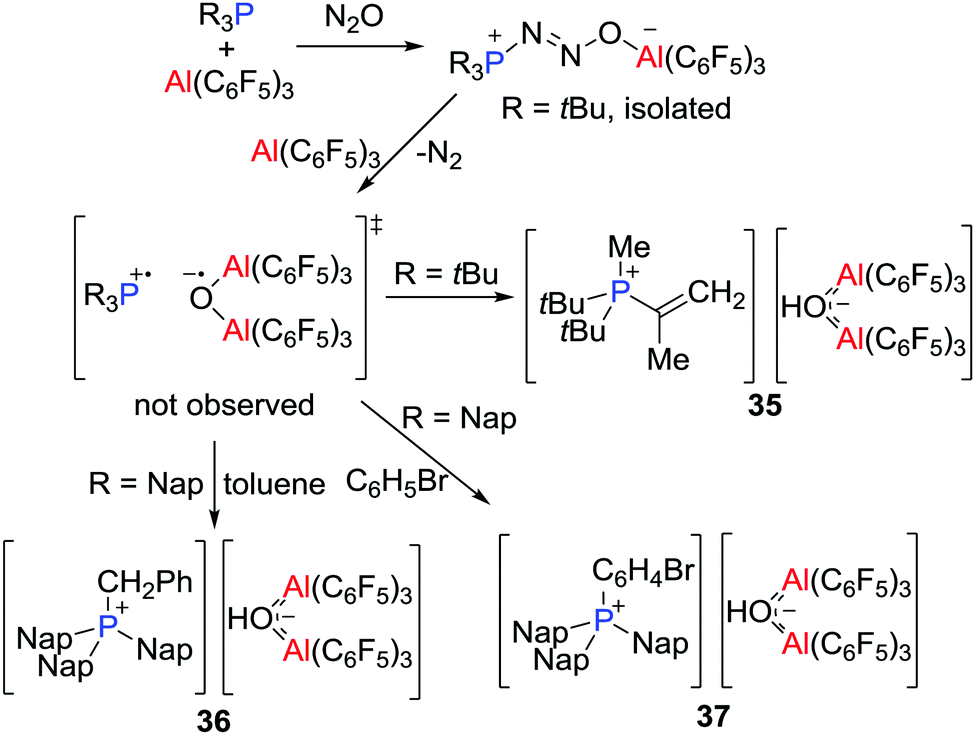
Prior to the recognition of the possibility of a radical pathway for FLP reactivity, it was noted that combination of B(C6F5)3 and PMes3 generated a faintly purple solution. While early efforts to support a radical pathway were unsuccessful, in 2013, unequivocal evidence of highly reactive transient FRPs of the form [R3P˙][˙O(Al(C6F5)3)2] (R = tBu or Nap, Nap = naphthyl) was obtained.31 Generated from the reactions of R3P and Al(C6F5)3 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with N2O (1 atm) (Scheme 10), these FRPs were shown to effect C–H bond activation. In the case of R = tBu, a C–H bond of the tBu group was readily activated to form [tBu2PMe(C(CH2)Me)][(μ-OH)(Al(C6F5)3)2] 35, whereas for R = Nap, the intermolecular activation of toluene or bromobenzene afforded [Nap3PCH2Ph][(μ-OH)(Al(C6F5)3)2] 36 or [Nap3PC6H4Br][(μ-HO)(Al(C6F5)3)2] 37, respectively (Scheme 10). These reactions proceeded via the corresponding FLP-N2O adducts, as supported by isolation of the species tBu3P(N2O)Al(C6F5)3 using the equal molar portion of tBu3P and Al(C6F5)3. The H atom in the anion [(μ-HO)(Al(C6F5)3)2]− was derived from hydrogen atom abstraction from the toluene solvent by the transient radical anion [(μ-O˙)(Al(C6F5)3)2]−.
:2 with N2O (1 atm) (Scheme 10), these FRPs were shown to effect C–H bond activation. In the case of R = tBu, a C–H bond of the tBu group was readily activated to form [tBu2PMe(C(CH2)Me)][(μ-OH)(Al(C6F5)3)2] 35, whereas for R = Nap, the intermolecular activation of toluene or bromobenzene afforded [Nap3PCH2Ph][(μ-OH)(Al(C6F5)3)2] 36 or [Nap3PC6H4Br][(μ-HO)(Al(C6F5)3)2] 37, respectively (Scheme 10). These reactions proceeded via the corresponding FLP-N2O adducts, as supported by isolation of the species tBu3P(N2O)Al(C6F5)3 using the equal molar portion of tBu3P and Al(C6F5)3. The H atom in the anion [(μ-HO)(Al(C6F5)3)2]− was derived from hydrogen atom abstraction from the toluene solvent by the transient radical anion [(μ-O˙)(Al(C6F5)3)2]−.
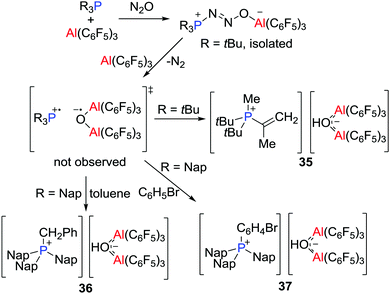 |
| | Scheme 10 C–H bond activation by transient FRPs. | |
Borocyclic radicals via H2 activation
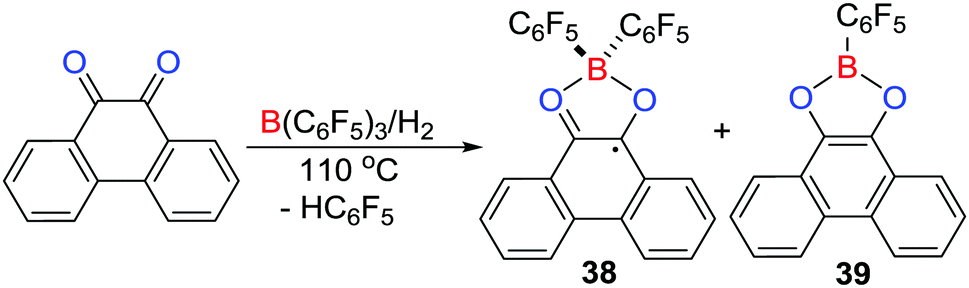
It is well known that carbonyl–oxygen atoms form adducts with Lewis acids.32 Nonetheless, the equilibrium access to Lewis pairs provides a pathway to reaction with H2 resulting in carbonyl hydrogenation.33 In a similar sense, Stephan et al. in 2016 reported that the reaction of phenanthrene-9,10-dione with B(C6F5)3 in toluene led to formation of a Lewis acid–base adduct (C6F5)3B(O2C14H8) featuring a tetracoordinated boron centre.34 Remarkably, treating the above reaction mixture with H2 (4 atm) at 110 °C resulted in the formation of two products, the major one being [(C6F5)2B(O2C14H8)]˙ 38 together with the minor product (C6F5)B(O2C14H8) 39 (Scheme 11). The paramagnetic nature of 38 was confirmed by EPR spectroscopy which revealed hyperfine coupling to the 11B atom (aB = 2.58 G) and to hydrogen atoms with aH = 3.39, 0.00, 2.43, and 1.01 G. These data were consistent with DFT calculations of the spin density showing the delocalization of the unpaired electron over the phenanthrene backbone.
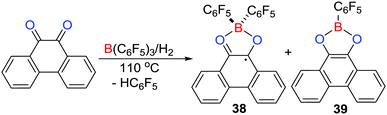 |
| | Scheme 11 Synthesis of a borocyclic radical 38via an FLP hydrogenation. | |
A mechanistic study showed that initially the FLP-hydrogenation of the dione proceeds giving phenanthrene-9,10-diol. This diol reacts with the dione to afford 9,10-phenanthrenequinhydrone radical that readily coordinates B(C6F5)3 to form a paramagnetic Lewis acid–base adduct, followed by protonation of one of the C6F5 rings affording 38 and HC6F5. Alternatively, if the reduced diol reacts with B(C6F5)3, double protodeborylation affords the minor product 39.
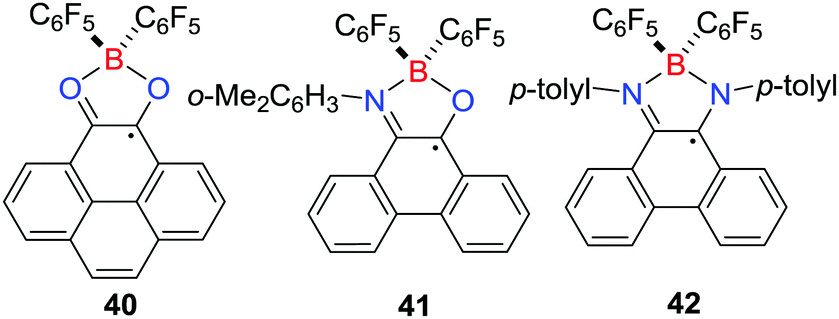
The analogous reactions of B(C6F5)3 with pyrene-4,5-dione,34N-(2,6-dimethylphenyl)-phenanthreno-iminoquinone35 or N,N-di-p-tolylphenanthrene-9,10-diimine35 under the atmosphere of H2 (4 atm) at 110 °C led to the formation of the related stable borocyclic radical 40–42, respectively (Scheme 12). Notably, the borocyclic radicals 38 and 40–42 are stable towards moisture and oxygen, presumably due to the delocalization of the unpaired electron over the aromatic backbone. Nevertheless, the one-electron chemical reduction of 38 or 40 with cobaltocene generated the corresponding cobaltocenium borate salts.34
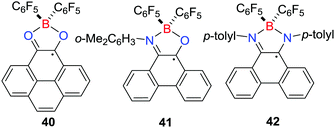 |
| | Scheme 12 Stable borocyclic radicals prepared via FLP hydrogenations. | |
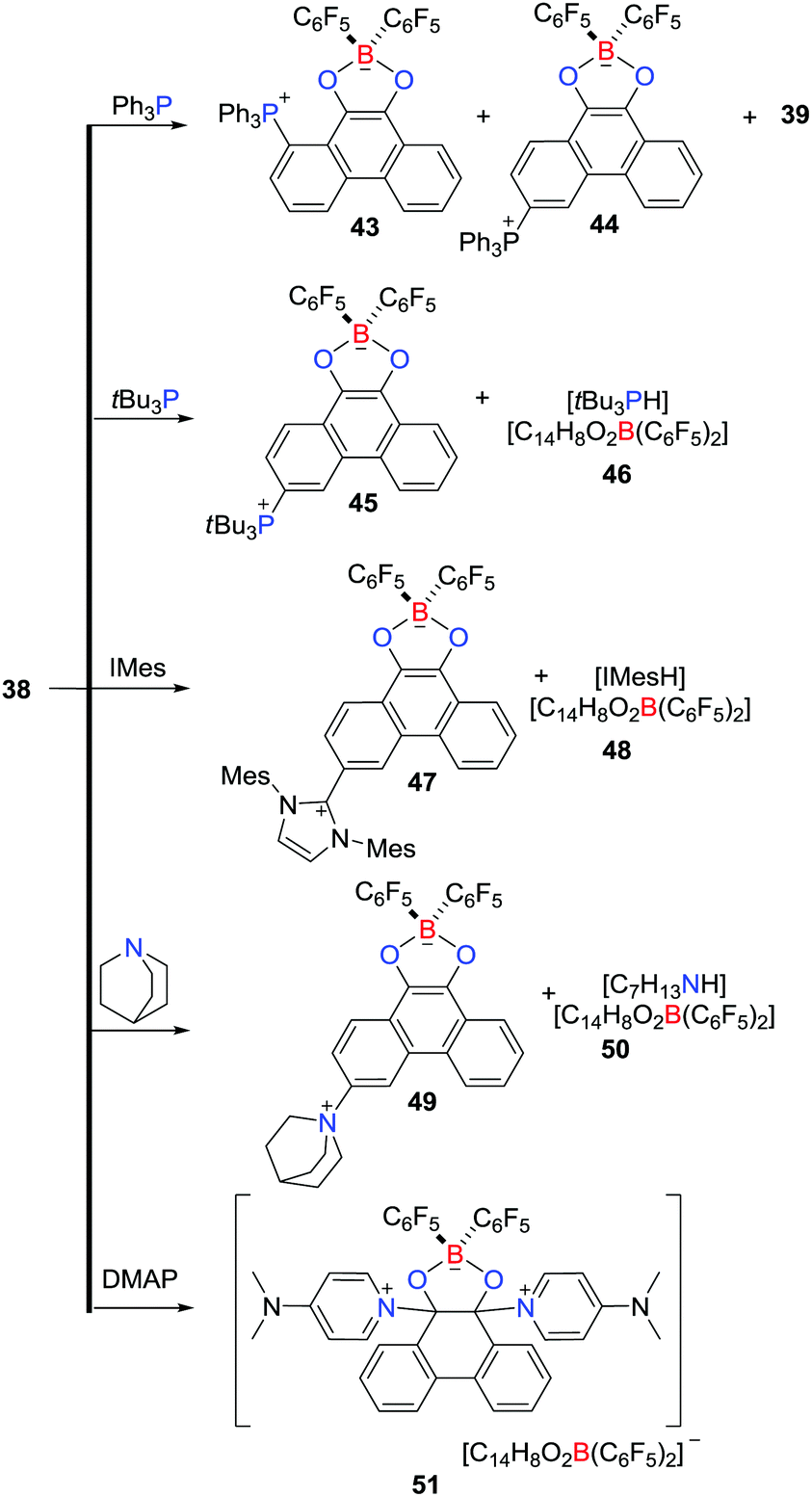
In an ensuing work, the reactivity of 38 with a variety of nucleophiles was probed (Scheme 13).36 The LUMO of 38 primarily involves π*-antibonding orbitals over the phenanthrene backbone, prompting a reaction with Ph3P to give two regioisomeric zwitterions, 1-(Ph3P)C14H7O2B(C6F5)243 and 3-(Ph3P)C14H7O2B(C6F5)244, coupled with the related boronic ester 39 and HC6F5. Similarly, the reaction of 38 with tBu3P afforded the zwitterion 3-(tBu3P)C14H7O2B(C6F5)245 and the salt [tBu3PH][C14H8O2B(C6F5)2] 46, while reaction with N-heterocyclic carbene IMes or quinuclidine gave 3-(IMes)C14H7O2B(C6F5)247 and [IMesH][C14H8O2B(C6F5)2] 48 or 3-(C7H13N)C14H7O2B(C6F5)245 and [H(NC7H13)2][C14H8O2B(C6F5)2] 50, respectively (Scheme 13). In contrast, treatment of 38 with DMAP led to attack at the carbonyl carbon atoms, affording [9,10-(DMAP)2C14H8O2B(C6F5)2][C14H8O2B(C6F5)2] 51 (Scheme 13).
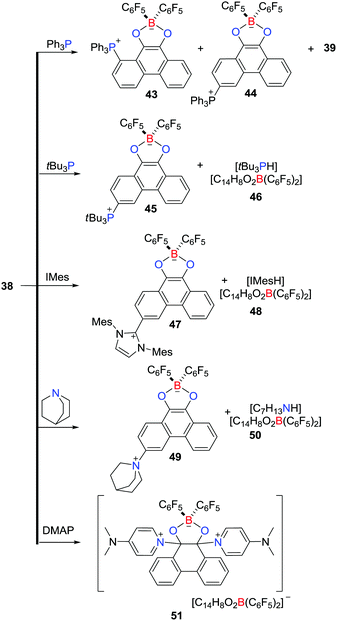 |
| | Scheme 13 Reactivity of 38 toward nucleophiles. | |
A neutral aminyl radical from disulfide activation
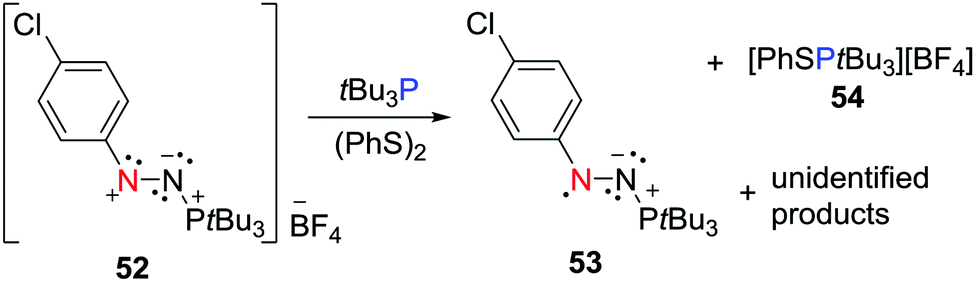
The FLP system consisting of the nitrogen based Lewis acidic nitrenium cation 52 with tBu3P has been shown to react (PhS)2 to afford the persistent neutral aminyl radical, [(p-ClC6H4)N2(PtBu3)]˙ 53 and the salt [PhSPtBu3][BF4] 54 (Scheme 14).37 This is reminiscent of the dione/borane reaction with H2 described above, suggesting that the concerted FLP action on the disulphide prompts electron transfer. It is noteworthy that the radical 53 was also accessible via a more conventional reduction of 52 with cobaltocene or PhSNa.
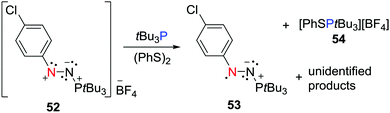 |
| | Scheme 14 Synthesis of an aminyl radical 53via FLP activation of diphenyl disulfide. | |
Transient donor–acceptor radicals
In 2016, the Erker group described the selective oxidation of the intramolecular amine/borane FLP, [C5H6Me4N(CH2)3B(C6F5)2], with dioxygen (Scheme 15).38 In this case, the resulting tetrahydroisoxazolium borate salt [C5H6Me4N(CH2)3O] [C5H6Me4N(CH2)3B(C6F5)2OB(C6F5)3] 56 was formed via a series of transient radical intermediates. Initially an endergonic generation of alkyl radicals prompts the propagation step generating a borylperoxide intermediate, leading to the formation of 56 in an overall thermodynamically favourable process (−59.5 kcal mol−1) (Scheme 15).
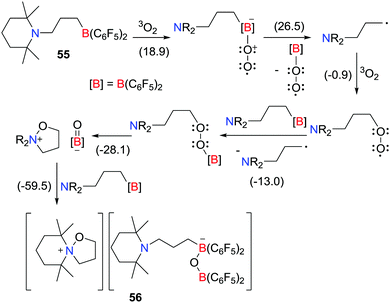 |
| | Scheme 15 Oxidation of an intramolecular amine/borane FLP with dioxygen. Free energies are given in kcal mol−1. | |
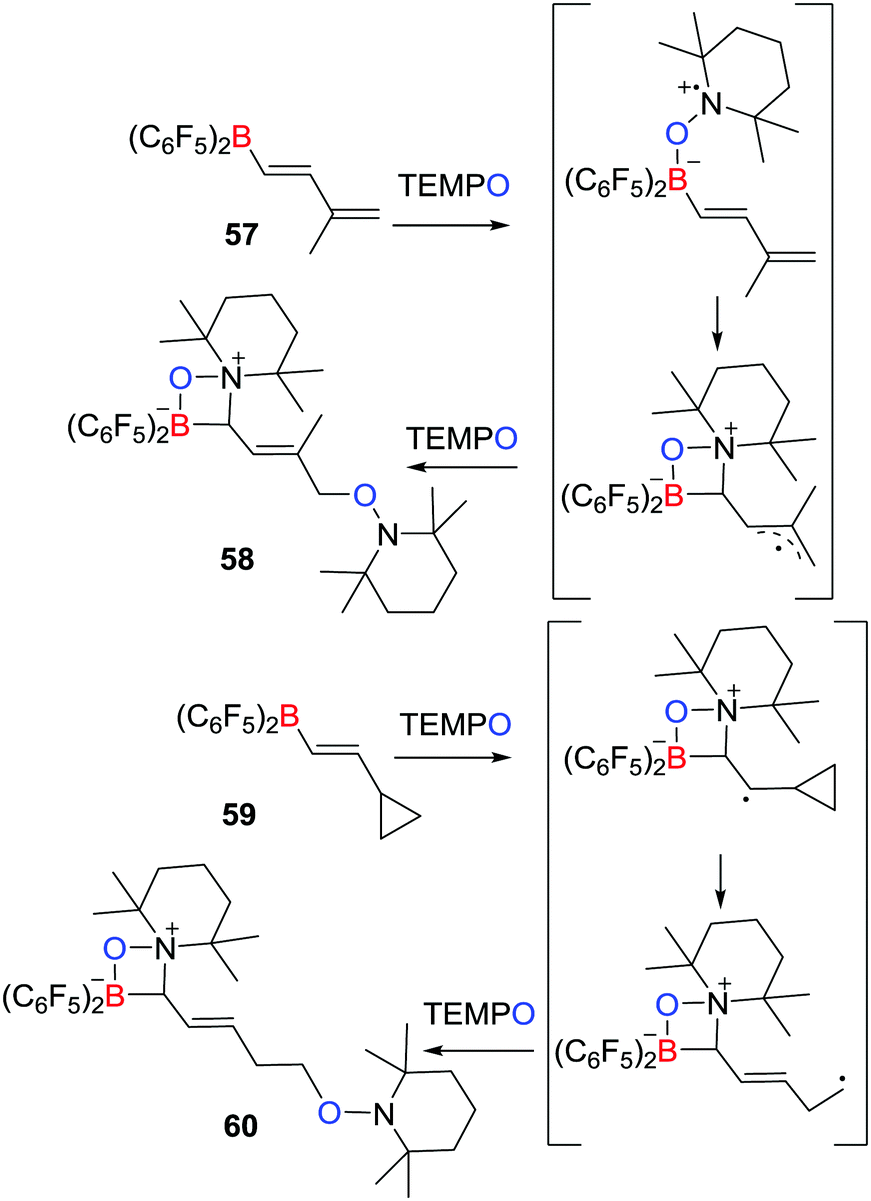
A related work described by Erker, Studer et al. described that the reaction of the boryldiene CH2C(Me)CHCHB(C6F5)257 with two equivalents of TEMPO yielded species 58 featuring a zwitterionic four-membered ring, in which two TEMPO radicals added to the boryldiene starting material (Scheme 16).39 The authors proposed that the initial coordination of TEMPO to the Lewis acidic boron centre affords a transient radical that undergoes cyclization and then couples a second TEMPO to give the product 58. Trapping of TEMPO at the terminal position is presumably sterically driven. Similarly, the cyclopropyl-substituted alkenyl borane, C3H5CHCHB(C6F5)259 was shown to react with two equivalents of TEMPO affording the ring-opening product 60 (Scheme 16). This observation supported the generation of the analogous radical intermediates. Indeed the scope of analogous reaction with TEMPO radical was extended in 2017.40
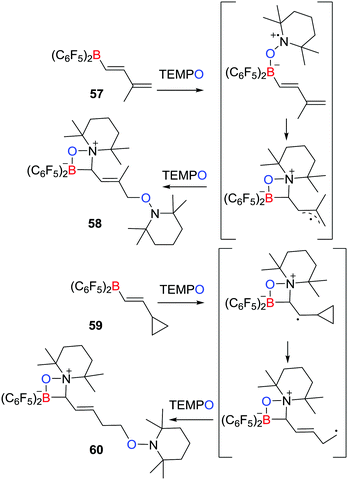 |
| | Scheme 16 Synthesis of 58 and 60via transient radicals. | |
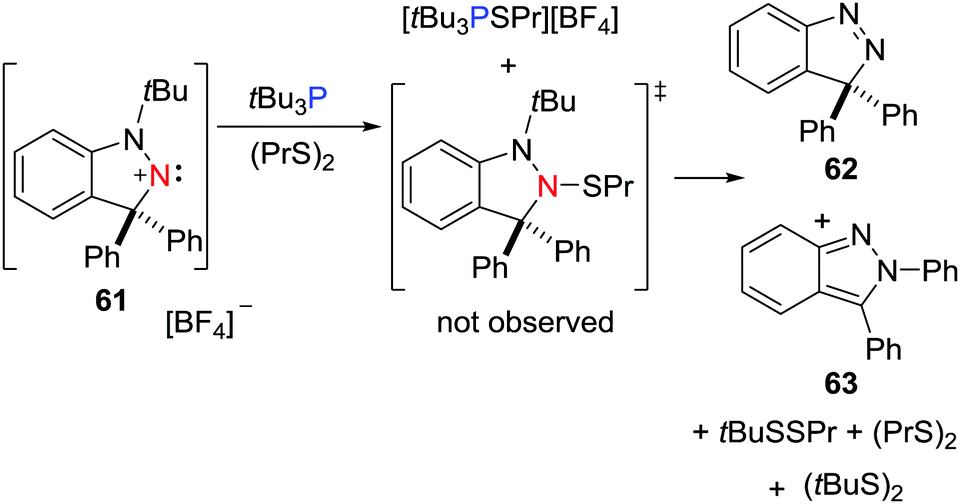
In a recent work, the FLP derived from a nitrogen-centered LA, namely a cyclic (alkyl)(amino)nitrenium cation, [C6H4CPh2N2tBu][BF4] 61, and tBu3P was shown to react with dipropyl disulfide (PrS)2.41 One product is the salt [tBu3PSPr][BF4]. The other product derived from the heterolytic S–S cleavage, [(C6H4)(NtBu)N(SPr)CPh2], was not observed but instead, the reaction affords a mixture of (tBuS)2, (tBuSSPr), Pr2S, [C6H4CPh2N2] 62 and [C6H4CPhN2Ph] 63, consistent with a radical chain pathway (Scheme 17).
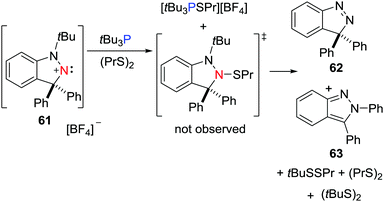 |
| | Scheme 17 FLP reactivity of tBu3P/61 with (PrS)2 leading to a spontaneous radical chain degradation. | |
Electron transfer to an acid/base adduct
Ferrocene/Lewis pair systems
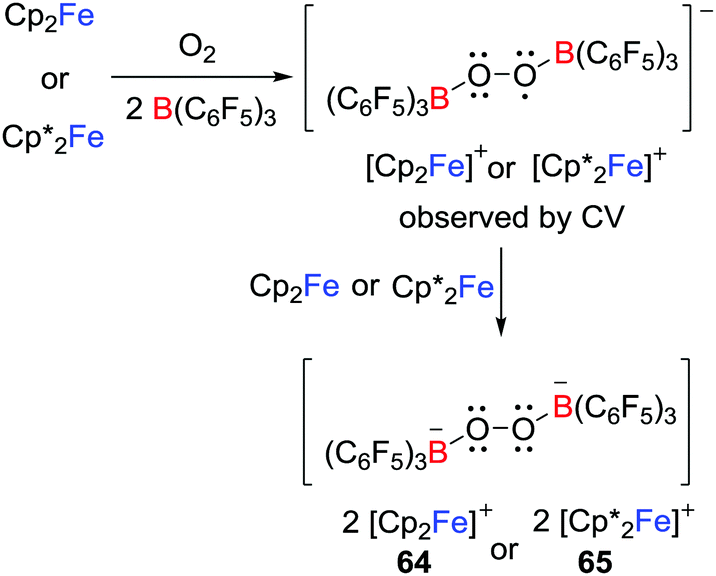
Another approach to reactive radicals is derived from the addition of a one-electron donor with a Lewis donor/acceptor combination. This concept was first demonstrated by Agapie and Henthorn who reported reactions of ferrocene/B(C6F5)3 pairs with dioxygen which afforded [Cp′2Fe]2[(C6F5)3BOOB(C6F5)3] (Cp′ = Cp 64 or Cp* 65); the first examples of dianionic boron peroxide salts (Scheme 18).42 Although B(C6F5)3 is inert to 3O2 over 24 h, the presence of B(C6F5)3 appeared to facilitate the reduction of 3O2 by ferrocenes. A cyclic voltammetry (CV) study suggested the reaction proceeded via the transient B2O2 superoxide intermediate although attempts to access this intermediate were unsuccessful.
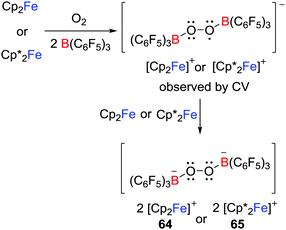 |
| | Scheme 18 Reactivity of ferrocene/B(C6F5)3 toward dioxygen. | |
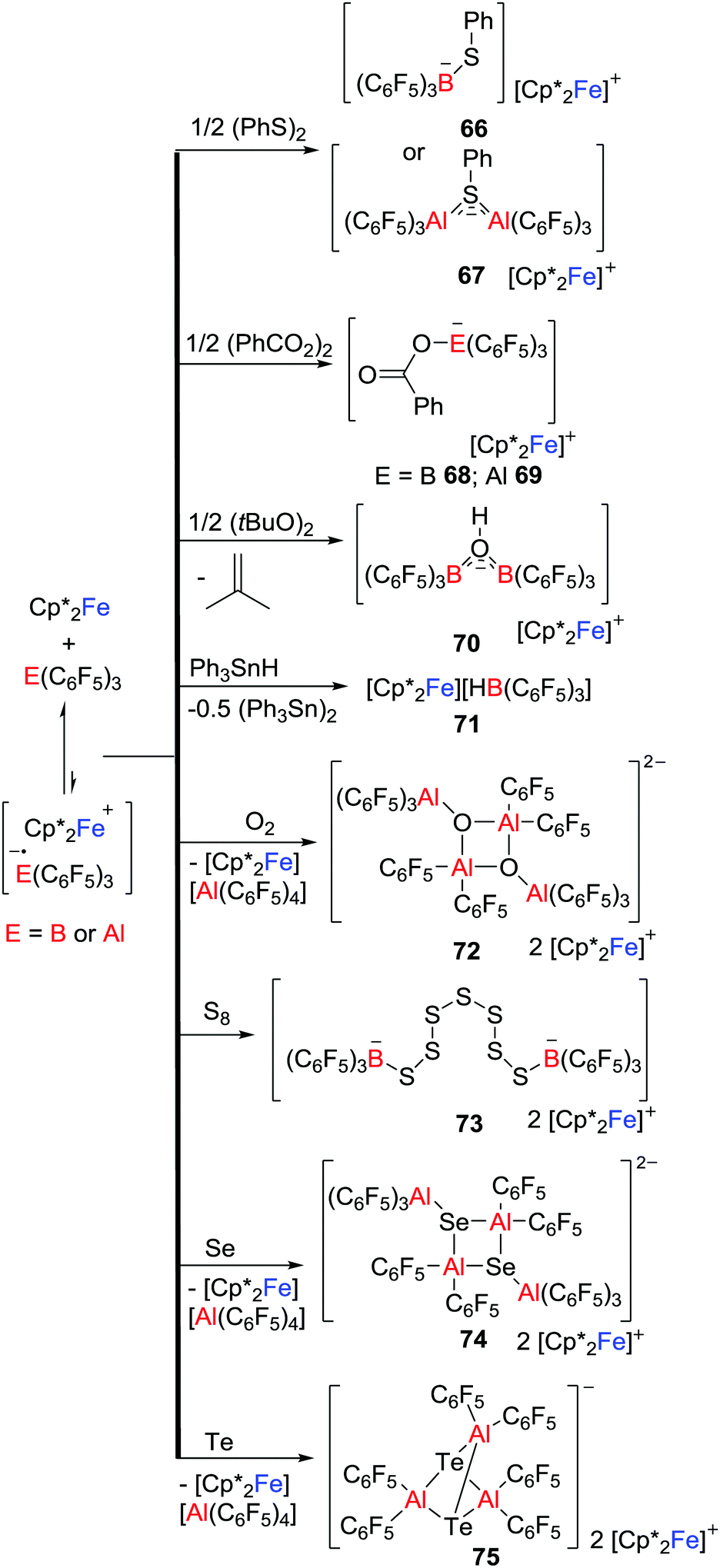
Exploiting a related strategy, Stephan and co-workers exploited such single electron delivery from Cp*2Fe in the presence of a Lewis acid (E(C6F5)3, E = B or Al) for small molecule activation.43 In this fashion, O–O, S–S, Se–Se, Te–Te, C–H, C–O and Sn–H bonds were cleaved in a homolytic fashion, affording a variety of ferrocenium salts 66–75 featuring novel B- and Al-containing anions (Scheme 19). For example, exposure of a solution of Cp*2Fe/Al(C6F5)3 to dry 3O2 immediately resulted in the O–O bond cleavage, giving the salt [Cp*2Fe]2[((C6F5)3AlOAl(C6F5)2)2] 72 with a dianionic Al2O2 core. This stands in contrast to the corresponding reaction of Cp*2Fe/B(C6F5)3 that yields 65 in which the O–O bond remains intact (Scheme 18). The corresponding reactions of Cp*2Fe/E(C6F5)3 with elemental Te and S8 afford a unique tricyclic Al3Te2 anion and a dianion in which two boron centers are linked by seven sulfur atoms, respectively (Scheme 19).
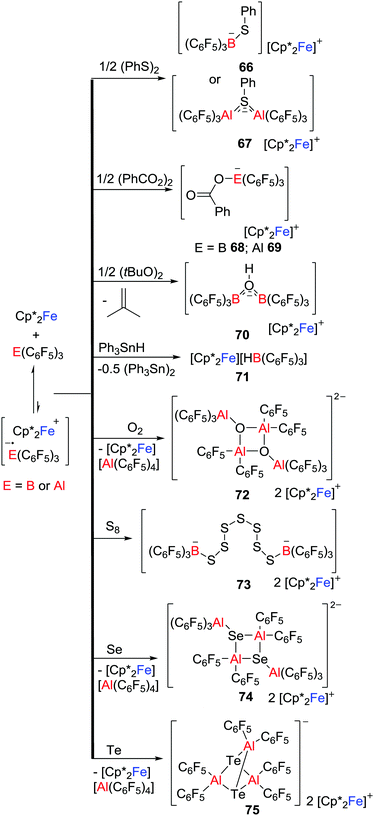 |
| | Scheme 19 Reactivity of Cp*2Fe/E(C6F5)3 toward small molecule substrates. | |
Control reactions showed the slow generation of ferrocenium cation in the absence of substrate molecules, consistent with SET from Cp*2Fe to E(C6F5)3, suggesting that the generated [˙E(C6F5)3]− reacts with substrate. Alternatively, SET to the Lewis acid-substrate adduct is also possible. It is noteworthy that Chen also proposed the generation of the fleeting [˙Al(C6F5)3]− that was followed by decomposition during thermolysis of a C6D5Br solution of Al(C6F5)3 with Cp2Fe at 100 °C for 3 days,44 although the radical anion [˙Al(C6F5)3]− was not observed.
In a recent work, this concept of SET was extended to systems based on the transition-metal-based LA; Zn(C6F5)2 (Scheme 20).45 In this case, the combination of Zn(C6F5)2 and [(PhC(S)S)2] gave a weak CLA, while the cleavage of the S–S bond of [(PhC(S)S)2] was achieved immediately upon mixing the substrate with Zn(C6F5)2 and Cp*2Fe, leading to a salt [Cp*2Fe][PhCS2Zn(C6F5)2] 76. No SET reaction from Cp*2Fe to Zn(C6F5)2 was observed in the absence of [(PhC(S)S)2], inferring that single electron delivery from Cp*2Fe to the CLA prompts the rapid cleavage of the S–S bond.
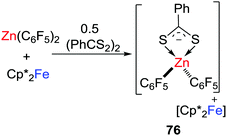 |
| | Scheme 20 Reaction of Cp*2Fe/Zn(C6F5)2 with [(PhC(S)S)2]. | |
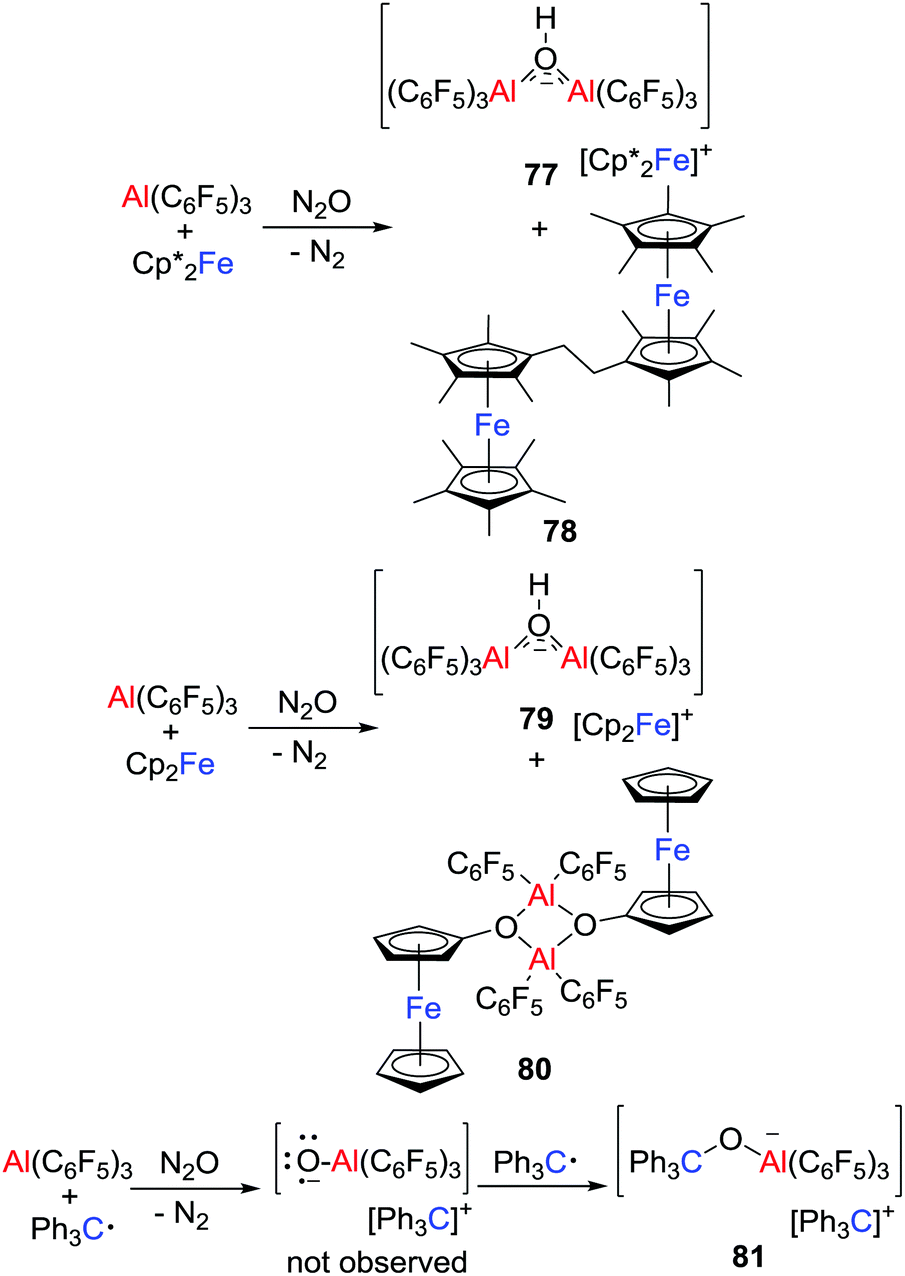
More recently, the group of Severin reacted Cp′2Fe/Al(C6F5)3 (Cp′ = Cp* or Cp) with N2O (Scheme 21), affording salts [Cp’2Fe][(μ-HO)(Al(C6F5)3)2] (Cp′ = Cp*, 77; Cp, 79).46 At the same time, neutral species 78 and 80 involving the C–H activation of the metallocenes were isolated. These observations indicated the formation of the transient oxyl radical anion [˙OAl(C6F5)3]− that spontaneously abstracts hydrogen atoms from the metallocenes. The ensuing metallocene-based radicals underwent radical/radical coupling reactions leading to 78 and 80. This is further evidenced by the reaction of Ph3C˙/Al(C6F5)3 with N2O, wherein [˙OAl(C6F5)3]− was trapped by the excess Ph3C˙ to give the salt [Ph3C][Ph3COAl(C6F5)3] 81.
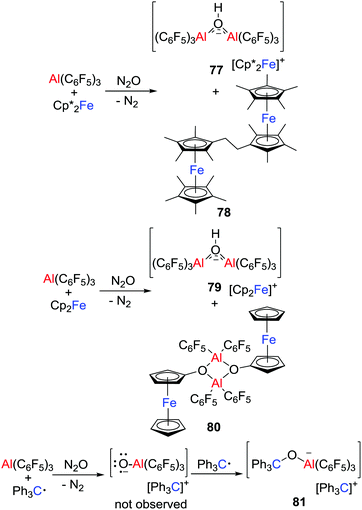 |
| | Scheme 21 Reaction of Cp′2Fe/Al(C6F5)3 or Ph3C˙/Al(C6F5)3 with N2O. | |
Zinc-containing radical anions
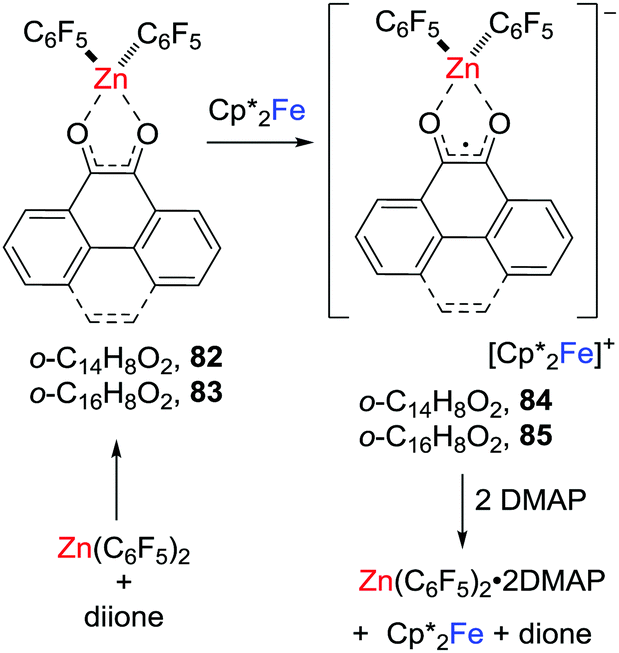
In 2018, related zinc-dione-containing radical anions were prepared via a single electron transfer to donor–acceptor system consisting of dione/Zn(C6F5)2 (Scheme 22).45 Specifically, the phenanthrene-9,10-dione or pyrene-4,5-dione donor–acceptor adduct [(C14H8O2)Zn(C6F5)2] 82 or [(C16H8O2)Zn(C6F5)2] 83 was shown to feature low-lying LUMOs relative to those of the corresponding free diones. One-electron reduction of these adducts with Cp*2Fe gave the persistent zinc-cyclic radical anions 84 and 85 with counterions of the ferrocenium cation.
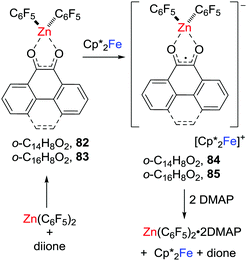 |
| | Scheme 22 Synthesis and reactivity of zinc-containing radicals. | |
These salts proved labile undergoing reaction with DMAP to give [Zn(C6F5)2(DMAP)2] with the regeneration of Cp*Fe and the free corresponding dione (Scheme 22).
Semiquinone radical anions
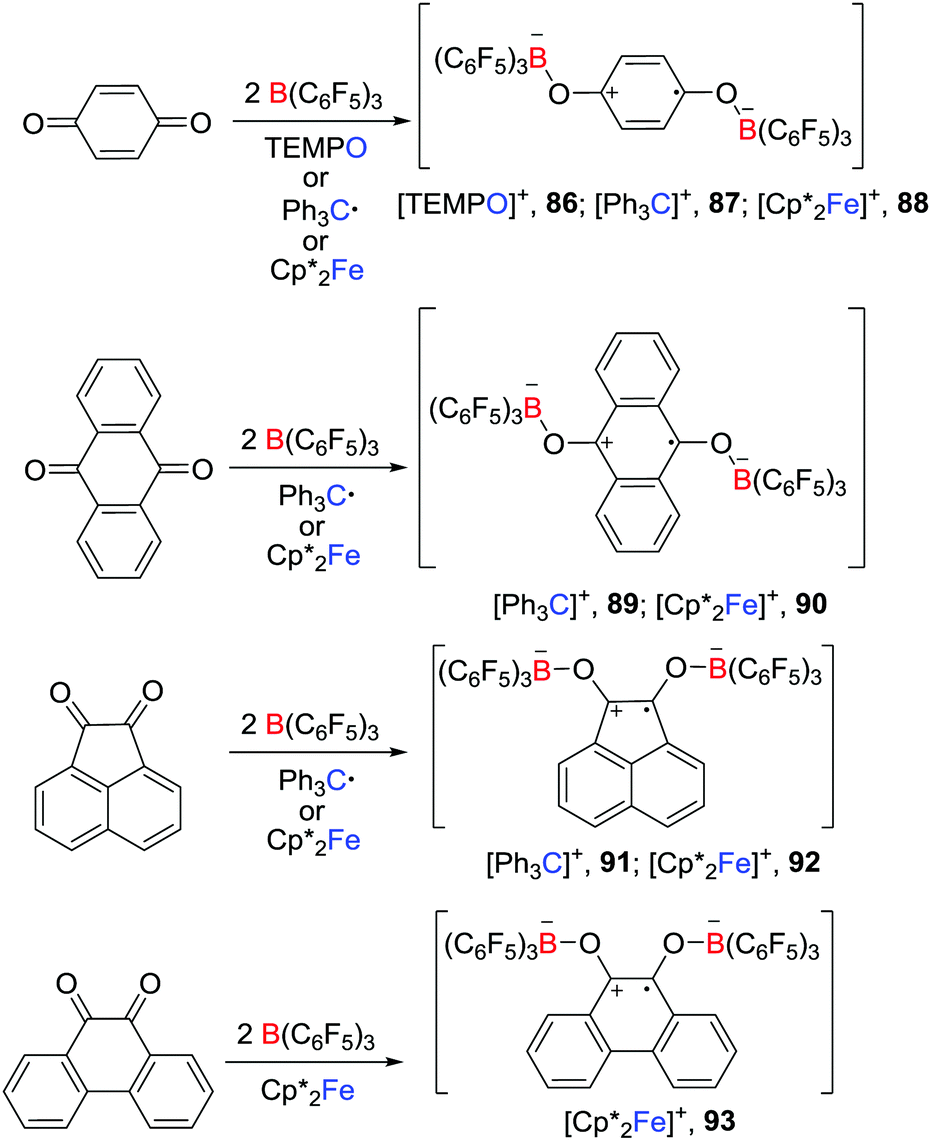
In a recent and related work, Erker et al. reported that, in the presence of two molar equivalents of B(C6F5)3, p-benzoquinone, 9,10-anthraquinone, acenaphthenequinone or 9,10-phenanthrenequinone reacted with a one-electron donor such as TEMPO, Gomberg dimer and Cp*2Fe, leading to the formation of a series of isolable doubly O-borylated semiquinone radical anion salts 86–93 (Scheme 23).47 In the absence of B(C6F5)3, the corresponding reaction of the studied quinones with TEMPO, Gomberg dimer or Cp*2Fe did not react. The presence of B(C6F5)3 appeared to facilitate the reduction of the quinones.
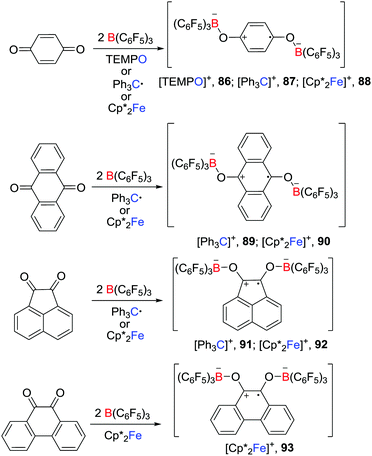 |
| | Scheme 23 Synthesis of semiquinone radical anions. | |
Bis(borane)superoxide radicals
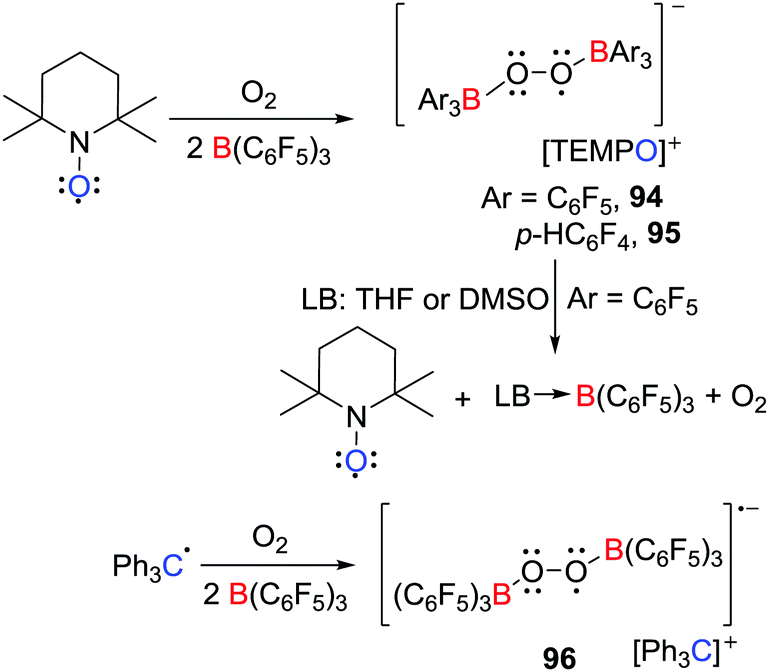
In 2017, the group of Erker demonstrated that electron transfer can be effected from TEMPO to boranes in the presence of O2 (Scheme 24),48 affording the salts of the bis(borane)superoxide radical anion [C5H6Me4NO][˙O2[B(C6F5)3]2] 94 and [C5H6Me4NO][˙O2[B(p-C6F4H)3]2] 95 respectively. These species are analogous to the [Cp*2Fe]+ salts reported by Agapie and Henthorn (Scheme 18) (vide supra).42 Interestingly, the Gomberg dimer of the trityl radical reacted in a similar fashion to give the salt [Ph3C][˙O2[B(p-C6F4H)3]2] 96 (Scheme 24). These results showcase the advantage of application of TEMPO or the trityl radical for the isolation of these bis(borane)superoxide radical anions. Interestingly, THF or DMSO reacted with 94 prompted the regeneration of TEMPO and O2 and formation of (THF)B(C6F5)3 or (DMSO)B(C6F5)3, respectively. This observation indicates that displacement of B(C6F5)3 moiety from the anion alters the redox potential of the transient superoxide radical prompting electron transfer to the oxoammonium cation to generate TEMPO and liberating O2.
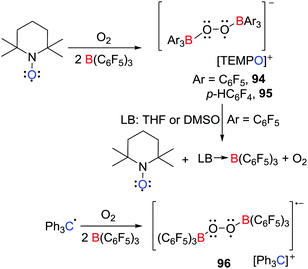 |
| | Scheme 24 Synthesis and reactivity of a bis(borane)superoxide radical anion. | |
Conclusion and perspectives
The generation and characterization of radical species in main group chemistry has become more prevalent in the literature recently. Indeed, this tutorial has described a number of strategies involving the combinations of Lewis acids and bases that result in radical reactants or products. These include the induction of element–element bond homolysis, electron transfer between donor and acceptor molecules, the use of FLPs to stabilize or generate reactive radicals, and electron transfer to donor–acceptor adducts. The systems presented herein, also serve to highlight the notion that donor–acceptor interactions are not be limited to classical dative two-electron adduct formation. Thus, while many combinations of Lewis acids and bases form either CLA and FLP combinations and react via a diamagnetic, two-electron pathway, judicious choice of donor–acceptor combinations can afford radical pathways, offering new routes and strategies for the exploitation of main group chemistry in reactivity and synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
NSERC of Canada is thanked for financial support. DWS is grateful for the award of a Canada Research Chair and a Visiting Einstein Fellowship at TU Berlin.
Notes and references
-
G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalog Com., New York, 1923 Search PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- B. M. Hoffman and T. B. Eames, J. Am. Chem. Soc., 1969, 91, 5168–5170 CrossRef CAS.
- J. J. Scepaniak, A. M. Wright, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 19350–19353 CrossRef CAS PubMed.
- B. M. Hoffman and T. B. Eames, J. Am. Chem. Soc., 1971, 93, 3141–3146 CrossRef CAS.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- G. Wang, H. Zhang, J. Zhao, W. Li, J. Cao, C. Zhu and S. Li, Angew. Chem., Int. Ed., 2016, 55, 5985–5989 CrossRef CAS PubMed.
- L. L. Cao and D. W. Stephan, Organometallics, 2017, 36, 3163–3170 CrossRef CAS.
- L. Zhang and L. Jiao, Chem. Sci., 2018, 9, 2711–2722 RSC.
- L. Zhang and L. Jiao, J. Am. Chem. Soc., 2017, 139, 607–610 CrossRef CAS.
- A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7567–7571 CrossRef CAS.
- M. Sajid, A. Stute, A. J. P. Cardenas, B. J. Culotta, J. A. M. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Fröhlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156–10168 CrossRef CAS PubMed.
- M. Sajid, G. Kehr, T. Wiegand, H. Eckert, C. Schwickert, R. Pöttgen, A. J. P. Cardenas, T. H. Warren, R. Fröhlich, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 8882–8895 CrossRef CAS PubMed.
- R. Liedtke, F. Scheidt, J. Ren, B. Schirmer, A. J. P. Cardenas, C. G. Daniliuc, H. Eckert, T. H. Warren, S. Grimme, G. Kehr and G. Erker, J. Am. Chem. Soc., 2014, 136, 9014–9027 CrossRef CAS PubMed.
- C. J. Harlan, T. Hascall, E. Fujita and J. R. Norton, J. Am. Chem. Soc., 1999, 121, 7274–7275 CrossRef CAS.
- C. J. Beddows, A. D. Burrows, N. G. Connelly, M. Green, J. M. Lynam and T. J. Paget, Organometallics, 2001, 20, 231–233 CrossRef CAS.
- X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, J. Am. Chem. Soc., 2013, 135, 14912–14915 CrossRef CAS PubMed.
- E. J. Lawrence, V. S. Oganesyan, G. G. Wildgoose and A. E. Ashley, Dalton Trans., 2013, 42, 782–789 RSC.
- L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, Chem, 2017, 3, 259–267 CAS.
- L. L. Liu, L. L. Cao, D. Zhu, J. Zhou and D. W. Stephan, Chem. Commun., 2018, 54, 7431–7434 RSC.
- X. Pan, X. Chen, T. Li, Y. Li and X. Wang, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed.
- Z. Dong, H. H. Cramer, M. Schmidtmann, L. A. Paul, I. Siewert and T. Muller, J. Am. Chem. Soc., 2018, 140, 15419–15424 CrossRef CAS.
- G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2016, 138, 2500–2503 CrossRef CAS PubMed.
- K. L. Bamford, L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, Dalton Trans., 2017, 46, 5308–5319 RSC.
- L. E. Longobardi, P. Zatsepin, R. Korol, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 426–435 CrossRef CAS PubMed.
- A. Waked, R. O. Memar and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 11934–11938 CrossRef CAS PubMed.
- T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302–4305 CrossRef CAS PubMed.
- F. Türkyilmaz, G. Kehr, J. Li, C. G. Daniliuc, M. Tesch, A. Studer and G. Erker, Angew. Chem., Int. Ed., 2016, 55, 1470–1473 CrossRef PubMed.
- X. Tao, F. Türkyilmaz, C. G. Daniliuc, G. Kehr and G. Erker, J. Organomet. Chem., 2017, 847, 167–172 CrossRef CAS.
- J. Zhou, L. L. Liu, L. L. Cao and D. W. Stephan, Angew. Chem., Int. Ed., 2018, 57, 3322–3326 CrossRef CAS PubMed.
- J. T. Henthorn and T. Agapie, Angew. Chem., Int. Ed., 2014, 53, 12893–12896 CrossRef CAS PubMed.
- L. L. Liu, L. L. Cao, Y. Shao and D. W. Stephan, J. Am. Chem. Soc., 2017, 139, 10062–10071 CrossRef CAS PubMed.
- J. Chen and E. Y. X. Chen, Dalton Trans., 2016, 45, 6105–6110 RSC.
- L. L. Cao, K. L. Bamford, L. L. Liu and D. W. Stephan, Chem. – Eur. J., 2018, 24, 3980–3983 CrossRef CAS PubMed.
- Y. Liu, E. Solari, R. Scopelliti, F. Fadaei Tirani and K. Severin, Chem. – Eur. J., 2018, 24, 1–8 CrossRef CAS.
- X. Tao, C. G. Daniliuc, R. Knitsch, M. R. Hansen, H. Eckert, M. Lubbesmeyer, A. Studer, G. Kehr and G. Erker, Chem. Sci., 2018, 9, 8011–8018 RSC.
- X. Tao, C. G. Daniliuc, O. Janka, R. Pöttgen, R. Knitsch, M. R. Hansen, H. Eckert, M. Lübbesmeyer, A. Studer, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2017, 56, 16641–16644 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Douglas W.
Stephan
and
Douglas W.
Stephan