
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gelation enabled charge separation following visible light excitation using self-assembled perylene bisimides†
Charlotte L.
Smith
a,
Laura L. E.
Mears‡
a,
Benjamin J.
Greeves
a,
Emily R.
Draper
 b,
James
Doutch
c,
Dave J.
Adams
*b and
Alexander J.
Cowan
*a
b,
James
Doutch
c,
Dave J.
Adams
*b and
Alexander J.
Cowan
*a
aStephenson Institute for Renewable Energy and the Department of Chemistry, University of Liverpool, Liverpool, L69 7ZF, UK. E-mail: acowan@liverpool.ac.uk
bSchool of Chemistry, College of Science and Engineering, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
cSTFC ISIS Neutron and Muon Source, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, OX11 0QX, UK
First published on 27th November 2019
Abstract
Perylene bisimides (PBIs) can be functionalised to enable controlled aggregation into complex supramolecular structures and are promising materials for photovoltaic and solar fuel applications. Amino acid appended PBIs such as PBI-alanine (PBI-A) have been found to form photoconductive films containing worm-like structures that enable charge transport. However, despite being strong chromophores in the visible region, when PBI-A films are prepared by drying down solutions, activity only occurs under UV illumination. This limits potential applications. The requirement for UV illumination has previously been suggested to be due to the large ion-pair energy in the low dielectric environment of the dried samples. Hydrogel films, rehydrated xerogels and dry xerogels of PBI-A can also be prepared offering an ideal sample set to examine the influence of hydration on charge-separation. Using transient absorption (TA) spectroscopy, we demonstrate a correlation between water content and efficiency of generation of long-lived charge separated states within the PBI-A materials, highlighting their potential, particularly for light-driven water splitting.
Introduction
PBIs are interesting materials for use in a range of solar energy technologies, including in organic photovoltaics, as photocatalysts, and as photoelectrodes for water splitting,1–6 due to their visible light harvesting properties, excellent thermal and electrochemical stabilities and ability to act as both electron donors and acceptors. A particularly attractive feature of PBIs is that they readily aggregate into structures consisting of π-stacked molecules, which can in principle enable both exciton and long-range charge transport through the structure.7–10 Designing functionality onto the PBIs that directs self-assembly offers a route to control the nature of the aggregates formed. This is important as changes in aggregate type, size or in the interlayer distance between the PBI units of the aggregate can have a profound effect on the photophysics and charge transport properties of the materials.2,10–12Recently a number of researchers, including some of us,1,13–15 have studied the photophysical properties and structures of PBIs functionalised at the imide sites with amino acids, Fig. 1a. A wide range of many different amino acid functionalised PBIs have now been synthesised,16 and it has been shown that these materials are also able to act as low molecular weight gelators (LMWGs). At pH values above those of the apparent pKa (or the lowest apparent pKa in cases where there are multiple values) of the pendant amino acid functionality, supramolecular structures form and are stable in solution even at very low (<10−5 M) concentrations.15 The self-assembled structures are typically long worm-like micelles, which consist of bundles of PBI fibres that persist on the micron length scale.16 At pH values below the apparent pKa, hydrogel networks form which contain high concentrations of water within the films containing the self-assembled fibres.17 Early studies on dried solutions of the amino acid modified PBIs at high pH, and on dried hydrogels (xerogels) demonstrated that the supramolecular structures formed gave rise to photoconductive materials.1,13 UV illumination leads to the formation of a radical anion (PBI˙−) state that can then migrate through the supramolecular structure.1 Although these were highly promising results, particularly as the system was insensitive to O2, it was notable that the materials only became photoconductive under UV illumination, potentially limiting application.1,16
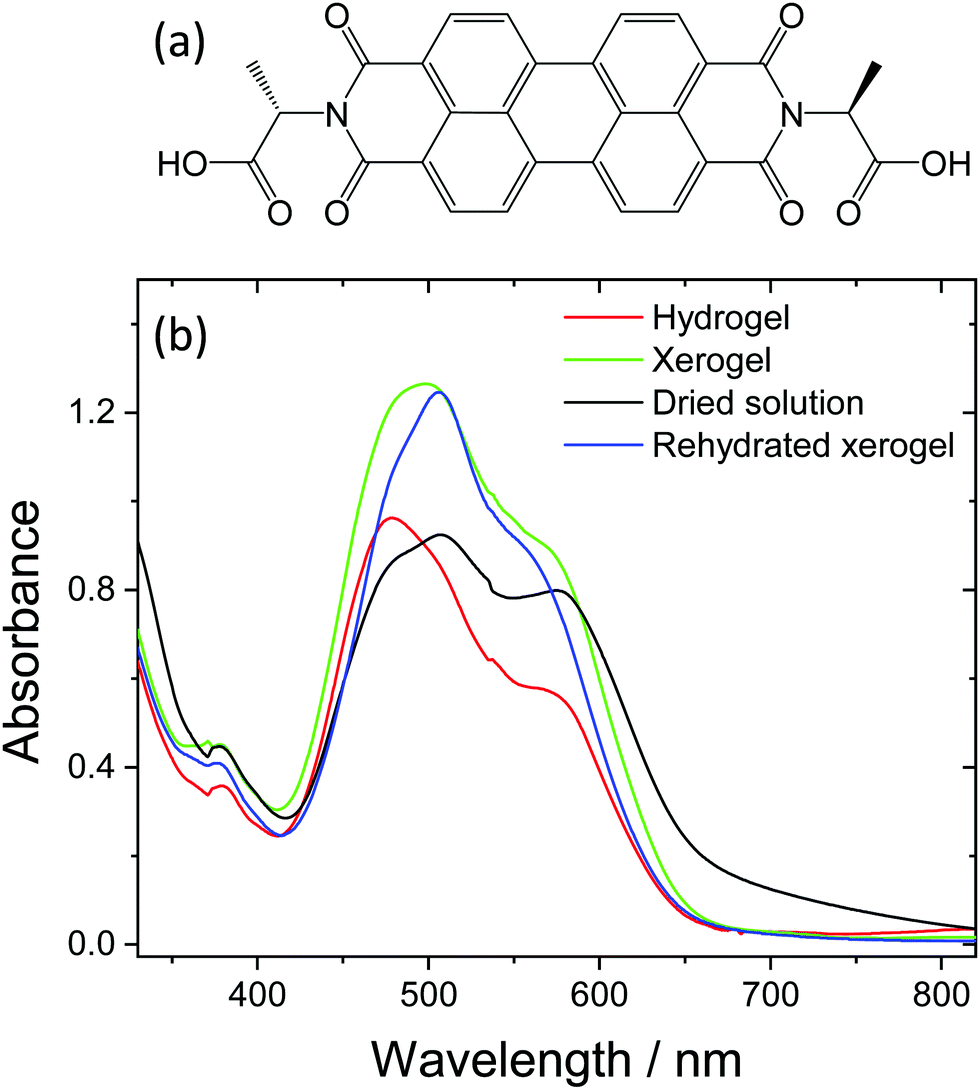 | ||
| Fig. 1 Structure of PBI-A (a) and UV/Vis spectra of the films used within the study (dried solution, hydrogel, xerogel and rehydrated xerogel) (b). The hydrogel and rehydrated xerogel samples are immersed in pH 4 aqueous solution to maintain hydration levels. All samples are prepared on conductive glass (FTO). | ||
In a previous study on alanine-functionalised PBI (PBI-A) using TA spectroscopy and DFT calculations, we demonstrated that the minimal activity under visible light was due to the lack of charge separation (to form the PBI˙− state) in the low dielectric environment of the dried PBI-A film.15 Experiments and calculations demonstrated that the energy required to generate a charge separated state was greater than that available under visible illumination, and could only be achieved using higher energy UV photons. In contrast, visible light excitation led to charge separation and PBI˙− formation when the PBI-A supramolecular structure was dissolved in water at high pH (9). The change in behaviour was assigned to a combination of the high dielectric constant of the aqueous solution (ε ∼ 80), which screens the free charge of the excess electron and hole and reduces the ion pair energy, and a calculated solvent induced change in the site of hole localisation. For PBI-A structures in aqueous solution, experimental electrochemical evidence and DFT calculations suggested that in the charge separated state the electron localised on the PBI core and the hole was localised on the alanine group.15 With dried films of PBI-A prepared at high pH (likely to exist as PBI-A2−), the site of electron accumulation was the same but it was predicted that the hole would partially localise on both the alanine (50%) and the PBI core (50%), placing it in closer proximity to the electron. Solvent controlled light-induced charge separation has been reported elsewhere with PBI based materials18,19 and it represents a potentially valuable way to achieve improved device activity. However, for use in many applications a solution-based absorber is undesirable. The generation of PBI-A hydrogels offers a route to forming self-supporting materials that contain the desired self-assembled structures whilst in a highly solvated environment, which may be beneficial for enabling formation of long-lived charge separated states, the formation of which is critical for solar energy conversion devices.
Hydrogels are also beginning to be explored for use as photocatalysts and photoelectrodes in water.20–23 Perylene monoimide gels have been studied as photocatalysts for hydrogen production in the presence of a sacrificial electron donor by the group of Stupp.20,21 Some of us have also reported the use of L-phenylalanine functionalised PBI (PBI-F) as a photocatalyst and in pH dependent studies shown that gelation at pH ∼5 correlates to a sudden increase in photocatalytic activity.22 A PBI-based organic photoelectrode for water oxidation has also been reported24 and the current densities are amongst the highest achieved with an organic material.25,26 In that study, an aqueous solution of N,N′-bis(phosphonomethyl)-3,4,9,10-perylenediimide (PMPDI) was spin-coated onto fluorine doped tin oxide (FTO) glass. Although the nature of the structures formed within the electrode were not described in detail, it is likely that by briefly dipping the films of dried PMPDI solution in acid, that either an outer gel layer was formed or that the entire structure was gelled. It is apparent that the development of highly active PBI hydrogel photocatalysts and photoelectrodes is feasible. These recent studies on light-driven water splitting further reinforce the need to develop an understanding of the factors controlling light driven charge separation within PBI hydrogel supramolecular structures. Here, we report a TA spectroscopic study of PBI-A dried solutions, hydrogels, air-dried hydrogels (xerogels) and rehydrated xerogels to explore the role of water content on charge separation yields and lifetime within the PBI supramolecular structures. Significantly, we demonstrate that self-supporting hydrogels and rehydrated xerogels are able to undergo visible light induced charge separation to form long-lived PBI˙− species, likely with the alanine acting as an electron donor, in a manner similar to the PBI-A structures in aqueous solution. The change in activity for the hydrated materials is proposed to be due to a decrease in the energy of the ion-pair state. The results for the hydrogel materials presented here have interesting wider implications for the design of organic photocatalysts and photoelectrodes. In contrast to inorganic materials, where effective screening of the mutual Coulomb attraction occurs enabling facile generation of free electron–hole pairs at room temperature, inefficiency of charge separation is commonly cited to be a limiting factor with many organic solar to fuels materials.
Results
Dried solution, hydrogel, xerogel and rehydrated xerogel PBI-A samples were prepared from a 7.5 mg mL−1 PBI-A solution on FTO glass. Hydrogel and xerogel samples were prepared using a HCl vapour pH trigger and xerogels are dried in air for at least 17 hours. Rehydrated xerogels were soaked in pH 4 solution for a minimum of 1 hour. Thermogravimetric measurements (Fig. S1, ESI†) and room temperature mass loss experiments provide estimations of water contents for the different samples. As would be anticipated, the highest water content by weight is recorded for the hydrogel (hydrogel ∼96%, xerogel 5%, rehydrated xerogel ∼72%, dried solution 5%).We initially studied the UV/Vis spectra of the prepared PBI-A structures, Fig. 1. UV/Vis spectroscopy probes the electronic structure of the PBI-A structures and is sensitive to changes in the molecular level environment as the water content of the PBI-A sample is changed. In the visible region, a broad electronic transition (S0 to S1) dominates, with a series of vibronic modes (0–0′, 0–1′ and 0–2′) apparent between 400 and 650 nm. In all cases, the spectra of the PBI films are broadened when compared to in solution at low concentration (Fig. S2, ESI†), indicating extensive π-stacking and increased aggregation. A recent combined DFT, NMR and UV/Vis study of PBI-A at a range of pH values demonstrated that aggregates form in both solution and upon gelation.27 Furthermore the thin films formed by drying the deprotonated PBI-A solutions (referred to here as dried solutions) were predicted to contain similar structures to the hydrogel films but the structures would be present at different densities. In line with this, we observe that the dried solution, hydrogel, xerogel and rehydrated xerogel spectra are broadly similar, with only slight shifts in the UV/Vis maxima and the relative intensities of the vibronic modes. We therefore conclude that aggregated structures which contain extensive π-stacked PBI-A units are present in all the samples, regardless of water content. However, the small changes in the UV/Vis spectra, see in Fig. 1b, are still potentially significant as the changes in ratio of vibronic modes may indicate a different degree of overlap between PBI-A units within the structure, which could lead to a change in the supramolecular structure.
To further investigate the possible differences between the supramolecular structures of the gel materials as the water content is changed, small angle neutron scattering (SANS) measurements were carried out, Fig. 2 and Fig. S4–S7 (ESI†). SANS can be applied to both wet and dry samples, the only requirement is a contrast in neutron scattering length density28 between the structures and their environment. The technique has been used extensively to study the nature of the aggregated structures within PBI materials on the nanometre to microns scale, thus complementing the UV/Vis spectroscopic data which addressed the molecular level environment.16,29 The SANS data could be fitted equally well to either a flexible elliptical cylinder model or a flexible cylinder combined with a power law. In brief, both of the flexible cylinder models are parameterised by the cylinder radius, the flexible segment (Kuhn) length and the contour length. Focussing first on the use of the flexible elliptical cylinder model, (Fig. 2) consistent with our previous data for the gel state,1,16 the fit to the model implies that there is little change in radius or axis ratio across the samples, with the radius being 2.5 ± 0.1, 3.1 ± 0.3 and 3.0 ± 0.03 nm respectively and the axis ratio being 2.7, 2.7 and 2.8 respectively. The lengths are all very long as expected from the microscopy (Fig. S3, ESI†). For the fit to the data using the flexible cylinder combined with a power law (Fig. 2 and Fig. S4–S6, ESI†), drying of the hydrogel to form the xerogel apparently changes the primary fibre dimensions with the radius of fibres decreasing from 5.9 ± 0.2 nm to 3.1 ± 0.3 nm. Rehydration of the xerogel also led to partial, but not complete, recovery of the fibre radius (5.0 ± 0.5 nm). A detailed discussion regarding the two model fits is available in the ESI,† but irrespective of the model used, the radii in all cases are greater than the molecular size and so SANS can be used to show the presence of long, anisotropic structures persisting across the hydrogel, xerogel and re-hydrated xerogel states.
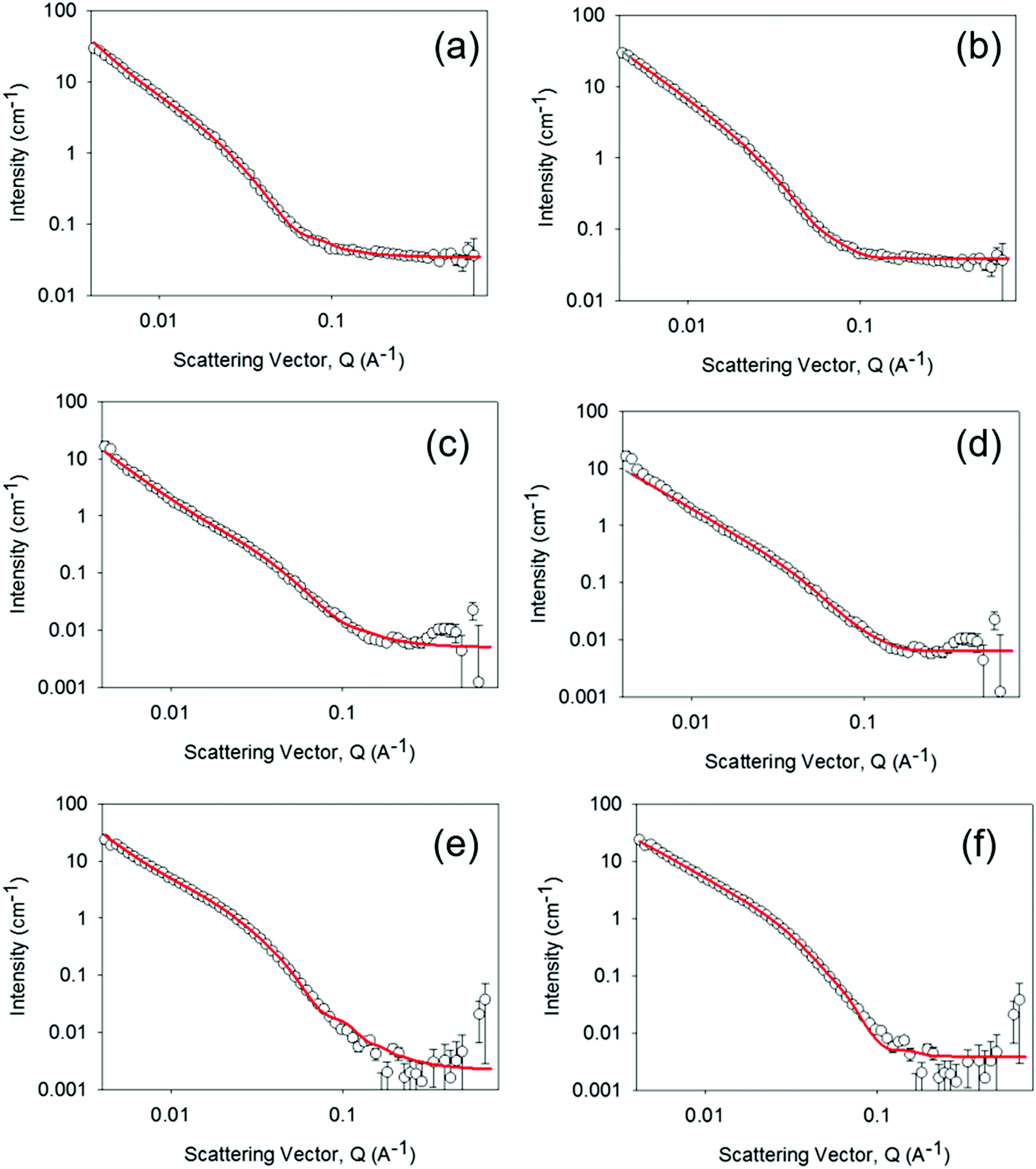 | ||
| Fig. 2 SANS of 1 mm thick hydrogel (a and b), a xerogel prepared from a 1 mm thick hydrogel (c and d) and the rehydrated xerogel (e and f) following rehydration in 0.1 M KCl electrolyte, pD 4, in D2O for 50 hours. Fit lines are shown in red and the data can be fitted to either a custom flexible cylinder combined with a power law model (a, c and e) or a flexible elliptical cylinder model. All fitting data parameters and the relative contributions within the custom model are given in Tables S1, S2 and Fig. S4–S6 (ESI†). For the rehydrated xerogel sample of the remainder of the 1 mm demountable cell thickness was filled with 0.1 M KCl electrolyte in D2O pD 4 and the hydrogel was prepared in the same electrolyte. | ||
As our structural characterisation (SANS, UV/Vis) measurements confirm the persistent presence of self-assembled aggregate structures, regardless of the water content of the PBI film, we have used TA spectroscopy to study if the changing water content of the sample changes the photophysical behaviour of the structures. The TA spectra recorded following 490 nm excitation of the PBI-A dried solution, hydrogel, xerogel and rehydrated xerogel samples are shown in Fig. 3 and 4. The TA spectra of the dried solution, Fig. 3(a) and 4(a), are markedly different from those of the hydrogel and the rehydrated xerogel samples, Fig. 4(b–d). The spectra recorded of the dried solution of PBI-A are very similar to those previously reported for a PBI-A dried solution prepared from a different concentration solution and are only briefly discussed.15 Within 1 ps of excitation, broad positive absorptions centred at 740 and 840 nm are observed, which were previously assigned to the formation of a PBI excited state, Fig. 3(a).15 The new transient bands decay rapidly (t50% = 3.5 ps at 740 nm, Fig. S8, ESI†) and by 3 ns only a weak TA signal is present with maxima at 746 nm (718 (sh)) and ca. 860 nm (br.), Fig. 3a. Comparison of the TA data at 3 ns to the spectrum of electrochemically generated PBI˙−-A (Fig. 3a, λmax = 544, 715 and 805 nm) and to the previously reported spectrum of photochemically generated PBI˙−-A following UV excitation of a dried solution (ca. 550, ca. 725 and 810 nm)15 shows minimal agreement. The shoulder at 718 nm in the TA spectrum here may indicate the formation of a fraction of PBI˙−, however if present, the yield of PBI˙− formed in the dried solution is low. In line with our past studies15 we find here the formation of a charge separated state is not the dominant process following visible light (490 nm) excitation of dried solutions of PBI-A.
 | ||
| Fig. 3 Comparison between the TA spectra of PBI-A dried solution (a), hydrogel (b), rehydrated xerogel (c) and xerogel (d) recorded at 1 ps (dashed line) and 3 ns (solid line) following visible light excitation (490 nm) to the UV/Vis spectrum of electrochemically generated PBI˙−-A (red line). | ||
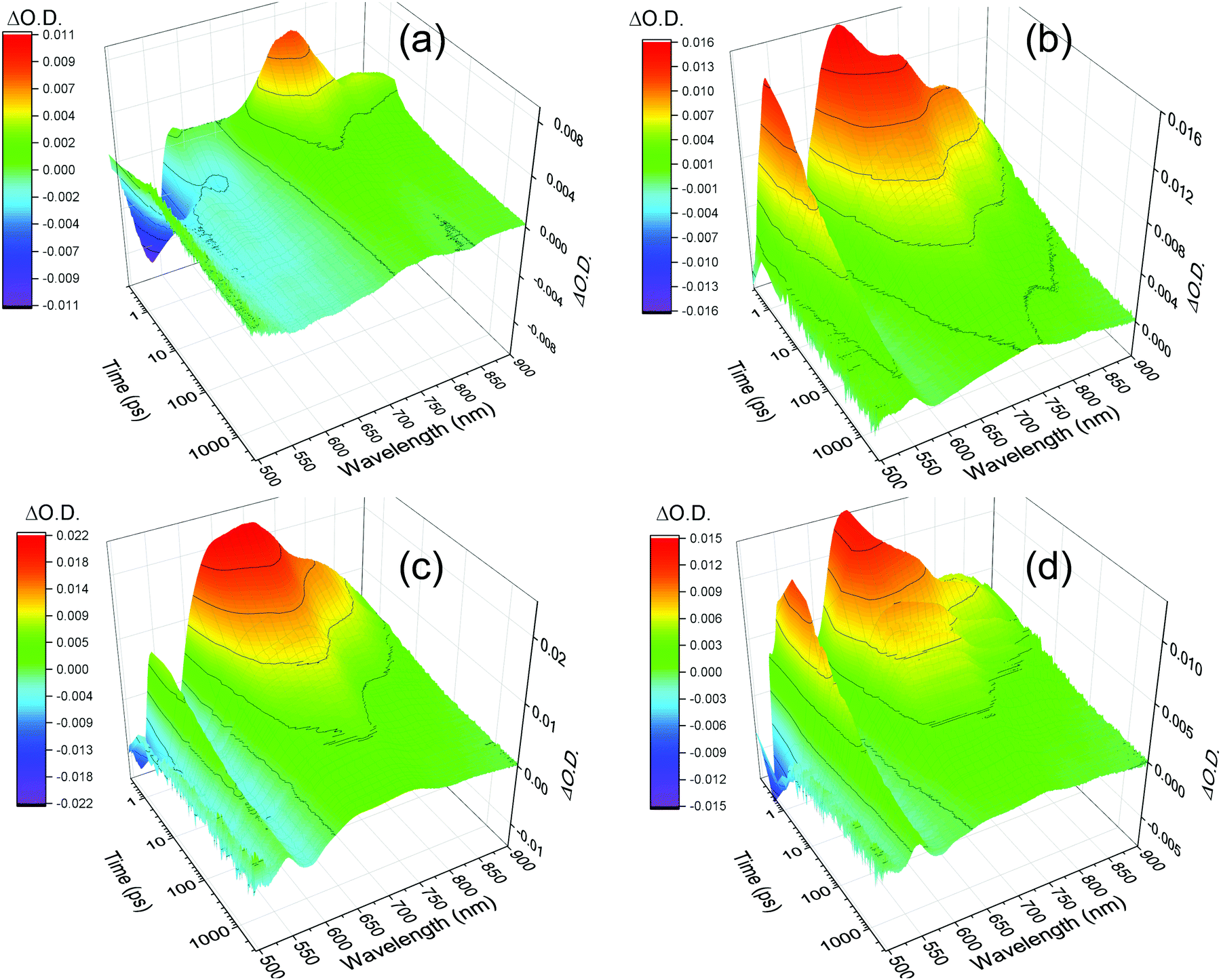 | ||
| Fig. 4 TA spectra of PBI-A dried solution (a), hydrogel (b), xerogel (c) and rehydrated xerogel (d) recorded following visible light excitation (490 nm, 5 kHz excitation repetition rate). All samples are on FTO glass. The hydrogel and rehydrated xerogel samples are immersed in argon purged pH 4 aqueous solution to maintain water content. All other samples are under an argon gas atmosphere. | ||
Visible light excitation of the hydrogel gives rise to new positive bands within 1 ps at 543, 650, 722 and 814 nm, Fig. 3b and 4b. The TA spectrum of the hydrogel recorded at 3 ns is similar to that observed at very early times (1 ps shown) with strong bands remaining at 546, 730, 819 nm and a weak shoulder at ∼650 nm, Fig. 3(b). A comparison of the spectrum of electrochemically generated reduced hydrogel (PBI˙−-A, λmax = 544, 731 and 820 nm) and the TA spectrum recorded at 3 ns shows excellent agreement indicating that visible light can lead to the formation of a PBI˙− state. Previously, it has been reported electron accumulation occurs on the perylene core with PBI-A materials and here we find the TA and spectroelectrochemical (SEC) spectra to be in good agreement with those studies, Fig. 3.15,16 At the latest time that could be studied with our apparatus (3.2 ns), >10% of the signal assigned to the PBI˙− state remains (Fig. S9, ESI†), indicating that visible light excitation is able to lead to the formation of a long-lived reduced PBI within the hydrogel, a key step for solar energy conversion materials.
The excellent agreement between the TA spectrum of the hydrogel at 3 ns and the SEC spectrum suggests that by 3 ns only the charge separated state is present, Fig. 3b. To further explore the formation of the charge separated state, a lifetime analysis has been carried out. In principle by approximating the kinetics of the transient absorption data to a small number of discreet exponential functions it is possible to carry out a global lifetime analysis that yields decay associated spectra and lifetimes can be carried out. However, the complex self-assembled structures studied here are shown below to give rise to a distribution of lifetimes making approximation to a small number of discreet exponential functions inappropriate. Instead, we have employed a lifetime density analysis (LDA) within the Opitmus30,31 data analysis suite. LDA uses a semi-continuous sum of exponents function to analyse transient kinetics without the need for an assumed model.31,32 The lifetime distribution maps (LDM) that are generated using 100 lifetimes between 0.3 and 5 ns are shown in Fig. 5. Full details of the fitting procedure, comparison of raw and fitted TA data from which the LDM (Fig. 5) is generated and the associated L-curves accompany Fig. S10–S14 (ESI†). The LDM for the hydrogel shows that the initially generated feature at 650 nm, likely due to formation of a PBI based singlet excited state, decays with lifetimes ranging from ∼0.4 to 2 ps, Fig. 5a. On these timescales we also observe decay lifetimes with a negative amplitude (indicative of an increase in optical density) at wavelengths associated with the PBI˙− state (ca. 730 and 820 nm) and the ground state (580 nm), Fig. 5(a). This suggests ultrafast charge separation and rapid formation of PBI˙− as the 650 nm excited state decays in the water rich hydrogel. The PBI˙− state (730, 820 nm) then decays over three main timescales. Initial rapid loss mechanisms have lifetimes distributed between 1.5 and 5 ps, with subsequent processes between 25 and 300 ps and at >3 ns. Interestingly, the LDM shows that a small population of the 650 nm band survives the initial fast decay processes and it then undergoes decay by mechanisms (likely through relaxation to the ground state based on the partial recovery at 580 nm) with a distribution of lifetimes centred around 23 ps, Fig. 5a.
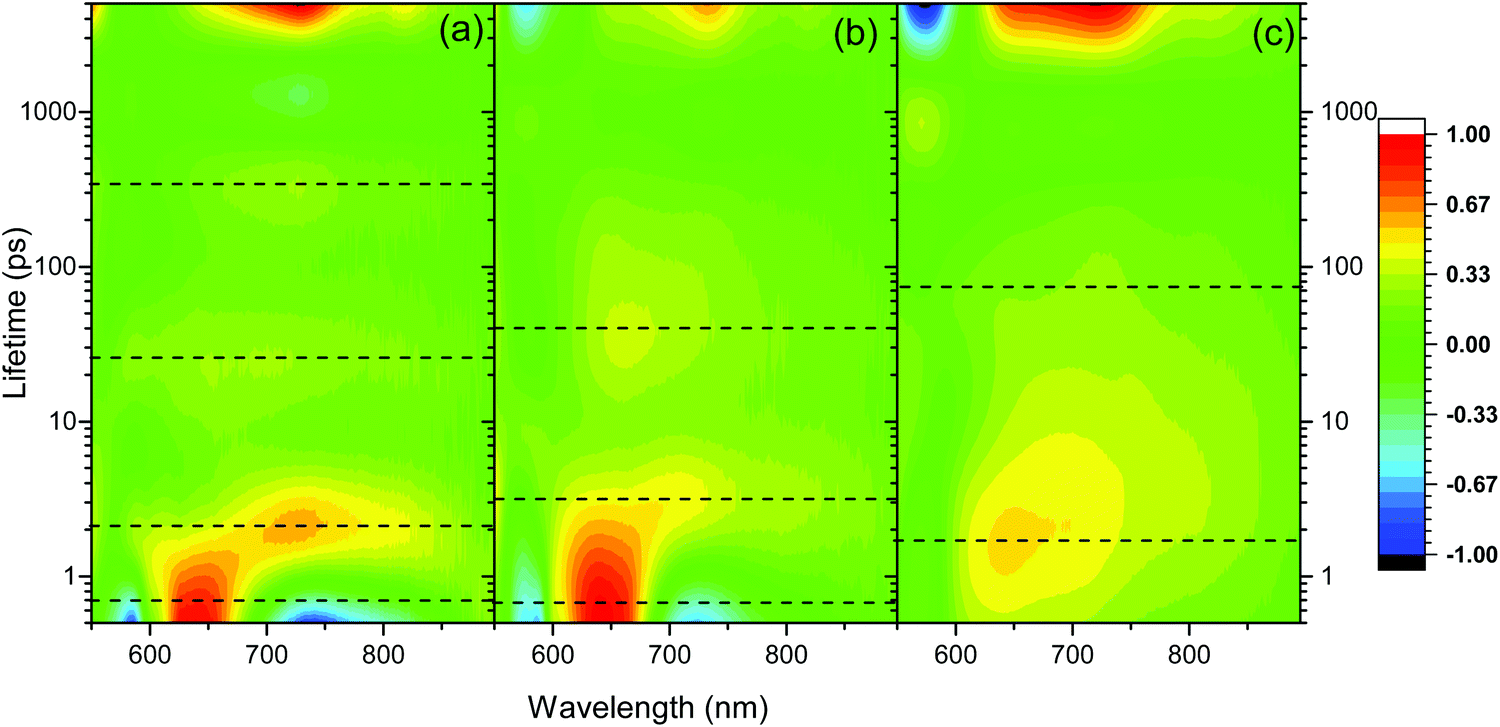 | ||
| Fig. 5 Lifetime distribution maps (LDM) for PBI-A hydrogel (a), rehydrated xerogel (b) and xerogel (c) following visible light excitation (490 nm). The scale shows the relative pre-exponential amplitude of the decay function. Generated using a quasi-continuous distribution of 100 lifetimes over the time-range shown. The dotted lines indicate regions of interest discussed in the main text. 3D plots are shown in Fig. S10 (ESI†). | ||
The TA spectra of the rehydrated xerogel following 490 nm excitation are very similar to the hydrogel and at 1 ps new bands are present at 543, 650, 736 and 831 nm, Fig. 3c and 4d. The TA spectrum at 3 ns shows maxima at 552, 736 and 830 nm and it is in good agreement with the spectrum of electrochemically reduced PBI-A, Fig. 3c. A shoulder at 650 nm is still present in the TA spectrum at 3 ns and is more pronounced than in the electrochemically reduced sample. However, overall it is apparent that rapid formation of a PBI˙− based state can occur following visible light excitation of the rehydrated xerogel and that a significant population of this state remains at >3 ns. The LDM of the rehydrated xerogel also shows behaviour similar to the hydrogel, Fig. 5b. The initially formed band at 650 nm undergoes a fast decay process with lifetimes between ∼0.4 to 3 ps as the bands assigned PBI˙− increase, also indicating that ultrafast charge separation can occur in the rehydrated xerogel. The 650 nm band of PBI˙− decays with lifetimes over the 2 to 5 and 10 to 100 ps range. In line with the observation that a very small quantity of the initially formed excited state persists to the longest time-scales studied (3.2 ns), we also find that at 650 nm the LDA analysis generates lifetime components of around 4 ns, Fig. 5b.
Analysis of the TA spectra recorded following visible light excitation of the xerogel is more complicated, Fig. 3d and 4c. It is not possible to carry out spectroelectrochemical studies of (dry) xerogels; as soon as they are placed in electrolyte they begin to rehydrate leading to structural changes. Therefore, we compare the TA data recorded to the spectroelectrochemical response of a rehydrated PBI-A xerogel as a best estimate (PBI˙−-A, λmax = 552, 738, 824 nm). The TA spectra of the xerogel shows bands at 542, 643, 721 and 833 (br.) nm in both the spectra recorded at 1 ps and 3 ns, Fig. 3d. The TA bands observed at 542, 721 and 833 (br.) nm are assigned to the formation of a quantity of the PBI˙− species, with the shift in maxima being due to solvation effects from the absence/low levels of water in the xerogel. However, it is unlikely that the strong band in the TA spectra at 643 nm at both 1 ps and 3 ns is due to a PBI˙− state on the basis of the spectroelectrochemical study. Therefore, the TA data demonstrates that at 3 ns the charge separated state is not the sole species present, and indicates that it is not the dominant, species. Compared to the rehydrated xerogel the efficiency of formation of a long-lived population of PBI˙− is significantly decreased in the dried xerogel.
The xerogel LDM is also markedly different to the hydrogel and rehydrated xerogel LDMs and offers insight into the lower yield of the PBI˙− species at 3 ns, Fig. 5(c). The initially formed excited state with a band at ∼643 nm is found to be stabilized in the xerogel and the distribution of initial decay lifetimes is ∼0.7 to 7 ps, peaking at 2 ps, compared to ∼0.4 to 2 ps for the hydrogel. As the 643 nm state decays, the LDM does not show a clear negative amplitude for the decay components at longer wavelengths where PBI˙− species would be expected to absorb (>700 nm). Instead, we only see evidence for the ground state recovery as indicated by the behaviour at 570 nm, Fig. 5c. However, from the LDM we can identify distributions of decay lifetimes (20 to 200 ps) that are common at both ∼720 and 815 nm. This indicates that a small population of a PBI˙− species is formed, likely with it being a minor pathway. This process occurs as the initial 643 nm band decays which would prevent the relatively small increase in optical density around 720 and 815 nm from being clearly observed. We therefore conclude that the lower yield of the charge separated state in the TA spectrum of the xerogel at 3 ns is due to a combination of both inefficient initial charge separation and significant levels of sub-ns recombination of those charges that do separate. Overall the TA spectroscopy shows the highest level of PBI˙− formation following visible light excitation for the samples that contain the highest water content (hydrogel, rehydrated xerogel).
Discussion
UV/Vis spectroscopy confirms the persistent presence of aggregated PBI-A in all samples, regardless of the water content. However it is notable that the drying induced changes upon going from a hydrogel to a xerogel are not fully reversible, Fig. 1b. The UV/Vis spectrum of the rehydrated xerogel does not match that of the pristine hydrogel. The measurements of water content show that the rehydrated xerogel (72% water by weight) still contains less water than the pristine hydrogel (96%). Therefore it is not surprising that UV/Vis spectroscopy, a probe of the electronic structure of the chromophore, which is sensitive to the local solvent-chromophore interactions, reports a difference between the spectra of the rehydrated xerogel and hydrogel. Significantly, SANS experiments demonstrate that the wider structural motifs are retained in all our PBI-A samples.Therefore, we conclude that the differences in the TA spectra of the PBI-A samples upon drying are primarily due to solvation effects as the water content of the samples is changed. It is striking that the highest yields of long-lived PBI˙− species and the fastest rates of charge separation following visible light excitation were measured with the water rich hydrogel sample. Furthermore, the lowest levels of long-lived charge separated states were measured in the samples with the lowest water content, the dried solution (5% water) and xerogel (5% water). Rehydration of the xerogel led to a recovery in the yield of charge separation, and in a manner similar to the hydrogel, PBI•- appears to dominate the TA spectrum at 3 ns.
Experimentally, the change in Gibbs energy for photoinduced electron transfer (ΔGET) in a donor–acceptor system can be estimated using eqn (1), providing a route to rationalising the observed TA trends. In eqn (1), EOX and ERED are the donor and acceptor oxidation and reduction potentials, E* the excited state energy, C the coulombic interactions between the two ions and S the difference in solvation energies between the sample under study and the solvent in which the oxidation and reduction potentials were measured.
| ΔGET = EOX − ERED − E* − C + S | (1) |
 | ||
| Fig. 6 Spectroelectrochemistry of the PBI-A hydrogel oxidations. Experiments are carried out at pH 4, 0.1 M KCl. The step at 350 nm is due to the change in light source and is an instrument artefact. Potential differences are reported versus a Ag wire pseudo reference electrode. The inset shows an expansion of the spectral changes occurring at the first oxidation. | ||
As described above spectroelectrochemical studies on the dry xerogel are not possible; as soon as they are placed in electrolyte they begin to rehydrate. This has prevented us from experimentally determining the electron acceptor and donor sites. By assuming that the donor and acceptor sites remain the same as the hydrated samples we can estimate the effect of the change in dielectric environment on the Gibbs energy of the proposed photoinduced electron transfer, again using eqn (1). In this case we took the effective constant to be ε ∼ 3, typical for a dried organic film.15 This gives rise to an estimated ion-pair energy of ca. 2.2 eV, significantly greater than in the hydrated hydrogel and rehydrated xerogel samples, and no driving force (ΔGET ∼ 0.06 eV) for charge separation following formation of the lowest energy singlet excited state. This result is readily understood as, in environments with a low dielectric permittivity, such as the dry xerogel, the excess free charge of the electron and hole are not effectively screened, increasing the ion pair energy. In contrast, in the water rich hydrogel (96% water) and rehydrated xerogel (72% water) PBI-A materials, the high dielectric permittivity of the environment enables screening of the excess free charge.
Our inability to carry out spectroelectrochemical measurements to assign the electron donor site in the dry xerogel means that the estimated Gibbs energy of photoinduced electron transfer needs to be treated with caution. Nonetheless the value of ca. 0 eV, arrived at assuming no change in the site of acceptor and donor sites upon film drying, is broadly in-line with our TA measurements where the level of PBI˙− formed following 490 nm excitation is significantly less than measured with the hydrogel and rehydrated xerogel. Within eqn (1) the solvation term does not take into account specific solvent (water)–solute (PBI-A) interactions. Therefore, it is notable that eqn (1) successfully models the charge-separation results through considering just the change in the local dielectric constant upon hydration, without the need for specific PBI-A/water interactions. This result indicates that the effect of the water within the structure can be modelled as a bulk solvent that is charge screening and lowering the ion-pair energy. The formation of a small quantity of PBI˙− within the xerogel, as measured by TA spectroscopy, is likely due to the small amount of residual water (5%) within the PBI-A structure, which we did not account for within the calculation. The ion-pair energy for PBI-A dried solutions prepared at high pH (9) has been previously calculated to be very large, ca. 4 eV. Again this is in agreement with our TA results which shows that minimal charge separation occurs following visible light excitation.15 Therefore, the transient spectroscopic behaviour and the yield of visible light generated PBI˙− can be largely rationalised by the presence or absence of water within the PBI-A structures.
Conclusions
Self-assembled PBI-A films can be prepared in a variety of structures and hydration levels, which makes them an ideal model material to examine the effect of hydration on charge separation yields and kinetics. Transient UV/Vis, electrochemical and spectroelectrochemical measurements show that PBI-A hydrogels can undergo visible light induced charge separation with the alanine acting as an electron donor and the PBI core as an electron acceptor. It is clear that in the water rich hydrogels high yields of long-lived charge separated states can be generated following visible light excitation. In contrast, dried films of solution and the dried hydrogel films (xerogels) of PBI-A show lower levels of charge separation under visible illumination. This result is rationalised by the high dielectric permittivity of the aqueous environment within the hydrated samples that facilitates both the formation of and stabilisation of the charge separated state. The observed facile charge separation in the hydrated samples is important, as a significant issue with many organic photocatalysts and photoelectrodes is that the low dielectric environment of the bulk polymer leads to generation of free charges only occurring at defect sites36 or at the polymer/water (or electron donor) interface.37,38 Therefore, these results coupled to the emerging reports of high levels of photocatalytic activity with similar hydrogel materials20,22,24 indicates that the development of water rich-hydrogel materials is a promising approach to generating useful materials for the production of solar fuels.Experimental methods
Materials
Milli-Q water (18.2 MΩ) was used throughout (Millipore Corp). All chemicals were obtained from Sigma-Aldrich and used as received, except for PBI-A which was synthesised according to literature procedures.1 Argon and nitrogen were purchased from BOC at pureshield grade.Preparation of PBI-A gel electrodes
FTO (TEC-15) substrates were cleaned by sonication for 20 minutes in three different solutions; 1% Helmanex solution, ethanol and water, and rinsed with water between each sonication. After drying of the FTO glass substrates with compressed air, they were heated in an oven to 400 °C (20 °C min−1) and held for 20 minutes before cooling. Quick drying epoxy resin was used to mask out a surface area of 150 mm2 (10 × 15 mm2). Gelation was carried out in a Petri dish within a humidified glove bag in order to ensure PBI-A gel electrodes did not dry out. FTO glass substrates were placed in a Petri dish containing 8 layers of blue roll dampened with water, and 20 μL of PBI-A (7.5 mg mL−1 dissolved with 1 molar equivalence of 0.1 M NaOH with the remainder made up with Milli-Q water) was deposited onto each FTO conductive surface. 40 drops of concentrated HCl were evenly dropped around both the outer corners of the Petri dish and near to the PBI-A FTO electrode to lower the pH. The lid was placed on the Petri dish and sealed with parafilm, allowing for an acidic atmosphere to build up within the Petri dish. Typically, a gel was formed after around ca. 20–40 minutes. Once gelled, the PBI-A electrode was transferred out of the Petri dish into a vial containing 0.1 M KCl electrolyte (pH 4 adjusted by 1 M HCl) in the glove bag to prevent drying. In the case of PBI-A xerogel electrodes, the electrode was removed from the glove bag, and allowed to dry in air for at least 17 hours. By this time only minimal change in water content occurs, consistent with previous reports (albeit at a slightly lower concentration of PBI-A).1 Rehydrated xerogels are defined as xerogels which are left to rehydrate in 0.1 M KCl pH 4 electrolyte for between 1–4 hours. The exception to this is for SANS experiments where the timescale of the measurement is longer and, in this case, the samples were left to rehydrate for 50 hours prior to the start of the experiment. Water contents for the xerogel and dry solution were obtained by TGA (thermogravimetric analysis) (Fig. S1, ESI†) with samples studied between 20 and 200 °C and held at 120 °C for 20 minutes. The hydrogel and rehydrated xerogel samples were found to dry immediately upon exposure to air and the transfer time from the FTO to the TGA apparatus prevented reliable water content data from being obtained in this manner. Instead water contents of hydrogel and rehydrated xerogel samples were measured by room temperature mass loss in air. Samples were placed directly onto a balance, surface water removed, and the mass monitored for 24 hours.Femtosecond transient absorption (TA) spectroscopy
TA spectroscopy was carried out using a HELIOS spectrometer (Ultrafast Systems) PHAROS laser (Light Conversion Ltd) operating at 10 kHz coupled to an ORPHEUS optical parametric amplifier (Light Conversion Ltd) in tandem with a LYRA harmonic generator (Light Conversion Ltd). The system has been described in detail elsewhere.15 The pump wavelength was 490 nm (275 μW) with samples excited at 5 kHz. Thin film PBI-A (20 μL PBI-A, 7.5 mg mL−1, gelled over 150 mm2 area on FTO) hydrogel and partially rehydrated xerogel samples were recorded in a 2 × 1 cm2 quartz cuvette containing KCl electrolyte (0.1 M KCl, pH 4 adjusted by 1 M HCl). LDM analysis was carried out using the module within the OPITMUS software.31UV/Vis spectroscopy, electrochemistry, spectroelectrochemistry
UV/Vis spectroscopy was carried out using a Shimadzu UV-2600 spectrophotometer. To study the reduced PBI-A samples a 2 × 1 cm2 quartz cell was used containing a 5 μL PBI-A FTO electrode, reference and counter electrodes in KCl electrolyte (0.1 M, pH 4 adjusted with 1 M HCl), spectra were recorded at 0.1 V steps negative from open circuit potential, data shown in Fig. 4 is for the first reductive process observed. To study the PBI-A oxidations an increased spectral window was needed therefore experiments were carried out using ITO coated SrF2 windows coated with PBI-A. Samples were placed within a Harrick cell with 1 mm pathlength with Ag and Pt wires placed close to the PBI-A electrode as the pseudo reference and counter electrode respectively.Electrochemical measurements were carried out in a custom-designed 3 electrode cell. The working electrode was the PBI-A sample on an FTO glass substrate connected via a crocodile clip and nickel wire. The reference electrode was Ag/AgCl (3.5 M KCl), a counter electrode of Pt wire, and KCl electrolyte (0.1 M, pH 4 adjusted by HCl) were used in all experiments. Palmsens potentiostat was used to carry out electrochemical methods. Prior to all experiments the cell was degassed under a stream of argon. Square wave voltammetry was performed under a nitrogen atmosphere at a frequency of 25 Hz with an equilibration time of 90 or 120 seconds. During DCVA measurements the change in transmission of the PBI-A sample is measured against time with a Shimadzu UV-2600 spectrophotometer. Transmission versus time measurements were carried out at 487 nm, 608 nm, 729 nm and 820 nm whilst running a CV at 100 mV s−1 between +1.4 V to −0.8 VAg/AgCl.
SEM
SEM was carried out using an Hitachi S-4800 FE-SEM. Silver conductive paste was used to adhere the FTO/PBI-A electrode to the surface of an SEM stub, and silver paste was coated around the top and outer edges of the electrode to create a connection to the SEM stub. Samples were measured in deceleration mode with an acceleration voltage of 1.5 kV, using an upper electron detector, and a working distance of 3 mm. Due to a large build-up of charge the SEM had to be taken immediately after focusing.Small angle neutron scattering (SANS)
SANS measurements were carried out on the SANS2D instrument at the STFC ISIS Pulsed Neutron Source (Didcot, UK). PBI-A solutions were prepared at 7.5 mg mL−1 as above, except D2O and NaOD were used. Gels were prepared on quartz discs with a 1 mm PTFE spacer creating a well for the gel. Here, the gels were thicker compared to that used for previous experiments (specifically 240 μL over 113 mm2), and were required to fill the cell and obtain a suitable signal from the scattering and were gelled as above using DCl vapour. Hydrated gels were covered with a second quartz disc before loading into aluminum cells, as were the xerogels once dried and the rehydrated xerogels were loaded into the same cells with the 1 mm space filled with KCl electrolyte (0.1 M, pD 4) made in D2O. All cells were housed in a temperature-controlled sample rack at 25 °C during the measurements. The datasets were background subtracted with the empty quartz cell and D2O as appropriate. Where necessary the amount of D2O background to be subtracted was scaled according to the transmission to account for small variations in cell spacer thickness.All raw analytical is freely available from the University of Liverpool data catalogue and can be found at http://datacat.liverpool.ac.uk/.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The UK Central Laser Facility (STFC) Laser Loan Pool is acknowledged for access to the TA spectrometer (EP/G03088X/1). AC, DA, BG acknowledge the Leverhulme Trust for funding (RPG-2018-013). Dr M. Forster and Dr K. Arnold are thanked for help with SEM measurements and Dr J. Lee for assistance with TA measurements. Prof. T. Veal is thanked for the ITO coated SrF2. DA thanks the EPSRC for a Fellowship (EP/L021978/1). ED thanks the Leverhulme Trust for funding (ECF-2017-223) and the University of Glasgow for an LKAS Leadership Fellowship. Experiments at the ISIS Pulsed Neutron and Muon Source were supported by a beamtime allocation (RB1720272) from the Science and Technology Facilities Council. This work benefitted from SasView software, originally developed by the DANSE project under NSF award DMR-0520547.References
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156–11162 CrossRef CAS PubMed.
- R. Marty, R. Nigon, D. Leite and H. Frauenrath, J. Am. Chem. Soc., 2014, 136, 3919–3927 CrossRef CAS PubMed.
- S. Wolter, K. Magnus Westphal, M. Hempel, F. Würthner, O. Kühn and S. Lochbrunner, J. Chem. Phys., 2017, 50, 184005 Search PubMed.
- R. J. Lindquist, B. T. Phelan, A. Reynal, E. A. Margulies, L. E. Shoer, J. R. Durrant and M. R. Wasielewski, J. Mater. Chem. A, 2016, 4, 2880–2893 RSC.
- P. E. Hartnett, E. A. Margulies, H. S. S. R. Matte, M. C. Hersam, T. J. Marks and M. R. Wasielewski, Chem. Mater., 2016, 28, 3928–3936 CrossRef CAS.
- S. Berardi, V. Cristino, M. Canton, R. Boaretto, R. Argazzi, E. Benazzi, L. Ganzer, R. Borrego Varillas, G. Cerullo, Z. Syrgiannis, F. Rigodanza, M. Prato, C. A. Bignozzi and S. Caramori, J. Phys. Chem. C, 2017, 121(33), 17737–17745 CrossRef CAS.
- H. Marciniak, X. Q. Li, F. Würthner and S. Lochbrunner, J. Phys. Chem. A, 2011, 115, 648–654 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. Más-Montoya and R. A. J. Janssen, Adv. Funct. Mater., 2017, 27, 1605779 CrossRef.
- K. M. Lefler, D. T. Co and M. R. Wasielewski, J. Phys. Chem. Lett., 2012, 3, 3798–3805 CrossRef CAS PubMed.
- S. Roy, D. Kumar Maiti, S. Panigrahi, D. Basak and A. Banerjee, RSC Adv., 2012, 2, 11053 RSC.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390–410 CAS.
- J. J. Walsh, J. R. Lee, E. R. Draper, S. M. King, F. Jackel, M. A. Zwijnenburg, D. J. Adams and A. J. Cowan, J. Phys. Chem. C, 2016, 120, 18479–18486 CrossRef CAS.
- E. R. Draper, L. J. Archibald, M. C. Nolan, R. Schweins, M. A. Zwijnenburg, S. Sproules and D. J. Adams, Chem. – Eur. J., 2018, 24, 4006–4010 CrossRef CAS PubMed.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143 RSC.
- A. Nowak-Król, B. Fimmel, M. Son, D. Kim and F. Würthner, Faraday Discuss., 2015, 185, 507–527 RSC.
- Y. Wu, R. M. Young, M. Frasconi, S. T. Schneebeli, P. Spenst, D. M. Gardner, K. E. Brown, F. Würthner, J. F. Stoddart and M. R. Wasielewski, J. Am. Chem. Soc., 2015, 137, 13236–13239 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, J. Am. Chem. Soc., 2015, 137, 15241–15246 CrossRef CAS PubMed.
- M. C. Nolan, J. J. Walsh, L. L. E. Mears, E. R. Draper, M. Wallace, M. Barrow, B. Dietrich, S. M. King, A. J. Cowan and D. J. Adams, J. Mater. Chem. A, 2017, 5, 7555–7563 RSC.
- J. Yang, H. Miao, Y. Wei, W. Li and Y. Zhu, Appl. Catal., B, 2019, 240, 225–233 CrossRef CAS.
- J. T. Kirner, J. J. Stracke, B. A. Gregg and R. G. Finke, ACS Appl. Mater. Interfaces, 2014, 6, 13367–13377 CrossRef CAS PubMed.
- J. T. Kirner and R. G. Finke, J. Mater. Chem. A, 2017, 5, 19560–19592 RSC.
- L. Yao, A. Rahmanudin, N. Guijarro and K. Sivula, Adv. Energy Mater., 2018, 8, 1802585 CrossRef.
- E. R. Draper, L. Wilbraham, D. J. Adams, M. Wallace and M. Zwijnenburg, Nanoscale, 2019, 11, 15917–15928 RSC.
- L. L. E. Mears, E. R. Draper, A. M. Castilla, H. Su, B. Dietrich Zhuola, M. C. Nolan, G. N. Smith, J. Doutch, S. Rogers, R. Akhtar, H. Cui and D. J. Adams, Biomacromolecules, 2017, 18, 3531–3540 CrossRef CAS PubMed.
- E. R. Draper, B. J. Greeves, M. Barrow, R. Schweins, M. A. Zwijnenburg and D. J. Adams, Chem, 2017, 2, 716–731 CAS.
- http://www.sasview.org .
- C. Slavov, H. Hartmann and J. Wachtveitl, Anal. Chem., 2015, 87, 2328–2336 CrossRef CAS PubMed.
- G. F. Dorlhiac, C. Fare and J. J. van Thor, PLoS Comput. Biol., 2017, 13, e1005528 CrossRef PubMed.
- F. Würthner, A. Sautter, D. Schmid and P. J. Weber, Chem. – Eur. J., 2001, 7, 894–902 CrossRef.
- F. Ronconi, Z. Syrgiannis, A. Bonasera, M. Prato, R. Argazzi, S. Caramori, V. Cristino and C. A. Bignozzi, J. Am. Chem. Soc., 2015, 137, 4630–4633 CrossRef CAS PubMed.
- X. Li, K. Xu, H. Li, S. Yao, Y. Li and B. Liang, RSC Adv., 2016, 6, 8395–8405 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- D. J. Woods, R. S. Sprick, C. L. Smith, A. J. Cowan and A. I. Cooper, Adv. Energy Mater., 2017, 7, 1700479 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05839g |
| ‡ Present address: Institute of Applied Physics, Vienna University of Technology, Vienna, 1040, Austria. |
| This journal is © the Owner Societies 2019 |