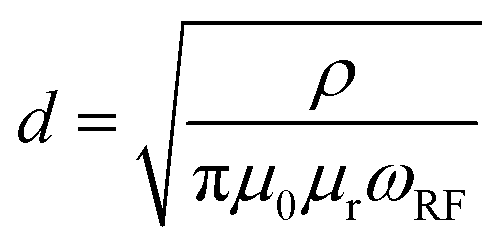In situ
7Li-NMR analysis of lithium metal surface deposits with varying electrolyte compositions and concentrations†
Received
27th September 2019
, Accepted 5th November 2019
First published on 20th November 2019
Abstract
A major challenge of lithium metal electrodes, in theory a suitable choice for rechargeable high energy density batteries, comprises non-homogeneous lithium deposition and the growth of reactive high surface area lithium, which eventually yields active material losses and safety risks. While it is hard to fully avoid inhomogeneous deposits, the achievable morphology of the occurring lithium deposits critically determines the long-term cycling behaviour of the cells. In this work, we focus on a combined scanning electron microscopy (SEM) and 7Li nuclear magnetic resonance spectroscopy (7Li-NMR) study to unravel the impact of the choice of conducting salts (LiPF6 and LiTFSI), solvents (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC, 3:7, DME:DOL, 1:1), as well as their respective concentrations (1 M, 3 M) on the electrodeposition process, demonstrating that lithium deposition morphologies may be controlled to a large extent by proper choice of cycling conditions and electrolyte constituents. In addition, the applicability of 7Li-NMR spectroscopy to assess the resulting morphology is discussed. It was found, that lithium deposition analysis based on the 7Li chemical shift and intensity should be used carefully, as various morphologies can lead to similar results. Still, our case study reveals that the combination of SEM and NMR data is rather advantageous and offers complementary insights that may provide pathways for the future design of tailored electrolytes.
:DEC, 3:7, DME:DOL, 1:1), as well as their respective concentrations (1 M, 3 M) on the electrodeposition process, demonstrating that lithium deposition morphologies may be controlled to a large extent by proper choice of cycling conditions and electrolyte constituents. In addition, the applicability of 7Li-NMR spectroscopy to assess the resulting morphology is discussed. It was found, that lithium deposition analysis based on the 7Li chemical shift and intensity should be used carefully, as various morphologies can lead to similar results. Still, our case study reveals that the combination of SEM and NMR data is rather advantageous and offers complementary insights that may provide pathways for the future design of tailored electrolytes.
Introduction
The current demand of energy storage solutions for portable electronic devices, electro-mobility but also for stationary (“grid”) storage systems requires availability of affordable, preferably fast charging and high energy density batteries.1–3 While lithium metal anodes offer high theoretical specific capacity (3860 mA h g−1), low weight, and also low redox potential (−3.04 V vs. standard hydrogen electrode),4–8 the often inhomogeneous lithium deposition upon cycling and its associated safety risks tend to impede widespread commercial application of lithium metal batteries (LMBs).4–7,9–16 Indeed, the explicit nature of micro-structured lithium deposits is critically determined by a variety of factors including environmental (e.g. pressure,17 temperature,18 and current density19,20) and chemical (electrolyte salt,21 solvents,22 and salt concentration23) conditions, where the electrolyte composition defines the solvation structure and actual reactivity of the electrolyte, including features of solid electrolyte interphase (SEI) layers on lithium anodes due to decomposition of electrolyte constituents.24 The nature of the SEI, for example its conductivity for lithium ions, its composition, and flexibility is therefore crucial for the deposition process.25,26 Ideally, the surface layer should have low resistance, facilitating fast lithium ion transport despite being electronically blocking. A promising concept to enhance the cycling performance in LMBs comprises the application of highly concentrated (≥2 M) electrolytes.23,27–29 Despite their significant costs, highly concentrated electrolytes mitigate operational safety risks due to lower flammability, allowing for lesser deposition of ‘needle-like’ lithium27 but rather growth of denser lithium deposits that have reduced specific surface areas compared to 1 M electrolyte solutions.23,28
Since the actually occurring microstructure of lithium deposits may constitute a major challenge for routine application of lithium metal anodes, in this work, the impact of conducting salts, the choice of solvent as well as the salt concentration of the electrolytes on the nature of lithium deposits was considered. While ex situ solid-state NMR of electrode materials with various elements (nuclei) is often used in battery research,30 applications of in situ NMR (where thin film pouch cells17,31–33 or other cell setups34 are operated within the NMR magnet) are reported less frequently. In favourable cases, spatial resolution of the lithium distribution may be gained via magnetic resonance imaging (MRI), though a more complex setup is required.35–39 In 7Li-NMR, corresponding signals for lithium ions within both the liquid electrolyte and the SEI appear at chemical shifts around 0 ppm, while metallic lithium due to Knight shifts has a higher 7Li-NMR chemical shift at about 250 ppm.17,31 Lithium deposits with morphologies different from (‘smooth’) bulk lithium and varying orientations with respect to the external magnetic field due to bulk magnetic susceptibility have higher chemical shifts in the range of 260–300 ppm.33 In addition to NMR-based methods, scanning electron microscopy14,23 constitutes a more qualitative but complementary method to determine the nature of lithium deposits, which typically is applied ex situ (particularly in the presence of volatile solvents), revealing localized insight into lithium deposits.
Herein, we monitor lithium deposition phenomena combining electrochemical measurements, in situ7Li-NMR spectroscopy and SEM data for various electrolyte compositions and salt concentrations, thereby discussing the applicability of 7Li-NMR spectroscopy to assess the morphology of lithium deposits. In addition, higher concentrations of the most promising electrolyte formulation are utilized to elucidate if changes of achievable lithium deposit morphologies towards smoother and larger particles could be obtained.23 All studies were performed in symmetrical Li‖Li cells to evaluate the influence of the electrolyte on the lithium deposition, even though it was shown, that also the choice of the cathode has an effect on the deposition behaviour.40
Experimental
Cell preparation and assembly
Symmetrical Li‖Li thin film pouch cells (Fig. 1b, a similar cell setup was also used by others17,32,33) prepared in a dry room with H2O concentrations below 200 ppm were used for all electrochemical investigations, except for impedance measurements, where a 2032 coin cell setup was used.41
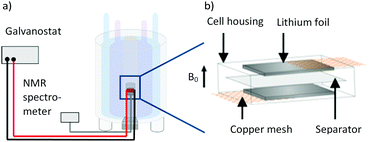 |
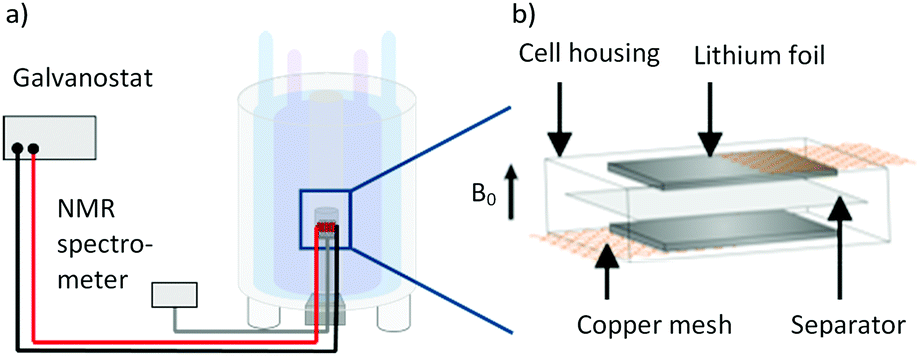
| | Fig. 1 (a) Schematic depiction of the measurement setup with a thin film pouch cell placed in the coil of a NMR device, connected to a battery cycler. Redrawn from ref. 33. (b) Schematic thin film pouch cell with the cell housing, 300 μm roll pressed lithium foil, the current collector copper mesh and PP-based separator (Celgard® 2500 separator). | |
Ethylene carbonate (EC, Powerlyte BASF, battery grade) and diethyl carbonate (DEC, Powerlyte BASF, battery grade) in a ratio of 3:7 by wt, as well as ethylene glycol dimethyl ether (DME, Alfa Aesar, 99+%, stab. with BHT) and 1,3-dioxolane (DOL, Alfa Aesar, 99.5%, stab.) in a ratio of 1:1 by wt were used as solvents for electrolyte formulations. Lithium hexafluorophosphate (LiPF6, BASF, battery grade) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, Solvionic, 99.9%) were employed as conducting salts. All chemicals were used as received, except for LiTFSI that was dried (120 °C, in vacuo) prior to use. The considered electrolyte formulations are summarized in Table 1.
Table 1 Electrolyte formulations and corresponding short names. The short names consist of: concentration in M – anion – solvents (C: carbonates, E: ethers)
| Electrolyte |
Salt concentration |
Salt |
Solvent |
| 1-PF6-C |
1 M |
LiPF6 |
EC:DEC (3:7) |
| 1-TFSI-C |
1 M |
LiTFSI |
EC:DEC (3:7) |
| 1-TFSI-E |
1 M |
LiTFSI |
DME:DOL (1:1) |
| 3-TFSI-E |
3 M |
LiTFSI |
DME:DOL (1:1) |
Lithium metal foils were prepared as described elsewhere15 proceeding from 500 μm thick lithium metal precursors (Albemarle, battery grade), which were pressed between two siliconized biaxial-oriented polyethylene terephthalate (boPET) foils with a roll press (Hohsen Corp., HSAM-615H) to final thicknesses of 300 μm in 25 μm decrements. The foil was then cut into dimensions of 25 mm × 5 mm (±5%) and pressed on a carbon coated copper mesh (Benmetal) with a roll press. Each cell consisted of two such electrodes with a Celgard® 2500 separator (polypropylene, PP, 25 μm thickness, 55% porosity, with average pore sizes of 0.209 μm × 0.054 μm)42 in between. For the in situ NMR measurements, a coffee bag foil (Moreno) was used as cell case, while multilayer foils consisting of polyamide (PA), aluminium (Al) and PP were used for long-term cycling. The foils were wrapped around the electrodes and separator, and hermetically sealed under vacuum. The respective electrolyte formulation was stored in a PP bag during the assembling process and released after sealing. For the coin cell setup, two roll pressed lithium metal foil electrodes with a diameter of 12 mm and a Celgard® 2500 separator with a diameter of 13 mm were used.
Electrochemical testing and determination of ionic conductivity
Long-term electrodeposition/-dissolution tests were performed on a battery cycler (MACCOR Series 4000, MACCOR INC.) in a temperature chamber (BINDER KB 400) at 20 °C. The cycling behaviour was observed for 300 cycles with 1 hour electrodeposition and subsequent 1 hour electrodissolution at a current density of 0.5 mA cm−2 for each cycle. For electrodeposition measurements, a current density of −0.5 mA cm−2 was applied to the cell for 8 h. A Solartron SI 1287 potentiostat in combination with a Solartron SI 1260 impedance/gain-phase analyser was utilized for in situ deposition experiments as well as for impedance analysis. The impedance spectra were obtained at an amplitude of 5 mV in a frequency range from 1 MHz to 0.1 Hz. All coin cells were stored at 20 °C for eight days and measured after various days. The data was fitted using a R–C equivalent circuit containing an electrolyte resistance (R1) in series with a parallel constant phase element (CPE) and another resistance, representing processes at the electrode surfaces (R2). The ionic conductivity was derived from impedance measurements at an amplitude of 5 mV from 1 MHz to 0.5 Hz in a two-electrode glass cell setup with platinum blocking electrodes; the cells were calibrated with a 0.01 M KCl solution (VWR Chemicals).
Scanning electron microscopy
The surface of the electrodes was investigated with an Auriga field emission scanning electron microscope (FE-SEM) Crossbeam workstation equipped with a Schottky field emission gun by Carl Zeiss AG. To obtain the images, an in-lens secondary electron detector at an acceleration voltage of 3 kV was used. All images were taken at a working distance of 5 mm. To remove electrolyte residues after disassembling the cells in a dry room, the electrodes in case of either EC:DEC or DME:DOL-based electrolytes were washed (2 × 0.5 mL) with DEC or DME, respectively. The electrodes were placed in a sealed sample holder and transported to the SEM device without air or moisture contact.
Nuclear magnetic resonance spectroscopy
The NMR experiments were performed on a Bruker AVANCE III 200 MHz (4.7 T) spectrometer with an in-house build broadband (1H, 19F)//(X = 6Li–7Li) cross-polarization double-resonance probe containing a Helmholtz-type coil at room temperature.43 The NMR spectra were referenced to 1 M LiCl solution in H2O (7Li peak set to 0 ppm). All measurements were performed at a resonance frequency of 77.79 MHz with a π/12 excitation pulse and relaxation delays of 2 s (256 scans were averaged for each spectrum); the data was recorded and processed with Bruker Topspin 3.5 package. The corresponding NMR spectra were analysed using MestReNova v12.0.0-20080, CasaXPS 2.3.16 PR 1.6, and OriginPro 2016G (64 bit) Sr2 b9.3.2.303 software. Note that 7Li-NMR of bulk lithium electrodes may suffer from a limited detection skin depth (that is the penetration depth of the excitation pulse) resulting in signal intensities governed from the upper surface rather than the total volume of lithium metal anodes, which are thicker than two times the skin depth.31 The skin depth d that a radio frequency (rf) pulse may penetrate is given by eqn (1),31| | 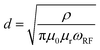 | (1) |
and is related to the resistivity of lithium (ρ = 92.8 nΩ m), the permeability of the vacuum (μ0 = 4π × 10−7 m kg A−2 s−2), the relative permeability of lithium metal (μr = 1.00002), as well as the Larmor frequency of the rf field (ωrf = 2π·77.8 MHz for 7Li at a 4.7 T magnet), affording a skin depth of d = 17.4 μm.17,31 Assuming that surface lithium deposits are on the order of few μm thus thinner than the experimental skin depth, the measured intensity of the 7Li-NMR peaks in principle reflects the total amount of deposited lithium.31
FT-Raman spectroscopy
A RAM II FT-Raman Module (Bruker) on a VERTEX 70 FT-IR spectrometer (Bruker) with a nitrogen-cooled Ge-diode detector and a 1064 nm laser source was used to perform FT-Raman spectroscopic investigations. The liquid samples were measured in 5 mm NMR-Tubes (Bruker) with OPUS 7.0. To fit the obtained spectra, CasaXPS 2.3.16 PR 1.6 was used.
Viscosity measurements
The viscosities of the electrolytes were measured at 25 °C (±0.5 °C) with an Anton Paar MCR 302 rheometer and a CP50.0.5 cone placed in a dry room with H2O contents below 200 ppm.
Results and discussion
A variety of electrolyte formulations were considered to monitor their impact on the observable lithium deposition phenomena in lithium metal batteries. All electrolytes (see Table 1, Experimental section) were evaluated with respect to achievable cell resistances and electrochemical performances, while the nature of the lithium deposits was determined from both in situ7Li-NMR and post mortem SEM analysis.
Variation of conducting salts and solvents
Ionic conductivity and viscosity.
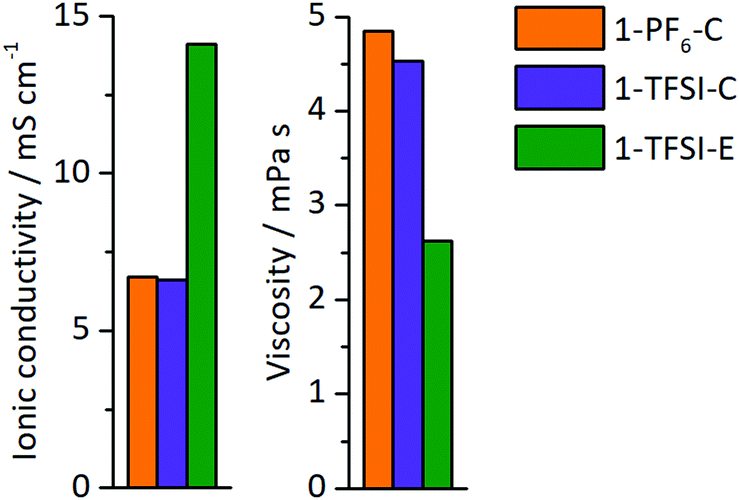
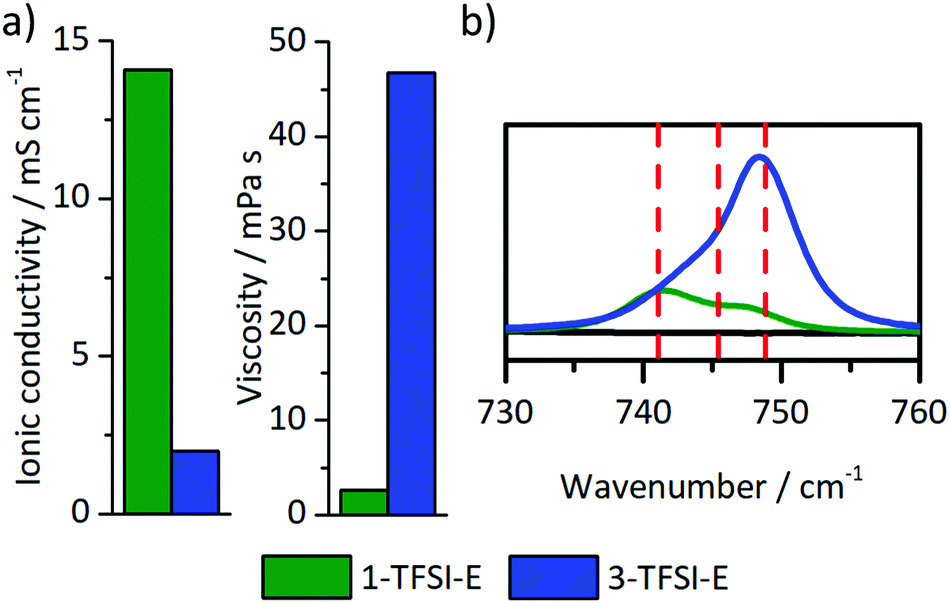
The ionic conductivity and viscosity of the electrolytes 1-PF6-C (1 M LiPF6 in EC:DEC, 3:7), 1-TFSI-C (1 M LiTFSI in EC:DEC, 3:7), and 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1) were measured (Fig. 2), and the results showed that both carbonate-based electrolytes have comparable ionic conductivities (1-PF6-C, 6.7 mS cm−1 and 1-TFSI-C, 6.6 mS cm−1) and viscosities (1-PF6-C: 4.9 mPa s, 1-TFSI-C: 4.5 mPa s). The electrolyte 1-TFSI-E exhibits an ionic conductivity (14.1 mS cm−1) that is twice as high and a viscosity (2.6 mPa s) only half of the carbonate-based electrolytes, indicating that present solvents rather than the nature of the anions are responsible for the observable properties of the considered electrolyte formulations, despite that specific anion solvation effects were suggested in case of for example adiponitrile-based electrolytes.44
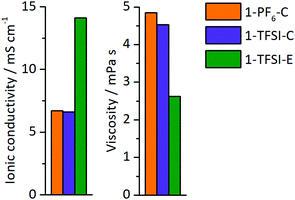 |
| | Fig. 2 Ionic conductivity and viscosity of 1-PF6-C (1 M LiPF6 in EC:DEC, 3:7, orange), 1-TFSI-C (1 M LiTFSI in EC:DEC, 3:7, purple), and 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green). | |
Cell resistance of Li‖Li coin cells in contact with the different electrolytes.
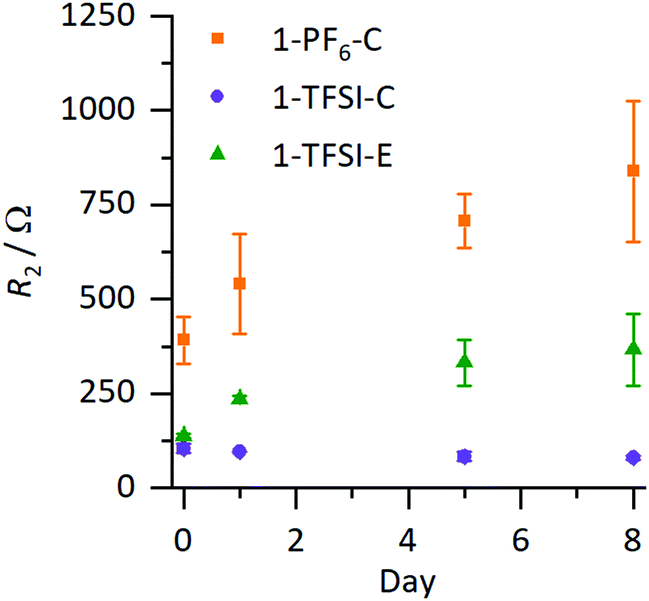
The evolution of the cell resistances was monitored via electrochemical impedance spectroscopy (EIS), where the resulting resistances of the coin cells are presented in Fig. 3 (corresponding Nyquist plots are shown in Fig. S1a–c, ESI†); the values are collected in Table S1 (ESI†). In Nyquist plots of symmetrical Li‖Li cells, the resistance R1 (mainly of the electrolyte) is derived from the intercept with the abscissa at high frequencies (>500 kHz), while lower frequencies (<100 kHz) reflect resistances of the surface films and charge transfer resistance, which are challenging to distinguish and therefore combined to R2.14 Changes in R2 are attributed to any variations of the SEI layer, assuming that resistances of other contributing processes are constant.14 Typically, the values of R2 in symmetrical Li‖Li cells are reported in a range of less than hundred to several thousand Ohm, depending on the electrolyte formulation and type of utilized separator within the cells, in agreement with our data (Table S1, ESI†).14,15R1 was comparable for all evaluated electrolyte formulations (3–14 Ω, Table S1, ESI†), while in case of 1-PF6-C electrolyte, R2 increased from (391 ± 63 Ω) to (838 ± 188 Ω) after eight days, strongly indicating continuous evolution of surface layers even under open circuit voltage conditions (OCV), reflecting that the formed SEI was not suitable to prevent further decomposition of the electrolyte. In contrast, for 1-TFSI-C, R2 did not change significantly during eight days after cell assembly (80–100 Ω), suggesting that the formed surface layer in the presence of 1-TFSI-C sufficiently passivates lithium metal from further reaction. Note that cells with ether-based electrolyte formulation 1-TFSI-E exhibited an initial increase of R2, which continued at slower pace compared to 1-PF6-C (evolving from 140 ± 5 Ω to 366 ± 95 Ω), in agreement with ongoing SEI growth.
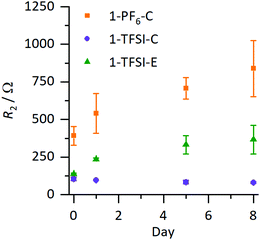 |
| | Fig. 3 Development of R2 under OCV conditions of Li‖Li coin cells containing 1-PF6-C (1 M LiPF6 in EC:DEC, 3:7, orange), 1-TFSI-C (1 M LiTFSI in EC:DEC, 3:7, purple), and 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green), derived from impedance measurements. The corresponding Nyquist plots are shown in Fig. S1 (ESI†). The error margins include contributions from local defects and small variations of the lithium surface as well as partially electrolyte evaporation. | |
Fig. 3 clearly illustrates trends of the development of R2, revealing comparable behaviour of the electrolytes 1-TFSI-C and 1-TFSI-E that is different from 1-PF6-C. The observation of higher initial R2 and its development under OCV conditions in case of 1-PF6-C may be attributed either to the formation of thicker SEI layers due to the reactivity of 1-PF6-C towards lithium compared to ether-based electrolytes, or to eventually different SEI compositions at grain boundaries.
Long-term electrodeposition/-dissolution behaviour.
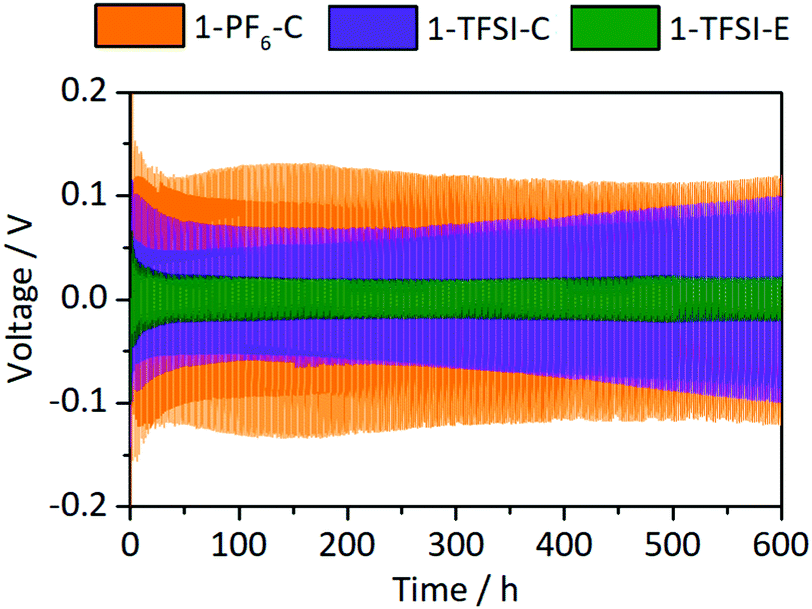
Constant current cycling of symmetrical Li‖Li thin film pouch cells with 1-PF6-C, 1-TFSI-C or 1-TFSI-E electrolytes was performed to evaluate the lithium deposition properties based on the corresponding overvoltage during lithium electrodeposition (negative scale) and -dissolution (positive scale) (Fig. 4). The overvoltage results from kinetic aspects such as ion transport or resistances of the SEI,14 and typically ranges from initially ±0.3 V or higher to ±0.025 V at later cycles, though transient changes of both the applied cycling conditions (e.g. due to cell aging) and the electrolyte may contribute to larger overvoltage.14,15 Note that in the considered cell-setup, the voltage between two lithium electrodes should be 0 V in the absence of disturbing effects, so that any voltages different from 0 V clearly reflect non-faradaic contributions. No external pressure, which was shown to decrease the amount of high surface area lithium (HSAL),17 was applied during the measurements, allowing for an assessment of the impact of increased surface area on the resulting overvoltage.
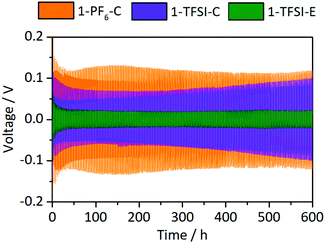 |
| | Fig. 4 Development of the overvoltages during lithium electrodeposition/-dissolution at a current density of 0.5 mA cm−2 in symmetrical Li‖Li thin film pouch cells containing 1-PF6-C (1 M LiPF6 in EC:DEC, 3:7, orange), 1-TFSI-C (1 M LiTFSI in EC:DEC, 3:7, purple), and 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green), respectively. An enlarged picture of the first discharge is given in Fig. S2 (ESI†). | |
Cells containing 1-PF6-C (orange) exhibited a rather high initial overvoltage of −0.67 V (after 24 h under OCV conditions, Fig. S2, ESI†), which is in agreement with the observed R2 after one day (Fig. 3 and Fig. S1, Table S1, ESI†), likely illustrating the presence of a thicker and/or poorly lithium conducting SEI. During the following cycles, the overvoltage decreased to ±0.13 V, indicative of both crack formation in the SEI and inhomogeneous lithium deposition,14 also corroborated by smaller R2 after cycling (Fig. S3a, ESI†). Notably, cells containing 1-TFSI-C (purple) showed a much lower initial voltage (−0.15 V), even decreasing to values about ±0.06 V after several cycles, consistent with lower surface resistances (Fig. 3 and Fig. S1, Table S1, ESI†). The overvoltage reached ±0.1 V in the 300th cycle, likely due to the formation of thicker SEI layers and/or parasitic consumption of solvent, yielding higher viscosity and lower ionic conductivity of the residual electrolyte. Indeed, increased R1 after cycling observed in the impedance spectra (Fig. S3a, ESI†) match with the occurrence of electrolyte degradation, while the barely decreasing surface resistance even in the presence of inhomogeneous lithium deposition (Fig. S4b, ESI†), reflects formation of rather thick SEI layers. In contrast, the overvoltage of cells with 1-TFSI-E electrolyte (green) reduces from initially ±0.1 V to ±0.02 V upon cycling, probably reflecting inhomogeneous lithium deposition and the substantial decrease of the surface resistance (Fig. S3a, ESI†). Note that unlike in case of 1-PF6-C and 1-TFSI-C, the electrolyte resistance remained almost constant (Fig. S3a, ESI†), indicating reduced electrolyte degradation.
Combined 7Li-NMR and SEM study
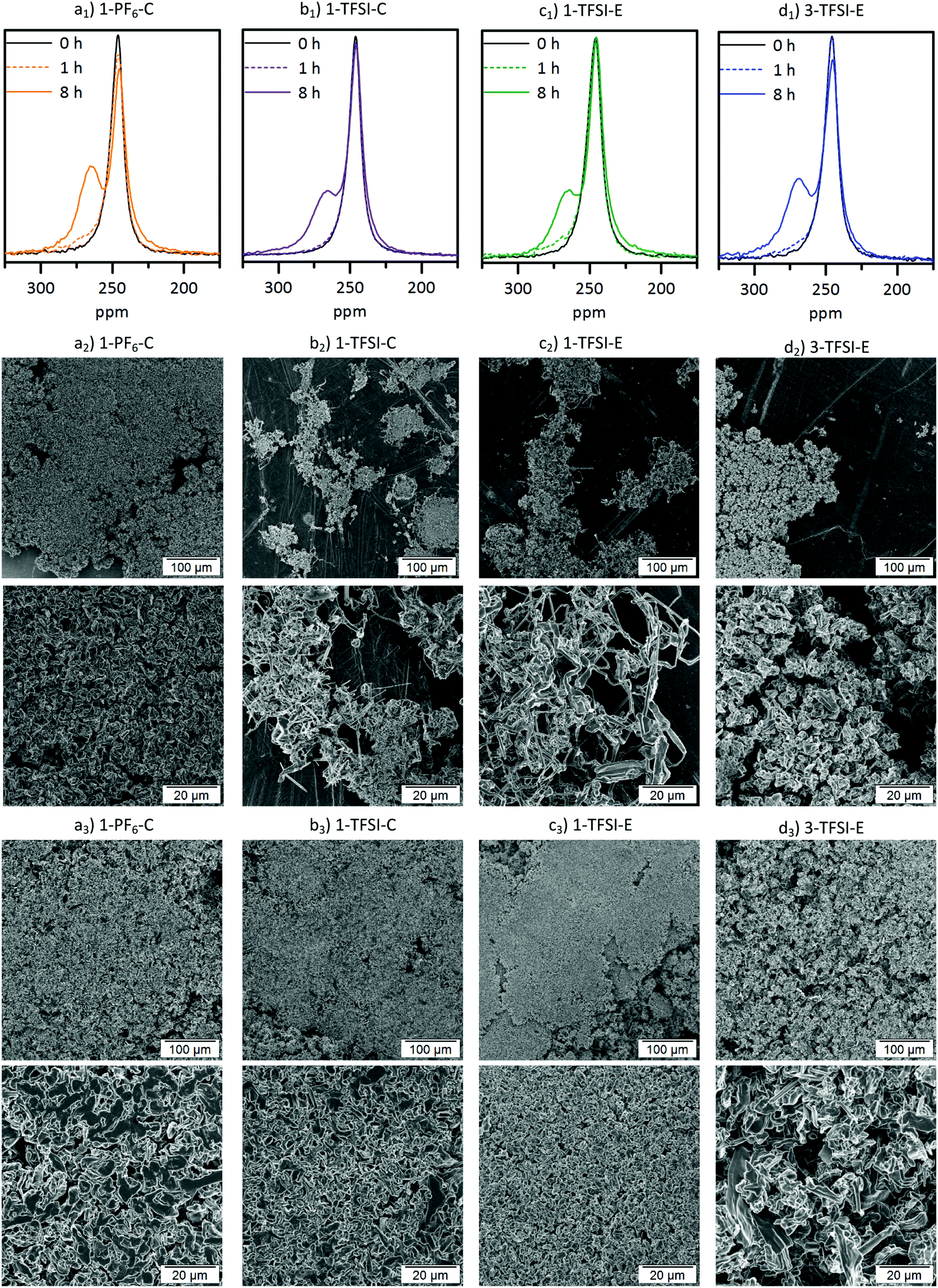
In addition to impedance and constant current cycling (CCC) data, the evolution of lithium deposits can be elegantly monitored by 7Li-NMR. The corresponding spectra and related SEM images after 1 and 8 hours of deposition at a current density of 0.5 mA cm−2 are collected in Fig. 5. All 7Li-NMR spectra (Fig. 5a1–c1) exhibited peaks at 246 ppm (±1 ppm), characteristic for bulk (‘smooth’) lithium, and peaks at 260–270 ppm, reflecting inhomogeneous lithium deposits.17
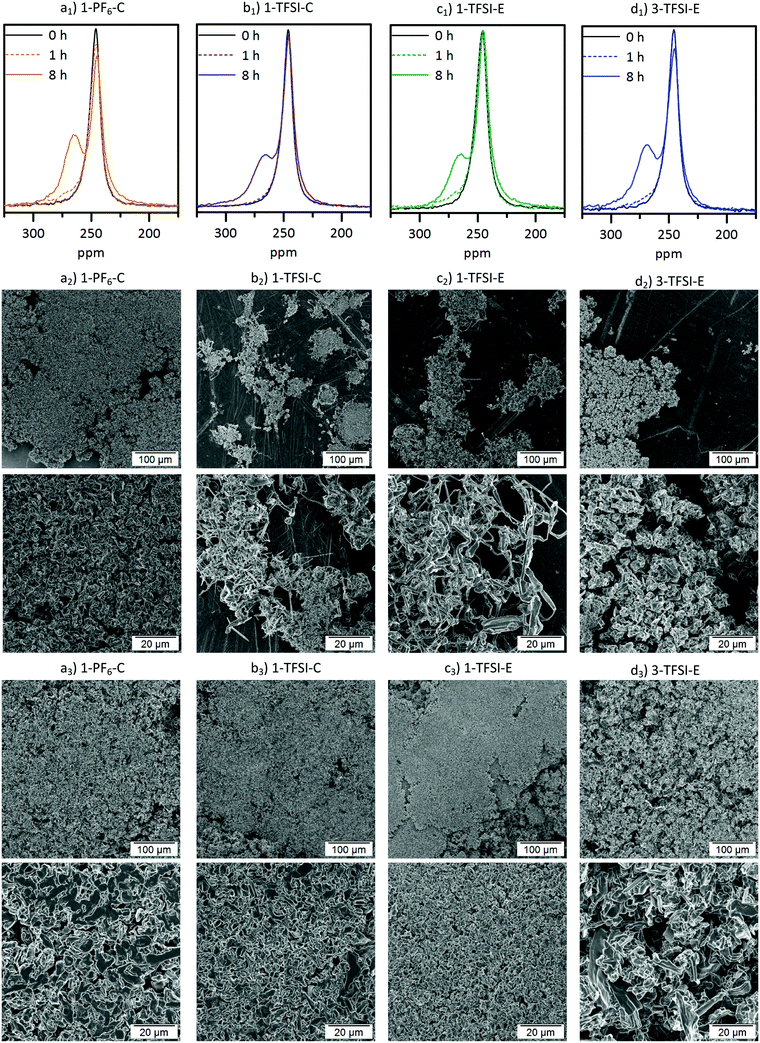 |
| | Fig. 5 (a1–d1) 7Li-NMR spectra of thin film pouch cells and SEM images of the corresponding electrodes after (a2–d2) 1 hour and (a3–d3) 8 hours of electrodeposition at 0.5 mA cm−2 containing (a) 1-PF6-C (1 M LiPF6 in EC:DEC, 3:7, orange), (b) 1-TFSI-C (1 M LiTFSI in EC:DEC, 3:7, purple), (c) 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green) and (d) 3-TFSI-E (3 M LiTFSI in DME:DOL, 1:1, blue). | |
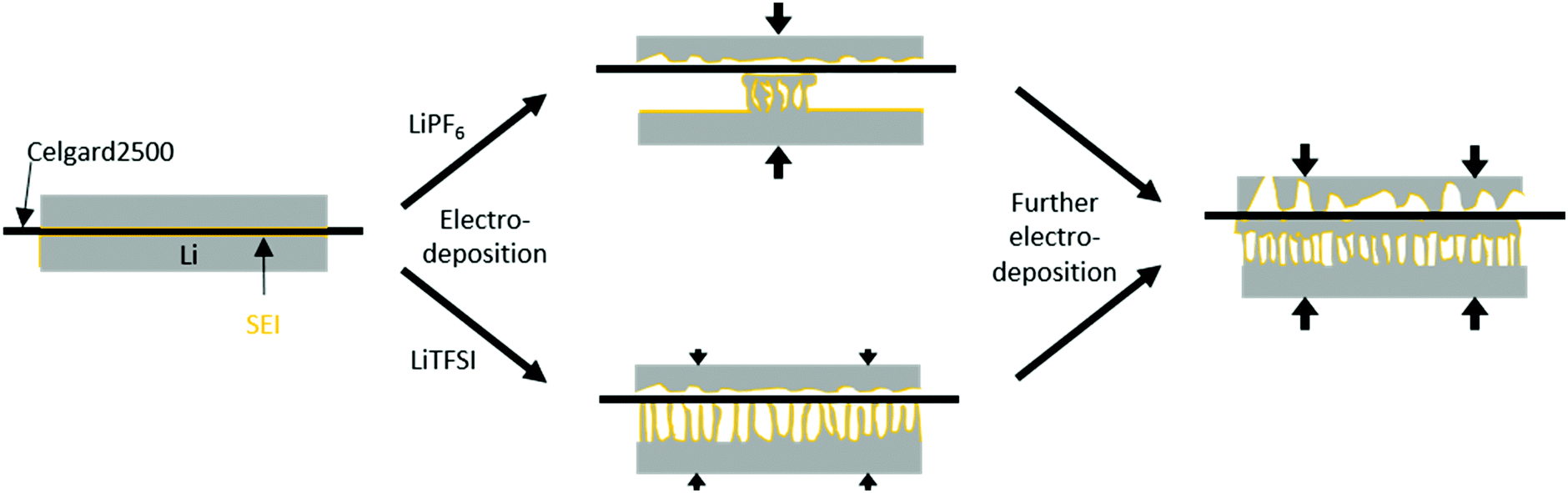
The 7Li-NMR spectra of Li‖Li thin film pouch cells containing 1-PF6-C (Fig. 5a1) exhibited an intensity decrease and broadening of the 7Li-NMR peak at 246 ± 1 ppm upon electrodeposition, in agreement with previous works considering 1 M LiPF6 in EC:DMC electrolytes,17,32 which may be attributed to both a gradual decrease of detectable bulk lithium based on its surface coverage and effects of the lithium deposits on the local magnetic field of the surface lithium.17 A peak at 265 ppm ± 1 ppm appeared upon lithium deposition, thereby revealing the potential nature of the deposited microstructures.17 Rather needle-like lithium deposits often have a 7Li-NMR peak centred at 270 ppm, whereas denser lithium structures, or structures that do not grow perpendicular to the lithium electrode surface are characterized by lower 7Li-NMR chemical shifts. Hence, the peak at 265 ppm ± 1 ppm in the 7Li-NMR spectra of cells with 1-PF6-C electrolyte after 8 hours deposition (Fig. 5a1) indicates formation of rather dense and ‘mossy’ lithium structures. The SEM images of electrodes after 1 hour of lithium deposition show, in good agreement with the 7Li-NMR data, dense mossy HSAL structures (Fig. 5a2), though most of the electrode surface appeared smooth, comparable to pristine electrodes (see ref. 15). After 8 hours of deposition (Fig. 5a3), the electrode surface was fully covered with quite dense lithium deposits, which upon higher magnification, revealed larger particles (>5 μm) compared to the structures observed after merely one hour of deposition time. The increased particle size clearly reflects that the lithium deposits not only grew in one direction, but also in length and width, thereby spreading onto the electrode surface. This observation is corroborated by the lower chemical shift for the lithium deposits, and the decrease of the peak for bulk lithium at 246 ppm. The dense lithium growth and formation of larger particles likely affected a significant fraction of the surface area of bulk lithium, in this way attenuating its response to the rf pulse.17 The 7Li-NMR spectra of Li‖Li thin film pouch cells containing 1-TFSI-C (Fig. 5b1), in contrast did not show a significant decrease of the peak at 246 ppm upon cycling, but rather an additional peak at slightly higher chemical shift (267 ppm ± 1 ppm), reflecting lower density, needle-like deposits. The latter peak did not decrease significantly, but exhibited a slight broadening, likely due to extensive surface coverage of bulk lithium with inhomogeneous deposits. The overall increase of peak intensities (between 175 and 325 ppm) for the cells containing 1-PF6-C and 1-TFSI-C were comparable (51% ± 6% vs. 45% ± 3%, Table 2). Still, considering only the peak of lithium deposits at about 265 to 267 ppm, 1-PF6-C reached 68% ± 6% while 1-TFSI-C only reached 52% ± 4% of the pristine intensity after 8 hours of electrodeposition. The higher intensity of the lithium deposit peak for the 1-PF6-C electrolyte could indicate larger fractions of inhomogeneous lithium deposits. However, one has to consider that also extensive SEI formation, and therefore loss of metallic lithium and intensity of the lithium deposit peak, can lead to a decreased total NMR intensity.31 Since the amount of SEI corresponds roughly to the SEI thickness, but more importantly to the surface of the lithium metal, HSAL as for example needle- but also moss-like lithium could yield much lower intensity of metallic lithium deposits compared to low surface area lithium (LSAL).31,45,46 In principle, SEI formation could be explicitly monitored based on diamagnetic Li+ at chemical shifts of 0 ppm ± 1 ppm, but due to its rather long relaxation times compared to metallic Li this was not considered,31 meaning that the obtained peak intensities of inhomogeneous lithium deposits could not be applied for quantitatively determining the individual fractions of lithium species and will therefore not further be discussed (at short relaxation times, the corresponding area fraction of peaks at 0 ppm will be underrepresented). In the SEM images of electrodes after 1 hour of electrodeposition in case of 1-TFSI-C, small areas with needle-like lithium deposits were visible (Fig. 5b2). After 8 hours of deposition (Fig. 5b3), in the SEM, lithium aggregates appeared quite dense, looking similar to the lithium deposits in case of 1-PF6-C. The denser lithium entities were in contrast to the observed 7Li-NMR chemical shift of 267 ppm ± 1 ppm (reflecting the lithium deposits) and the remaining intensity of the bulk lithium peak (at 245 ppm ± 1 ppm), indicating that the lithium ‘morphology’ beneath the surface was different to the one on the surface, in good agreement with needle-like lithium deposits after 1 hour of electrodeposition. A mechanism of the corresponding lithium deposition is proposed in Fig. 6. The application of pressure may allow for reduction or even suppression of needle-like lithium deposits in the presence of low-porous separators,17 but unlike both coin and Swagelok® T-cells, thin film pouch cells do not cause considerable pressure on the electrodes. Hence, particularly at the beginning of a current flow, needle-like deposits may appear on the electrode surface, if favoured by the considered electrolyte formulation and cycling conditions. After some deposition time (>1 hour), however, the electrode volume expansion due to inhomogeneous lithium deposits induces internal pressure on the electrodes, since the mechanical flexibility of commonly applied pouch foil cell housings is limited. The increased internal pressure in addition to low-porosity separator layers, such as Celgard® 2500 (55% porosity, 0.209 μm × 0.054 μm average pore size),42 eventually result in bending of needle-like lithium deposits, coalescence of lithium microstructures as well as the formation of denser ‘surfaces’. Different from this, e.g. in case of Whatman® separators that are glass fibre-based meshes with pore sizes of 1.0 μm, lithium deposits could grow into the separator so that needle-like lithium deposits are often favoured.17 The proposed bending of needle-like lithium deposits was monitored by the 7Li-NMR chemical shift evolution during longer deposition times, where the peak slowly moved from 268 ppm (8 hours) to 266 ppm (16 hours) (Fig. S5, ESI†). This is corroborated by the denser ‘structure’ of Li deposits obtained for 1-PF6-C, though formation of larger particles at the root of inhomogeneous Li deposits was also estimated via the decrease of the bulk lithium peak at 246 ppm. While during the first hour lithium deposits in case of 1-TFSI-C were quite homogeneously spread (Fig. 5b2), reflecting rather homogeneous pressure distribution, more localized lithium deposits on the electrode surface in case of 1-PF6-C electrolytes indicate higher pressures at the area of lithium deposition, consequently yielding denser surface ‘structures’ even after 1 hour of deposition (Fig. 5a2, scheme: Fig. 6 top). Different from 1-TFSI-C, which shows bending and coalescence of lithium microstructures only after an electrodeposition of more than hour (Fig. 5b2+3, scheme: Fig. 6 bottom), 1-PF6-C, with a locally increased pressure even after less than one hour, also shows a bending and coalescence of lithium microstructures after less than one hour (Fig. 5a2+3, scheme: Fig. 6 top). Note that locally preferred lithium deposition of 1-PF6-C might also be due to local differences of surface resistances based on the presence of surface defects with low resistances and associated high resistances (e.g. 540 ± 133 Ω after one day at OCV conditions, corresponding to the OCV time of the cells prior to current density application, see Fig. 3 and Table S1, ESI†) of formed SEI layers when 1-PF6-C is combined with lithium electrodes. In contrast, a resistance of 96 ± 1 Ω after one day (Fig. 3 and Table S1, ESI†) of the SEI formed in case of 1-TFSI-C likely facilitates faster lithium ion transport, thereby leading to more homogeneous lithium deposition. Commonly, lower resistances render smooth and ‘cracked’ areas of the SEI layers more comparable, so that lithium deposition at cracks is less pronounced, while high surface resistances almost always yield inhomogeneous lithium deposition, reflecting that defects in the SEI are
more favourable for lithium deposition compared to SEI covered areas. The 7Li-NMR spectra of cells with 1-TFSI-E (Fig. 5c1) exhibited similar features compared to 1-TFSI-C (Fig. 5b1). Both showed almost constant peak intensity at 246 ppm, and chemical shifts of 266 ± 1 ppm or 267 ± 1 ppm for lithium deposits. The SEM images in case of 1-TFSI-E were similar to 1-TFSI-C, exhibiting small areas of needle-like lithium deposits after 1 hour (Fig. 5c2) and denser ‘structures’ after 8 hours of deposition (Fig. 5c3), probably due to coalescence of lithium microstructures, bending of lithium deposits on the separator (similar to 1-TFSI-C), or even more homogeneous lithium deposition. Notably, thorough comparison of different electrolyte formulations revealed unlike reported literature44 that the solvent is primarily responsible for the overall viscosity and ionic conductivity, while the nature of anions apparently governs the achievable SEI whose resistance critically determines the rate of local lithium deposition. 1-TFSI-C appears rather promising regarding stable SEI formation and low resistances, while a higher ionic conductivity in case of 1-TFSI-E offers smaller overvoltage upon cycling. Lithium deposition in case of 1-PF6-C appears localized, likely reflecting the higher resistance of SEI layers and formation of larger, but denser lithium ‘structures’ due to locally increased pressures. However, for all considered electrolytes, the resulting SEI was insufficient to prevent continued inhomogeneous lithium deposition, as clearly reflected by a growth of needle-like deposits, in agreement with the observable 7Li-NMR chemical shifts.
Table 2 Comparison of the 7Li-NMR chemical shifts, normalized intensities, and the obtained morphology of lithium deposits from SEM for Li‖Li thin film pouch cells containing 1-PF6-C, 1-TFSI-C and 1-TFSI-E electrolytes after 8 hours of deposition at current densities of 0.5 mA cm−2
|
|
1-PF6-C |
1-TFSI-C |
1-TFSI-E |
| Chemical shift/ppm |
265 ± 1 |
267 ± 1 |
266 ± 1 |
| NMR norm. intensity |
151% ± 6% |
145% ± 3% |
145% ± 7% |
| Lithium morphology (SEM) |
Less dense, larger particles (>5 μm) |
Dense, smaller particles (<3 μm) |
Dense, smaller particles (<3 μm) |
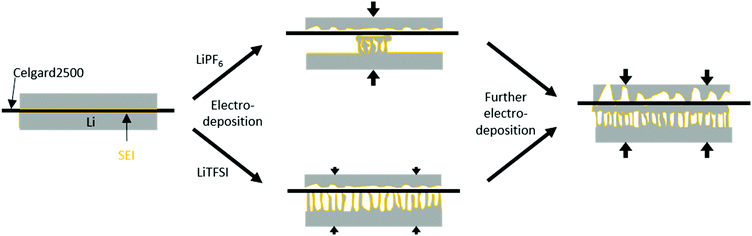 |
| | Fig. 6 Schematic deposition behaviour in Li‖Li thin film pouch cells with a low pressure upon application of a current density. With lithium (grey), covered by a SEI (orange), and a separator with a low porosity (black). The increasing pressure is illustrated by arrows. LiPF6-based electrolytes show a locally accumulated HSAL growth, resulting in locally increased internal pressure, bending and coalescence of lithium microstructures on the separator even after low amounts of electrodeposition (<0.5 mA h cm−2). LiTFSI-based electrolytes do not show local preferences, therefore induce only a low internal pressure and no bending and coalescence of lithium microstructures at low amounts of electrodeposition (<0.5 mA h cm−2). After further electrodeposition (4 mA h cm−2) all electrodes show bending and coalescence of lithium microstructures, induced by an increased internal pressure. | |
Variation of conducting salt concentration
The choice of electrolyte constituents such as the solvent or the conducting salt and its concentration critically determine the electrodeposition/-dissolution behaviour of a considered electrolyte.23,28,29,47–49 High salt concentrations are not only able to reduce aluminium current collector dissolution on the cathode side,47–49 but also yield denser lithium deposits and larger particle sizes.23,28,29 Here, the electrodeposition behaviour of highly concentrated electrolytes is considered, particularly focussing on the resulting microstructures of the lithium deposits compared to reported data. Among the considered electrolytes, 1 M (1-TFSI-E) and 3 M (3-TFSI-E) were compared, based on the achieved cycling performances and beneficial solubility of LiTFSI in DME:DOL mixtures. 1-TFSI-E exhibited increased ionic conductivity and lower viscosity compared to carbonate-based systems, even affording an ionic conductivity of 2 mS cm−1 and a viscosity of 45 mPa s at high salt concentrations (Fig. 7a). For highly concentrated (up to 7 M) LiTFSI- or LiFSI-based electrolytes with ether solvents, lithium deposition was previously reported as rather uniform, yielding lower surface roughness compared to diluted electrolytes,28,50 but more detailed 7Li-NMR studies were, to our knowledge, not yet presented.
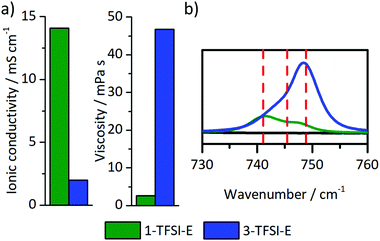 |
| | Fig. 7 (a) Ionic conductivity and viscosity, and (b) Raman spectra in the wavenumber region from 730 cm−1 to 760 cm−1 of 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green) and 3-TFSI-E (3 M LiTFSI in DME:DOL, 1:1, blue) electrolytes. | |
Ionic conductivity, viscosity and solution structure.
Notably, the relatively low ionic conductivity (2.0 mS cm−1 to 14.1 mS cm−1) as well as the substantially higher viscosity (45 mPa s to 2.6 mPa s, Fig. 7a) of 3-TFSI-E compared to 1-TFSI-E are correlated with different solution structures, which also in turn impact deposition phenomena on metal surfaces. In diluted electrolytes, lithium ions are typically coordinated by up to four solvent molecules, separating lithium ions from anions, e.g. by forming solvent separated ion pairs (SSIP). The lower ratio of lithium ions and solvent molecules at higher salt concentration results in the coordination of anions with one (contact ion pairs, CIPs) or even more lithium ions (cation–anion aggregates, AGG) as well as lower amounts of freely available solvent molecules.51 Note that TFSI− has a higher tendency to form coordination complexes with lithium ions compared to PF6−-based solutions (Fig. S6, ESI†).52 Due to increased ion coordination, the energy of the lowest unoccupied molecular orbital (LUMO) of the TFSI− is lowered affording higher reactivity in contact with lithium surfaces.53 The solution structures also govern the actual SEI formation based on lesser amounts of freely available solvent molecules and changing energy levels of the anions.53 Details of the solution structures can be derived from characteristic shifts of TFSI− in the Raman spectra of the considered electrolytes, provided that sufficient spectral resolution could be achieved (Fig. 7b). While 1-TFSI-E mainly consists of both SSIP (741 cm−1, 43%) and CIP (745 cm−1, 43%), the fraction of AGG (749 cm−1, 14%) is quite low.48,52,53 In contrast, 3-TFSI-E mainly contains AGG (69%) and CIP (31%), reflecting a much lower solvent-to-salt ratio. In addition, the amount of freely available DME was substantially reduced in 3-TFSI-E compared to 1-TFSI-E, as determined from the CO stretching and CH2 rocking modes of DME in the characteristic wavenumber region of 800 cm−1 to 870 cm−1 (Fig. S7, ESI†).54 Coordinated DME may be recognized based on the Raman peak at 875 cm−1 if LiTFSI is present in the solution.54 Notably, the higher reactivity of TFSI− and the lower amounts of freely available DME molecules in 3-TFSI-E electrolytes likely resulted in SEI layers that comprise higher amounts of salt degradation products compared to SEI layers formed in case of 1-TFSI-E, in agreement with data from literature.53 Based on the impedance data of cells operated with 3-TFSI-E (Fig. S1d and Table S1, ESI†) R2 evolved from (138 ± 13 Ω) to (174 ± 20 Ω) within one day, and to (158 ± 11 Ω) until the eighth day, thereby illustrating improved passivation abilities of 3-TFSI-E compared to 1-TFSI-E. Since the initial values of R2 were quite similar for both electrolytes (1-TFSI-E: 140 ± 5 Ω, 3-TFSI-E: 138 ± 13 Ω), the ability of lithium ion transport through the initially formed SEI appears highly comparable.
Long-term electrodeposition/-dissolution behaviour.
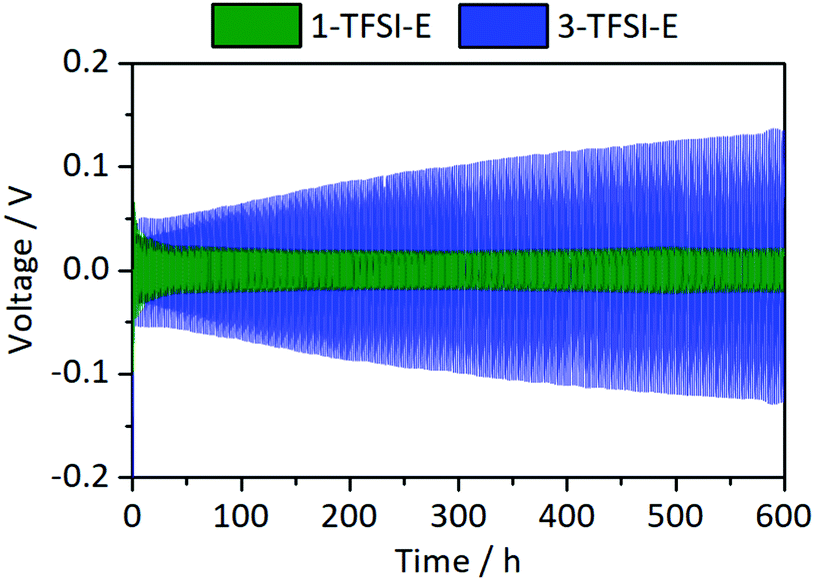
The cycling stability of Li‖Li thin film pouch cells with 1-TFSI-E and 3-TFSI-E electrolytes is shown in Fig. 8. Compared to 1-TFSI-E, the initial overvoltage in case of 3-TFSI-E is doubled (1-TFS-E: −0.1 V vs. 3-TFSI-E: −0.2 V, compare Fig. S2, ESI†), reflecting the lower ionic conductivity, and higher viscosity of 3-TFSI-E. The overvoltage evolved from ±0.05 V (similar to 1-TFSI-E) to ±0.13 V after 300 cycles for cells with 3-TFSI-E electrolyte (Fig. 8), likely due to electrolyte degradation, SEI formation as well as emerging fractions of ‘dead’ lithium. In view of the observed increase of R1 to 40 Ω after 300 cycles in case of 3-TFSI-E (Fig. S3b, ESI†), SEI formation due to electrolyte degradation appears as most significant factor, affording reasonable passivation abilities, despite that thicker inorganic layers of the SEI24 might have a lower mechanical flexibility and hence could induce more fractures during cell operation. However, the increase might also reflect the lesser surface area of lithium electrodes due to more homogeneous lithium deposition compared to cells with 1-TSFI-E.
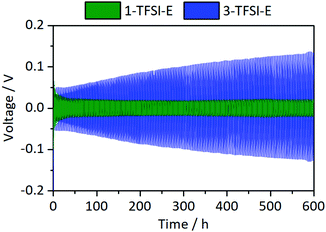 |
| | Fig. 8 Development of the overvoltage during lithium electrodeposition/-dissolution at a current density of 0.5 mA cm−2 in symmetrical Li‖Li thin film pouch cells containing 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green), and 3-TFSI-E (3 M LiTFSI in DME:DOL, 1:1, blue). An enlarged picture of the first discharge is given in Fig. S2 (ESI†). | |
Combined 7Li-NMR and SEM study
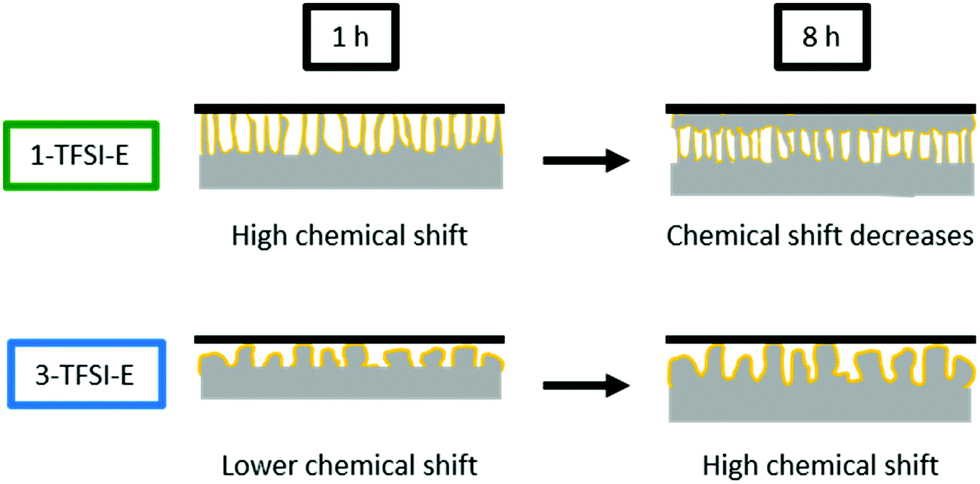
The NMR spectra of cells operated with 3-TFSI-E (Fig. 5d1) exhibited a peak at 268 ppm ± 1 ppm for electrodeposited lithium, slightly higher than 1-TFSI-E (266 ppm ± 1 ppm, Table 3), indicating lower density and needle-like lithium deposits in case of 3-TFSI-E. The ‘smooth’ lithium peak at 246 ppm decreased in case of 3-TFSI-E, probably resulting from a higher contact area between the bulk lithium and deposits, reflecting more mossy lithium deposition. In contrast to the observed 7Li-NMR chemical shift, but in good agreement with decreasing peak intensity for bulk Li, needle-like deposits could not be identified in the SEM images. Rather, mossy and porous lithium ‘structures’ were observed after 1 hour of deposition (Fig. 5d2), in addition to formation of quite large particles, though with lower density even after 8 hours of electrodeposition (Fig. 5d3).
Table 3 Comparison of the 7Li-NMR chemical shifts, normalized intensities, and the obtained morphology of lithium deposits from SEM for Li‖Li thin film pouch cells containing 1-TFSI-E and 3-TFSI-E electrolytes after 8 hours of deposition at current densities of 0.5 mA cm−2
|
|
1-TFSI-E |
3-TFSI-E |
| Chemical shift/ppm |
266 ± 1 |
268 ± 1 |
| NMR norm. intensity |
145% ± 7% |
150% ± 4% |
| Lithium morphology (SEM) |
Dense, small particles (>3 μm) |
Not dense, large particles (up to 40 μm) |
Note that the appearance of larger particles after 8 hours of deposition for higher concentrated electrolytes are in good agreement with literature data.23 The data clearly indicate that the peak intensity decrease at 246 ppm for 3-TFSI-E, probably due to larger contact area between bulk lithium and deposited Li, in addition to occurrence of larger particle sizes, is more important to actually assess the nature of formed lithium microstructures than observable 7Li-NMR chemical shifts. Similar to 1-PF6-C, the overall signal intensity between 175 and 325 ppm increased, though the peak intensity at 246 ± 1 ppm decreased more for 3-TFSI-E than for 1-TFSI-E. Despite that this might result from larger amounts of inhomogeneous lithium deposits compared to 1-TFSI-E, the smaller surface area and therefore lower losses of metallic lithium due to reduced SEI formation might be the determining step for this effect, again suggesting mossy or nodule-like lithium deposits.
7Li-NMR chemical shifts close to 270 ppm were often attributed to needle-like lithium deposits (which is counterintuitive in case of formation of larger particles), but is indeed in good agreement with the spatial orientation of the lithium deposits. While needle-like lithium deposits, e.g. from 1-TFSI-E (Fig. 5c2), in this system, are not only orientated perpendicular to the electrode surface (in favour of high chemical shifts) upon deposition, they might bend on the separator upon cycling, leading to a rather horizontal orientation with respect to the electrodes (Fig. 5c3), as reflected by decreasing 7Li-NMR chemical shifts. This could be confirmed by the observation of decreasing 7Li-NMR chemical shifts of the peak at 268 ppm ± 1 ppm after 8 hours electrodeposition compared to 266 ppm ± 1 ppm after 16 hours (Fig. S5, ESI†). Larger particles, on the other hand, typically yield lower 7Li-NMR chemical shifts at early stages of the deposition, since most of their volume is rather close to, and therefore affected by, the electrode surface, resulting in lower 7Li-NMR chemical shifts, comparable to more mossy lithium deposits.17 Nevertheless, upon cycling, larger particles are less likely to bend on the surface (Fig. 5d3) and thus maintain a perpendicular orientation to the electrode surface, as documented by higher 7Li-NMR chemical shifts. The likely nature of formed lithium microstructures and resulting 7Li-NMR chemical shifts are schematically depicted in Fig. 9.
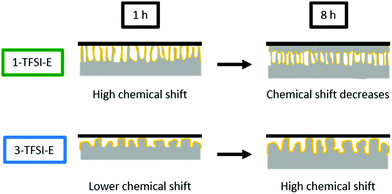 |
| | Fig. 9 Schematic display of the lithium deposition and the resulting 7Li-NMR shift for 1-TFSI-E (1 M LiTFSI in DME:DOL, 1:1, green), and 3-TFSI-E (3 M LiTFSI in DME:DOL, 1:1, blue). | |
Applicability of 7Li-NMR to monitor HSAL growth
Inhomogeneous lithium deposition was successfully monitored combining in situ7Li-NMR and SEM data, considering variations of solvents, lithium salt and the corresponding salt concentration in thin film pouch cells. 7Li-NMR data based on the skin depth typically afford (in principle quantitative) insight into lithium microstructures deposited across the entire surface of the electrode, whereas analysis of SEM data tend to yield more qualitative information about localized spots at the considered surfaces, rendering both techniques highly complementary. Minor changes of lithium deposits are often hard to identify, but 7Li-NMR is particularly beneficial in cases where a larger variation of lithium microstructures is present, which may be induced by application of pressure17 (see Fig. S8, ESI†) or selected utilization of ionic liquids.32 In both cases, small shoulders towards higher chemical shifts of bulk lithium peak (>250 ppm) could be observed, resulting in only a small increases of the overall intensity (for example 45% increase without and 15% increase with the application of pressure in a symmetrical Li‖Li thin film pouch cell with 1-TFSI-E, Table 3 and Fig. S8, ESI†).17,32
Conclusions
In this work, the impact of electrolyte constituents including salts, solvents and concentration on the achievable cell performance in lithium metal batteries and particularly on lithium electrodeposition was considered, combining 7Li-NMR and SEM data of symmetrical Li‖Li thin film pouch cells. Despite small variations of observable surface resistances as well as overvoltage for electrolyte formulations comprising different salts and solvents, the estimated amounts and morphology of lithium deposits monitored by 7Li-NMR did not change significantly when using thin film pouch cells where no external pressure control was applied. In addition, even though larger particle sizes of lithium deposits in case of highly concentrated electrolytes were reported in literature and identified from SEM data in this work, no substantial changes of 7Li-NMR chemical shifts reflecting pristine ‘smooth’ electrodeposited lithium could be observed. Most likely this results from the fact, that even though the particle sizes might be larger, the overall orientation of the deposited lithium on average is perpendicular to the bulk electrode surface, hence the impact of magnetic susceptibility yields higher 7Li-NMR chemical shifts. This clearly indicates that qualitative analysis of the nature of resulting microstructures of lithium deposits solely based on the 7Li-NMR chemical shift is rather challenging, particularly if only minor changes of the lithium microstructures occur. In addition to needle-like lithium morphology, also mossy lithium deposits as well as formation of larger particles eventually yield comparably high 7Li-NMR chemical shifts, even more so considering that electrodeposition often result in a distribution of lithium structures.55 Despite that unambiguous assignment of observable 7Li-NMR chemical shifts to explicitly occurring lithium microstructures was not feasible for most of the considered electrolyte formulations, a decrease of the bulk lithium peak at ca. 246 ppm could be clearly associated with the appearance of larger particle sizes of lithium deposits, as for example granule- or nodule-like lithium, and thus might serve to distinguish the present lithium microstructures. In addition, very small particles which only grow very close to the surface and do not expand perpendicular to bulk lithium, as for example mossy-lithium, cover more surface area of the bulk lithium metal, resulting in decreased intensity at 246 ppm, but also in a peak at lower chemical shift. An additional study and quantification of the SEI formation could yield hints to distinguish between microstructures with large surface areas (e.g. mossy or needle-like, large amount of SEI formation) and microstructures with smaller surface areas (e.g. nodule- or granule-like lithium, smaller amount of SEI formation). However, also different thicknesses of the locally present SEI based on the electrolyte properties determine the amount of SEI, so that unambiguous evaluation will be difficult. In the absence of spatial resolution (as e.g. exploited in case of chemical shift imaging – 1D profiling experiments35,37), the spectral resolution of the applied static solid-state NMR setup is limited, hence rendering differentiation of occurring microstructures within lithium deposits challenging. Nevertheless, different from imaging techniques such as SEM, 7Li-NMR provides in situ insight into growth of lithium deposits in application-related thin film pouch cells, including semi-quantitatively monitoring of lithium deposit yields, though likely correction for lithium losses due to SEI formation should be separately considered. It is therefore recommended to pursue the presented in situ7Li NMR technique complementary to any applied imaging techniques when the nature/morphology of lithium deposits is of concern, in this way allowing for a more complete picture of the processes involved. For future studies of microstructures of inhomogeneous lithium deposits with 7Li-NMR, it is important to keep in mind that small variations in the chemical shifts of the obtained peaks have to be analysed very carefully, since various lithium microstructures may yield comparable (average) 7Li-NMR chemical shifts. However, 7Li-NMR is highly promising for monitoring of lithium growth in polymer membrane-based (solid state) batteries, where SEM data is limited due to challenging sample preparation. Another opportunity to avoid variations of lithium growth patterns and resulting distribution of lithium microstructures might be consideration of separator-free cell setups. In summary, the combination of SEM and NMR data offers complementary insights and possibilities for the future development of sophisticated new methods.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful for BMBF (German Federal Ministry of Education and Research) funding of this work through the projects MEET Hi-End II (03XP0084A), MgMeAnS (03XP0140), and LiBEST (13XP0133A).
Notes and references
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- J. Betz, G. Bieker, P. Meister, T. Placke, M. Winter and R. Schmuch, Adv. Energy Mater., 2019, 9, 1803170 CrossRef.
- R. Wagner, N. Preschitschek, S. Passerini, J. Leker and M. Winter, J. Appl. Electrochem., 2013, 43, 481–496 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Chem., 2017, 21, 1939–1964 CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- J. Heine, S. Krüger, C. Hartnig, U. Wietelmann, M. Winter and P. Bieker, Adv. Energy Mater., 2014, 4, 1300815 CrossRef.
- J. Heine, P. Hilbig, X. Qi, P. Niehoff, M. Winter and P. Bieker, J. Electrochem. Soc., 2015, 162, A1094–A1101 CrossRef CAS.
- M.-H. Ryou, Y. M. Lee, Y. Lee, M. Winter and P. Bieker, Adv. Funct. Mater., 2015, 25, 834–841 CrossRef CAS.
- P. Bieker and M. Winter, Chem. Unserer Zeit, 2016, 50, 172–186 CrossRef CAS.
- P. Bieker and M. Winter, Chem. Unserer Zeit, 2016, 50, 26–33 CrossRef CAS.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- J. Becking, A. Gröbmeyer, M. Kolek, U. Rodehorst, S. Schulze, M. Winter, P. Bieker and M. C. Stan, Adv. Mater. Interfaces, 2017, 4, 1700166 CrossRef.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4270 CrossRef CAS PubMed.
- H. J. Chang, N. M. Trease, A. J. Ilott, D. Zeng, L.-S. Du, A. Jerschow and C. P. Grey, J. Phys. Chem. C, 2015, 119, 16443–16451 CrossRef CAS.
- R. Mogi, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, J. Electrochem. Soc., 2002, 149, A385–A390 CrossRef CAS.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- D. Aurbach, I. Weissman, A. Zaban and O. Chusid, Electrochim. Acta, 1994, 39, 51–71 CrossRef CAS.
- H. Ota, X. Wang and E. Yasukawa, J. Electrochem. Soc., 2004, 151, A427–A436 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- M. Winter, Z. Phys. Chem., 2009, 223, 1395–1406 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- J. Zheng, J. A. Lochala, A. Kwok, Z. D. Deng and J. Xiao, Adv. Sci., 2017, 4, 1700032 CrossRef PubMed.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- S.-K. Jeong, H.-Y. Seo, D.-H. Kim, H.-K. Han, J.-G. Kim, Y. B. Lee, Y. Iriyama, T. Abe and Z. Ogumi, Electrochem. Commun., 2008, 10, 635–638 CrossRef CAS.
- C. P. Grey and N. Dupré, Chem. Rev., 2004, 104, 4493–4512 CrossRef CAS PubMed.
- R. Bhattacharyya, B. Key, H. Chen, A. S. Best, A. F. Hollenkamp and C. P. Grey, Nat. Mater., 2010, 9, 504–510 CrossRef CAS PubMed.
- N. Schweikert, A. Hofmann, M. Schulz, M. Scheuermann, S. T. Boles, T. Hanemann, H. Hahn and S. Indris, J. Power Sources, 2013, 228, 237–243 CrossRef CAS.
- N. M. Trease, L. Zhou, H. J. Chang, B. Y. Zhu and C. P. Grey, Solid State Nucl. Magn. Reson., 2012, 42, 62–70 CrossRef CAS PubMed.
- J. Z. Hu, Z. Zhao, M. Y. Hu, J. Feng, X. Deng, X. Chen, W. Xu, J. Liu and J.-G. Zhang, J. Power Sources, 2016, 304, 51–59 CrossRef CAS.
- S. Klamor, K. Zick, T. Oerther, F. M. Schappacher, M. Winter and G. Brunklaus, Phys. Chem. Chem. Phys., 2015, 17, 4458–4465 RSC.
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed.
- H. J. Chang, A. J. Ilott, N. M. Trease, M. Mohammadi, A. Jerschow and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 15209–15216 CrossRef CAS PubMed.
-
K. Borzutzki and G. Brunklaus, in Annual Reports on NMR Spectroscopy, ed. G. A. Webb, Academic Press, 2017, vol. 91, pp. 115–141 Search PubMed.
- A. J. Ilott, M. Mohammadi, H. J. Chang, C. P. Grey and A. Jerschow, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10779–10784 CrossRef CAS PubMed.
- J. Betz, J.-P. Brinkmann, R. Nölle, C. Lürenbaum, M. Kolek, M. C. Stan, M. Winter and T. Placke, Adv. Energy Mater., 2019, 9, 1900574 CrossRef.
- R. Nölle, K. Beltrop, F. Holtstiege, J. Kasnatscheew, T. Placke and M. Winter, Mater. Today, 2019 DOI:10.1016/j.mattod.2019.07.002.
- S. S. Zhang, J. Power Sources, 2007, 164, 351–364 CrossRef CAS.
- F. Holtstiege, R. Schmuch, M. Winter, G. Brunklaus and T. Placke, J. Power Sources, 2018, 378, 522–526 CrossRef CAS.
- A. Narayanan Kirshnamoorthy, K. Oldiges, M. Winter, A. Heuer, I. Cekic-Laskovic, C. Holm and J. Smiatek, Phys. Chem. Chem. Phys., 2018, 20, 25701–25715 RSC.
- A. B. Gunnarsdóttir, S. Menkin, L. E. Marbella, A. S. Best and C. P. Grey, Meeting Abstracts, 2019, MA2019-04, 126.
-
L. E. Marbella, S. Emge and C. P. Grey, Meeting Abstracts, 2019, MA2019-04, 304.
- A. Heckmann, J. Thienenkamp, K. Beltrop, M. Winter, G. Brunklaus and T. Placke, Electrochim. Acta, 2018, 260, 514–525 CrossRef CAS.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416–426 RSC.
- K. Matsumoto, K. Inoue, K. Nakahara, R. Yuge, T. Noguchi and K. Utsugi, J. Power Sources, 2013, 231, 234–238 CrossRef CAS.
- R. Cao, J. Chen, K. S. Han, W. Xu, D. Mei, P. Bhattacharya, M. H. Engelhard, K. T. Mueller, J. Liu and J.-G. Zhang, Adv. Funct. Mater., 2016, 26, 3059–3066 CrossRef CAS.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Electrochem. Soc., 2012, 159, A1489–A1500 CrossRef CAS.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS.
- D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski and J. E. Desnoyers, Phys. Chem. Chem. Phys., 2002, 4, 6063–6071 RSC.
- L. Gireaud, S. Grugeon, S. Laruelle, B. Yrieix and J. M. Tarascon, Electrochem. Commun., 2006, 8, 1639–1649 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp05334d |
|
| This journal is © the Owner Societies 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Martin
Kolek
a,
Martin
Kolek