
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermodynamics of amyloid fibril formation from chemical depolymerization†
Nicola
Vettore
 a and
Alexander K.
Buell
*ab
a and
Alexander K.
Buell
*ab
aInstitut for Physical Biology, Heinrich-Heine-Universitaet Duesseldorf, Universitaetstrasse 1, Duesseldorf, Germany
bTechnical University of Denmark, Department of Biotechnology and Biomedicine, Lyngby, Denmark. E-mail: alebu@dtu.dk
First published on 19th November 2019
Abstract
Amyloid fibrils are homo-molecular protein polymers that play an important role in disease and biological function. While much is known about their kinetics and mechanisms of formation, the origin and magnitude of their thermodynamic stability has received significantly less attention. This is despite the fact that the thermodynamic stability of amyloid fibrils is an important determinant of their lifetimes and processing in vivo. Here we use depolymerization by chemical denaturants of amyloid fibrils of two different proteins (PI3K-SH3 and glucagon) at different concentrations and show that the previously applied isodesmic linear polymerization model is an oversimplification that does not capture the concentration dependence of chemical depolymerization of amyloid fibrils. We show that cooperative polymerization, which is compatible with the picture of amyloid formation as a nucleated polymerization process, is able to quantitatively describe the thermodynamic data. We use this combined experimental and conceptual framework in order to probe the ionic strength dependence of amyloid fibril stability. In combination with previously published data on the ionic strength dependence of amyloid fibril growth kinetics, our results provide strong evidence for the product-like nature of the transition state of amyloid fibril growth.
1 Introduction
Filamentous protein structures are ubiquitous in biology. They can fulfill functional roles, as in the case of the cytoskeletal proteins actin1 and tubulin,2 or be associated with diseases, as in the case of sickle hemoglobin polymers3 or amyloid fibrils.4 For the cytoskeletal filaments, both the mechanical and thermodynamic stability, as well as the molecular origin of their reversibility have been the subject of extensive studies in the last decades.5 Indeed, actin filaments were the first protein polymers the thermodynamic stability of which has been characterized in detail in seminal work by Oosawa.6 Aktin filaments can be described as helical polymers, whereby each monomeric building block interacts not only with its nearest neighbors in the polymer, but also with building blocks further away. Actin monomers undergo relatively minor structural changes upon polymerization,7 and the same is also true for tubulin and sickle hemoglobin.8 Amyloid fibrils, on the other hand, are protein polymers in which the protein building blocks usually adopt a very different structure inside the fibril compared to the isolated protein molecule.9 Amyloid fibrils are mostly known as being the hallmark of a wide range of diseases, but mounting evidence demonstrates that amyloid fibrils can also play functional roles in biology.10 Many different proteins entirely unrelated in structure (IDPs or folded proteins) and function (peptide hormones,11 lipid-binding proteins,12 milk proteins13) have been found to form amyloid fibrils, either in vivo or in the test tube. While much insight has been generated in the last two decades on the kinetics and mechanisms of amyloid fibril formation,14–17 much less is known about the thermodynamic stability of these structures,18 and to what extent common driving forces govern the amyloid formation of different proteins. It is very important to be able to quantify and rationalize the thermodynamic stability of amyloid fibrils because the thermodynamic stability is likely to be a decisive factor in determining whether amyloid fibrils of a certain protein can be cleared in a biological context. Such clearance could happen either through spontaneous binding of molecules, such as antibodies, to soluble or aggregated protein19 or else through active, energy consuming processes, such as the action of chaperone complexes on amyloid fibrils.20Experimental data of amyloid fibril stability21,22 has so far been analysed in the framework of the linear polymerization model.6 The simplest, so-called isodesmic form of this model contains only a single equilibrium constant, that for the addition of monomer to all possible species, including to another monomer. Under this assumption, the stability of the fibrils can be directly determined from a measurement of the free monomer concentration at equilibrium, for sufficiently high total concentrations. In many practical cases, the free monomer concentration is very low (nM range23) and difficult to determine accurately. In order to overcome this practical problem, amyloid fibrils can be destabilized by chaotropes, such as GndSCN or GndHCl.21,22 By assuming a linear relationship between the concentration of denaturant and the free energy difference ΔG between the monomeric and polymeric state, equivalent to the assumption in protein unfolding experiments,24 sigmoidal fibril depolymerization data can be fitted and the value of ΔG extrapolated to the absence of denaturant.21,22 While this method of determination of amyloid fibril stability is commonly used, its validity has not yet been thoroughly tested. Here we use intrinsic protein fluorescence to monitor the fraction of fibrillar vs. soluble protein. We investigate and rationalise the influence of the type of chemical denaturant, the ionic strength and the protein concentration on the depolymerization curves and show that a cooperative model provides a better description of the thermodynamics of amyloid fibril formation than the simple isodesmic polymerization model. The cooperative model, adapted from the field of supramolecular chemistry,25,26 describes the thermodynamics of polymerization by two different equilibrium constants: a nucleation constant and an elongation constant. We find that experiments in which the peptide concentration is varied allow a better discrimination between the isodesmic and cooperative models than the standard experiments in which only the denaturant concentration is varied.
We apply our insight and methodology to the analysis of the ionic strength dependence of amyloid fibril stability and compare it with the ionic strength dependence of the fibril growth kinetics.27 Our combined analysis of this data provides strong evidence for the fact that the transition state of the fibril growth reaction is highly product-like with respect to the distance of the newly adding peptide to the fibril end.
2 Experimental section
2.1 Proteins
The human glucagon employed in the study was a kind gift from Novo Nordisk. The bovine PI3K-SH3 domain was purified according to the protocol in ref. 28. The construct contains a 6xHis-tag linked to the protein by a thrombin cleavage site. The sequence of the WT protein after cleavage is the following, with the dipeptide Gly-Ser remaining as overhang from the cleavage:GS MSAEGYQYRA LYDYKKEREE DIDLHLGDIL TVNKGSLVAL GFSDGQEAKP EEIGWLNGYN ETTGERGDFP GTYVEYIGRK KISP
The protein was expressed in a BL21 E. coli strain with TB medium for auto induction containing 0.012% Glucose and 0.048% Lactose. The cells were grown for over 24 h and then harvested by centrifugation. After resuspension in sodium phosphate buffer (50 mM sodium phosphate pH 8, 5 mM Imidazole and 100 mM NaCl), the cells were disrupted by sonication, in the presence of protease inhibitors (cOmplete Mini EDTA-free, Roche) and DNAse (Sigma-Aldrich). The lysate was centrifuged, and the supernatant loaded on a Ni-NTA Superflow Cartridge (Qiagen, Venlo, Netherlands) equilibrated in 50 mM sodium phosphate pH 8, 5 mM Imidazole and 100 mM NaCl. The protein was eluted with a linear gradient from 5 to 300 mM imidazole in 50 mM sodium phosphate pH 8, 100 mM NaCl in 25 ml elution volume. Fractions containing the protein were collected and cleaved overnight at 7 °C with 1 unit of thrombin (from bovine plasma, Sigma-Aldrich Saint Louis, Missouri, USA) per 1 mg of protein. The cleaved solution was then concentrated and loaded on a SEC HiLoad 26/60 Superdex 75 column (GE Healthcare, Chicago, Illinois, USA) equilibrated with 5 mM ammonium acetate pH 7. Fractions containing the PI3K-SH3 domain were collected and lyophilised for further use.
2.2 Fibril preparation
Glucagon fibrils were formed from protein solutions prepared after resuspension of the lyophilized peptide in 10 mM glycine hydrochloride pH 2 at 1 mM final concentration. This solution was incubated under shaking at 37 °C for 1–2 hours. The fibrils obtained through this procedure were used as seeds for further solutions of monomeric protein, which were prepared at a concentration range between 1 and 2 mM and seeded at ca. 10% (monomer equivalents). The solution was left overnight at room temperature without shaking or stirring.PI3K-SH3 fibrils were formed from protein solutions prepared from the lyophilized protein (produced as described above), resuspended in 10 mM glycine hydrochloride pH 2 at 200 μM final concentration. This solution was incubated under shaking at 42 °C overnight. The fibrils obtained through this procedure were then used as seeds for further solutions of monomeric protein, which were prepared at a concentration range between 200 and 300 μM and then seeded at ca. 10% (equivalent monomer mass). The solution was left overnight at room temperature without shaking or stirring.
Such high concentrations of fibril stock solutions are needed, as the fibrils are strongly diluted upon addition of the denaturant, which is necessary to achieve denaturant concentrations high enough for depolymerization. Before preparing the samples, the fibril preparations were sonicated with a VialTweeter-sonotrode (Hielscher, Teltow, Germany). Glucagon fibril solutions were sonicated in a volume of at least 700 μl, twice for 3 seconds, 100% amplitude, with a pause of ca. 30 s. PI3K-SH3 fibrils were sonicated for 10 s at 100% amplitude in a volume of at least 700 μl. The fibril preparations were imaged by AFM, both before and after the sonication protocol, to evaluate the effect of the sonication on the lengths of the fibrils.
2.3 Atomic force microscopy imaging
Atomic force microscopy (AFM) was performed on dried fibril samples. The fibril samples were diluted to ca. 3 μM in the same buffer, then 20 μl were placed on freshly cleaved mica and incubated at room temperature for 10 min. After 10 min the mica was washed with MilliQ water. Imaging was performed with a Bruker Multimode 8 (Billerica, Massachusetts, USA) using OMCL-AC160TS cantilevers (Shinjuku, Tokyo, Japan).2.4 Depolymerization experiments
The fibril samples were mixed with different volumes of urea or GndHCl stock solutions and buffer. In order to maintain a solution pH of 2 constant throughout the whole denaturation series, an 8 M urea stock solution was prepared by dissolving 12 g of urea (SigmaAldrich) in 16 ml of concentrated buffer, to yield a final concentration of 10 mM glycine hydrochloride and 16 mM HCl. The HCl is necessary, as the urea has a weak buffering capacity. In order to keep the ionic strength constant in all samples, NaCl was added to the 10 mM glycine HCl buffer to a final concentration of 16 mM. In order to ensure that the samples have reached equilibrium when they are analyzed, we assessed the time of equilibration (see Fig. S5, ESI†) and based on these assessments left the samples to equilibrate for one week in the case of glucagon fibrils and two weeks in the case of PI3K-SH3 fibrils.2.5 Fluorescence measurements
Fluorescence spectra were recorded on a Tecan M1000pro instrument using Greiner UV-transparent 96 well plates. The wells were filled with 140 μl of solution and the temperature was maintained constant at 27 °C. The fluorescence spectra were measured by top reading, exciting at 280 nm for glucagon samples and 290 nm for PI3K-SH3 samples (5 nm bandwidth), while the emission was recorded between 300 nm and 420 nm (5 nm bandwidth). A blank spectrum (buffer) resulting from the average of 10 different spectra is subtracted before analysis. In order to determine the relative populations of soluble vs. fibrillar protein, we computed the ratios of the fluorescence intensities at 340 and 320/310 nm (glucagon/PI3K-SH3) for each spectrum. This choice of wavelengths was based on the difference between the fibrillar and monomeric spectra in each case (Fig. S2, ESI†).2.6 Absorbance measurements
In order to measure the degree of aggregation of the fibrillar samples, they were centrifuged at 16.100 g for 1 hour. The supernatant was removed and its absorbance was measured between 220 and 350 nm with a Nanodrop 2000 (ThermoFisher) spectrophotometer. The extinction coefficients at 280 nm are 8480 M−1 for glucagon and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 930 M−1 for PI3K-SH3.
930 M−1 for PI3K-SH3.
3 Results
In order to establish a reliable methodology for the determination of the thermodynamic stability of amyloid fibrils, we chose two polypeptide systems that have been shown to form amyloid fibrils under acidic conditions, the peptide hormone glucagon29,30 and the protein PI3K-SH3.28,31 Both of these polypeptides contain the amino acid tryptophan and therefore the use of intrinsic protein fluorescence can be explored for the distinction between monomeric and aggregated protein32 (Fig. S2, ESI†). In several previous studies of amyloid fibril stability based on chemical depolymerization, the equilibrated samples were subjected to centrifugation and the protein concentration in the supernatant was determined.22,33 Experimental data confirming the equivalence between the data from concentration measurements and the intrinsic fluorescence data can be found in Fig. S6 and S7 (ESI†).For both PI3K-SH3 and glucagon, homogeneous fibril preparations without appreciable amounts of non-fibrillar material (as judged by AFM imaging, see Fig. S1, ESI†) can be produced by adjusting the solution conditions appropriately.32,34 In our experiments of fibril depolymerization we started from fully equilibrated fibrillar samples that we prepared by seeding, i.e. the addition of preformed fibrils to monomeric samples. We then homogenized the samples and shortened the average fibril length by subjecting the samples to ultrasonication (Fig. S1, ESI†). Details about the sample preparation can be found in the methods section.
3.1 Choice of denaturant
Having chosen the polypeptide systems to investigate, we next proceeded to the choice of the chemical denaturant to be used to destabilise the fibrils. Previous reports mostly used strong denaturants, such as GndHCl or GndSCN21,22,33 rather than milder ones, such as urea. It is well established that the ionic denaturants are more powerful than urea in the unfolding of proteins.35 However, we found that in our case the fibrils could be dissociated by urea, and even that in the case of PI3K-SH3 amyloid fibrils, urea was a more powerful denaturant than GndHCl (see Fig. S4, ESI†). This is in contrast to the unfolding of monomeric PI3K-SH3 that was unfolded at lower concentrations of GndHCl compared to urea, at neutral pH (see Fig. S4, ESI†). Based on these initial results, we decided to chose urea as the denaturant of choice for our amyloid fibril depolymerization experiments, as this choice of a neutral denaturant allows us to explore the role of electrostatic interactions in amyloid fibril stability in more detail. The use of GndHCl would lead to an almost complete screening of the electrostatic interactions at the high denaturant concentrations used in these experiments. It has to be noted that prolonged incubation of proteins in high urea concentration can lead to carbamylation reactions.36 The acidic pH conditions employed in the present study strongly disfavour this reaction. We showed by mass spectrometric analysis (Fig. S9, ESI†) that even after 9 days incubation in 6 M urea, no sign of carbamylation was observed for PI3K-SH3, which required the longest equilibration times.3.2 Analysis of chemical depolymerization with an isodesmic model
Equilibrium denaturation curves of folded proteins are often analysed with a two state model, whereby it is assumed that the denaturant linearly shifts (with proportionality constant m) the free energy difference between the folded and the unfolded polypeptide.37 Due to the large number of aggregate species of different sizes, a two state model is not appropriate in the case of protein polymerization. It has first been proposed by Goto and coworkers21 to apply the isodesmic form of the linear polymerization model6 to fit equilibrium depolymerization curves of amyloid fibrils, and this method of analysis has been exclusively used to-date. The mathematical formulation of this model can be found in Section S2 (ESI†). Individual sigmoidal depolymerization curves can be fitted and free energy differences between the fibrillar and the soluble states, as well as m-values, can be determined. The absolute concentration of peptide is an important parameter in the model that determines the shape of the depolymerization curve (see Section S2, ESI†). If depolymerization curves are acquired at different peptide concentrations, the extracted free energies and m-values are expected to be identical, within error. We have fitted data from experiments at three (glucagon) and two (PI3K-SH3) different peptide concentrations. It is convenient to display such data in a normalised way, i.e. as a fraction of depolymerised protein, rather than as an absolute concentration (Fig. 1). In Fig. S8 (ESI†), we show the same data without normalisation. We find that while all the individual curves can be well-fitted (Fig. 1, left panel), in the case of glucagon the fits at the three different concentrations yield significantly different values for the free energy difference ΔG0 (−36.7 vs. −38.8 vs. −42.2 kJ mol−1 for the lowest, intermediate and highest concentrations, respectively). In the case of PI3K-SH3, the difference is similar (−64.7 vs. −71.1 kJ mol−1), despite the fact that here the difference in concentrations is less significant. The reason for the difference between the individual fits is that both ΔG0 and m are free parameters and can both vary between fits to the data sets at different protein concentrations. If, however, the data at the different concentrations are fitted globally (Fig. 1, right panel), we find that in the case of PI3K-SH3, a satisfactory fit result is achieved and the value for the free energy of fibril stability is intermediate between the two values of the individual fits (−66.5 kJ mol−1). On the other hand, a global fit to the data for glucagon is not as good and yields a value for the free energy comparable to that from a local fit of the lowest concentration (−36.7 kJ mol−1). Tables S1 and S2 in the ESI† show all the free energy and m-values, when neither, either of these or both parameters are globally fitted. Therefore, already a global fit to denaturation curves at only two or three different protein concentrations reveals potential inadequacies of the isodesmic model for the description of chemical depolymerization experiments of amyloid fibrils.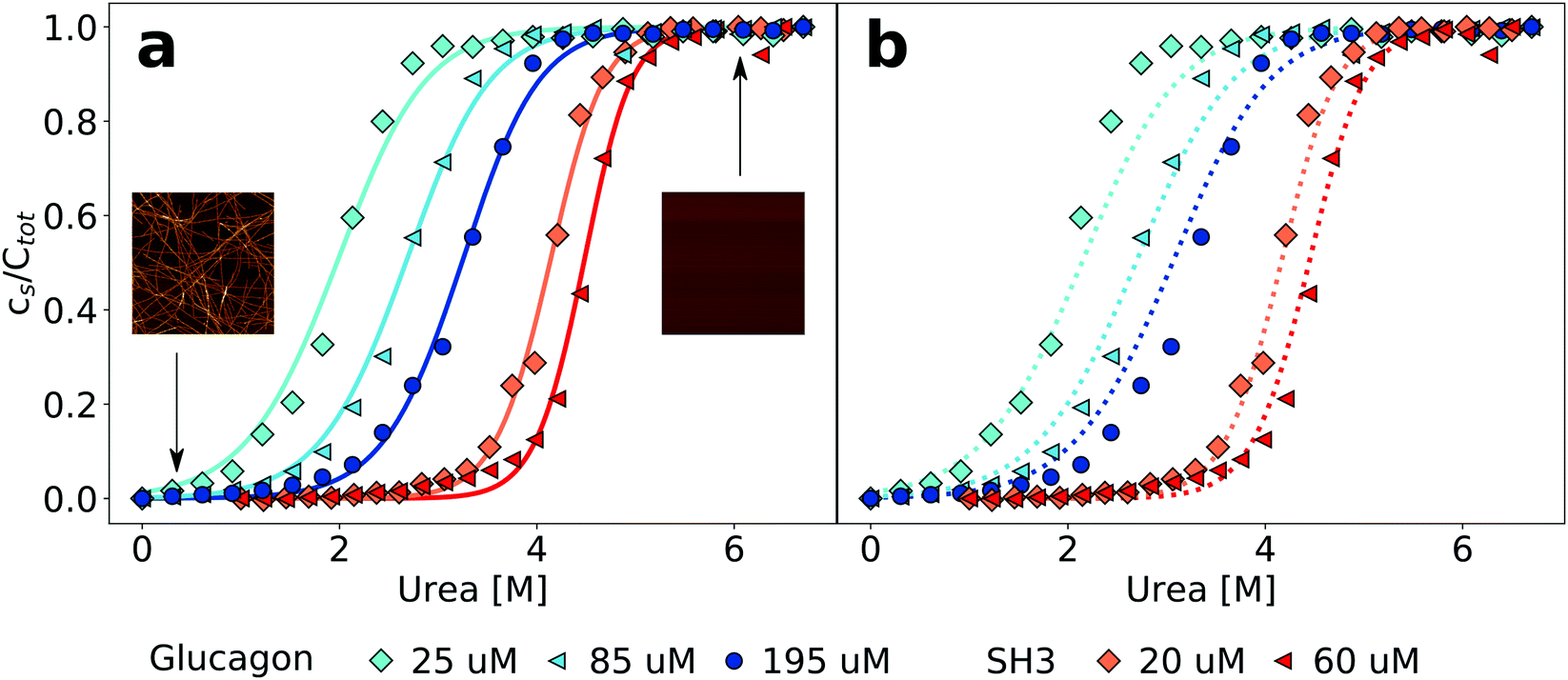 | ||
| Fig. 1 Equilibrium depolymerization profiles of glucagon and PI3K-SH3 fibrils at different peptide concentrations. (a) The data for each peptide concentration have been fitted individually (continuous lines). The insets are AFM images showing the sample at low denaturant and at high denaturant concentrations. The image size is 5 × 5 μm. (b) Global fits to all concentrations simultaneously for each peptide (dotted lines). | ||
3.3 Extension of the isodesmic model
The isodesmic version of the linear polymerization model assumes that all equilibria in a solution of polymers have the same equilibrium constant. However, this assumption is clearly not in agreement with the known mechanistic features of amyloid fibril formation. Amyloid fibrils form through a nucleated polymerization process,14,16 whereby the formation of the initial oligomeric nucleus is energetically less favourable than the addition of a monomer to a fully grown fibril.38 We hypothesised that this simplification could be at the origin of the inability of the isodesmic model to quantitatively describe the concentration dependence of chemical depolymerization curves of amyloid fibrils (Fig. 1). We therefore increased the complexity of the model by allowing a different equilibrium constant for monomer association to any aggregate below a certain aggregation number n, i.e. a distinct equilibrium constant of nucleation. The nucleation process is thus defined as a series of less favourable interactions between a monomer and any species up to an n-mer, where n defines the size of the nucleus. By allowing for two distinct equilibrium constants with a ratio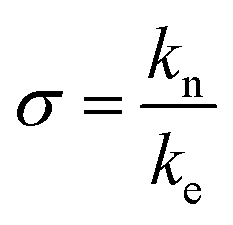 , it is possible to account for the fact that the formation of a pre-fibrillar structure is thermodynamically less favourable than its growth.
, it is possible to account for the fact that the formation of a pre-fibrillar structure is thermodynamically less favourable than its growth.
This cooperative model, which is slightly more general than the helical polymerization model of Oosawa5 (see Section S2 for details on possible extensions of the isodesmic polymerisation model, ESI†) has been successfully used to describe the aggregation process of supramolecular non-covalent polymers.25 It has for example allowed to explain the differential effects of a gradual change in solvent conditions on the stability of several supramolecular polymer systems.39 The building blocks of supramolecular polymers are usually simpler molecules than polypeptides, with much fewer degrees of freedom and less potential for polymorphism, allowing the systems in some cases to be characterised very accurately and in great detail.40 Our concentration-dependent measurements of amyloid fibril depolymerization allow us for the first time to test the applicability of this type of model also in the case of amyloid fibrils. Fig. 2 shows a comparison of fits of the same data sets as in Fig. 1 to both the isodesmic and cooperative models. Compared to the isodesmic model, the cooperative model has two additional free parameters, σ and n. We performed fits for different fixed values of n, ranging from 1 to 50. The resulting fit parameters are recorded in Tables S3 (PI3K-SH3) and S4 (glucagon) in the ESI.†σ and n are not independent of each other, the higher the fixed value of n, the closer σ is to unity. We also compared the model, whereby all species other than the monomer have the same spectroscopic signature (oligomer same as aggregate, ‘osaa’), to the model whereby all species up to size n have the same spectroscopic signature as the monomer (oligomer same as monomer, ‘osam’). Given the intrinsically disordered nature of both PI3K-SH328 and glucagon41 under these solution conditions, we think it more likely that the oligomers will display a signature of intrinsic fluorescence closer to that of the fibrillar aggregate than to that of the monomer. Furthermore, the osaa model yields more consistent values for both ΔG0 and σ. The best fits are achieved for small to intermediate values of n(2–5), whereby the difference between the fit to the isodesmic model and the best fit to a cooperative model is more significant in the case of glucagon compared to PI3K-SH3 (Fig. 2). The physical significance of n and σ are discussed in more detail in the ESI.†
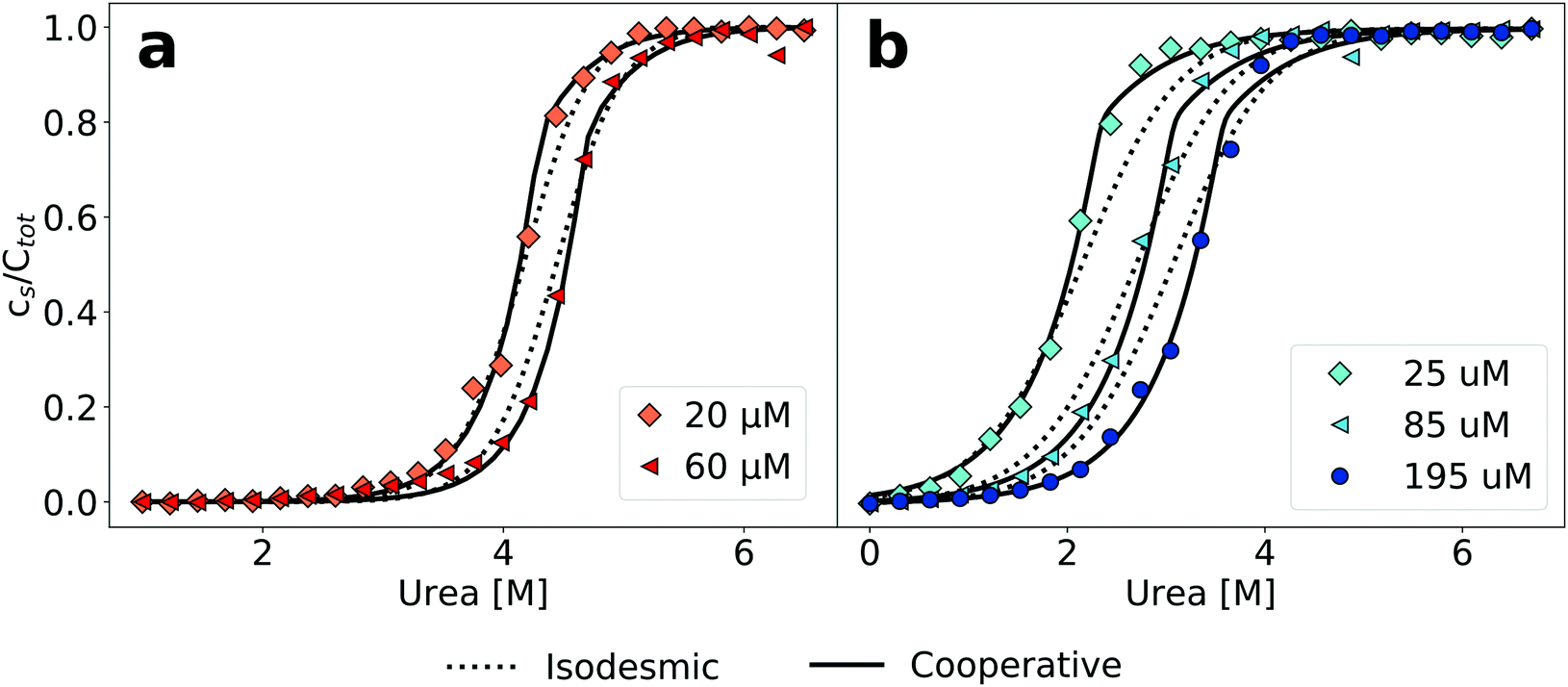 | ||
| Fig. 2 Cooperative polymerisation describes the concentration dependence of chemical depolymerization profiles of PI3K-SH3 (a) and glucagon (b) amyloid fibrils. The black lines show the best global fits of the two different linear polymerization models, corresponding to n = 4 in the case of the cooperative model for both proteins (see ESI† for a more detailed discussion of the effect of changes in n). | ||
3.4 Exploring the concentration dimension in chemical depolymerization
The finding that an increase in the concentration of urea allows to gradually depolymerize amyloid fibrils reflects that the denaturant shifts the equilibrium in a concentration dependent manner towards the soluble state. Therefore, while the critical concentration in the absence of denaturants can be very low and difficult to measure, at higher denaturant concentrations, it will eventually approach the total concentration of the sample. In order to explore this behaviour, we investigated the dependence of both glucagon and PI3K-SH3 amyloid fibril dissociation on the protein concentration at fixed concentrations of denaturant. A suitable denaturant concentration for each protein was chosen based on the data in Fig. 1 and 2; we chose 3 M urea for glucagon and 4 M urea for PI3K-SH3. In Fig. 3, we plot the concentration of soluble protein as a function of the total protein concentration. We performed these measurements by using the more conventional method of sample centrifugation, followed by measurement of the supernatant concentration. In Fig. S7 (ESI†), we show that for both proteins, the results from fluorescence and absorbance measurements are very similar, in particular at higher concentrations. We fit these data to both the isodesmic and the cooperative model and find that for both proteins, the cooperative model provides a significantly better fit than the isodesmic model. This is in particular also true for PI3K-SH3, for which both models gave very similar fits when two denaturant-dependent depolymerization curves were globally fitted at two different monomer concentrations (Fig. 2, left panel).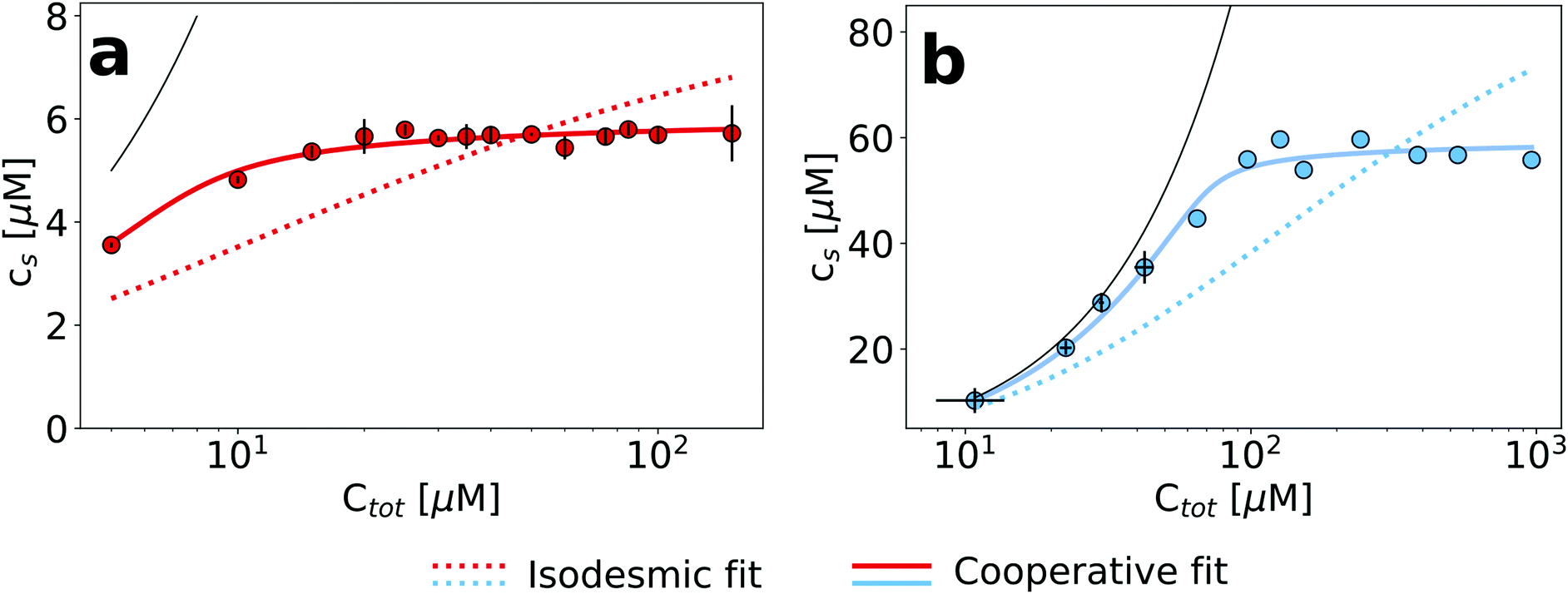 | ||
| Fig. 3 The dependence of the concentration of soluble protein on the total protein concentration at constant denaturant concentration. (a) PI3K-SH3 in 4 M urea. (b) Glucagon in 3 M urea. The data was obtained from direct concentration measurements of the supernatant after centrifugation. The solid black line indicates the total protein concentration in both cases. | ||
Therefore, the exploration of the protein concentration in addition to the denaturant concentration in amyloid fibril depolymerization experiments represents a powerful combination, allowing a more rigorous test and comparison of different models compared to an exploration of the denaturant concentration dimension alone.
3.5 Influence of ionic strength on amyloid fibril stability
Having established an experimental and conceptual framework in which to analyze the thermodynamic stability of amyloid fibrils quantitatively, we then proceeded to apply this methodology in order to probe the dependence of amyloid fibril stability on the ionic strength of the solution, by adding different concentrations of NaCl. We found that an increase in the concentration of NaCl stabilizes the amyloid fibrils of both PI3K-SH3 and glucagon (Fig. 4a and b), manifest through a shift of the depolymerization midpoint towards higher denaturant concentrations. This is expected, given the net positive charge of both proteins at the acidic pH of the study. The net charge carried by each monomer disfavors the polymerization process, and screening of the charges decreases this unfavorable contribution. We fitted the depolymerization curves by fixing the parameters of σ and n to the values determined in our previous best fits and plotted the logarithms of the equilibrium constants for elongation obtained from those fits against the square root of the total ionic strength of the solution (Fig. 4c and d, see Section S3 for the mathematical analysis, ESI†). We found that the stability of PI3K-SH3 amyloid fibrils depends more strongly on the solution ionic strength than the stability of glucagon fibrils, as evaluated from linear fits to these plots. This difference is consistent with the different (formal) net charge of PI3K-SH3 (+12) and glucagon (+5) at pH 2.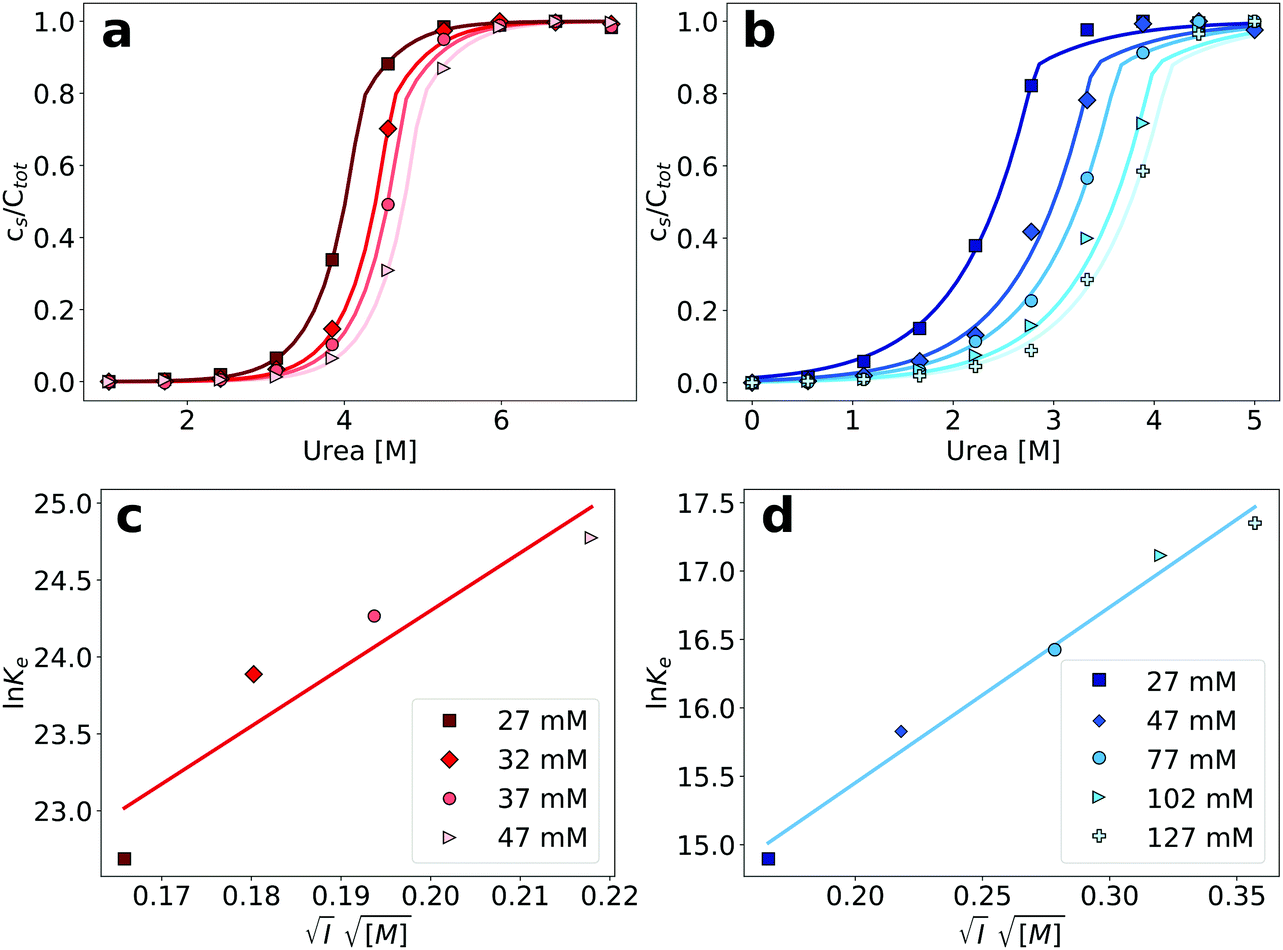 | ||
| Fig. 4 (a) Depolymerization profiles of PI3K-SH3 amyloid fibrils at different ionic strengths, adjusted by the addition of different NaCl concentrations. (b) Denaturation profiles of glucagon amyloid fibrils at different ionic strengths. (c) Plot of the logarithm of the equilibrium constants obtained from the fits in the top left panel, as a function of the square root of the ionic strengths for PI3K-SH3 (see Section S3 for details, ESI†). Parameters of the linear fit: slope = 37.5, y-intercept = 16.8. (d) Plot of the logarithm of the equilibrium constants obtained from the fits in the top-right panel, as a function of the square root of the ionic strengths for glucagon. Parameters of the linear fit: slope = 12.84, y-intercept = 12.88. | ||
We have previously performed a detailed study of the dependence of the amyloid fibril elongation kinetics of PI3K-SH3 on the concentration of NaCl at a low background ionic strength of 10 mM27 and we are now able to compare the ionic strength dependencies of both kinetics and thermodynamics of PI3K-SH3 amyloid fibril growth. The use of high urea concentrations in our chemical depolymerization experiments, however, requires a higher buffer concentration than that employed in the kinetic experiments and therefore the ionic strength ranges explored in the two cases overlap only slightly. However, by comparing the slope from the three highest ionic strength values explored in the kinetic experiments and the three lowest ionic strength values explored in the thermodynamic experiments, it is possible to semi-quantitatively compare the ionic strength dependence of kinetics and thermodynamics (Fig. 5a). We find that the fitted slopes are very similar for the kinetic (43.5) and thermodynamic (57.2) plots.
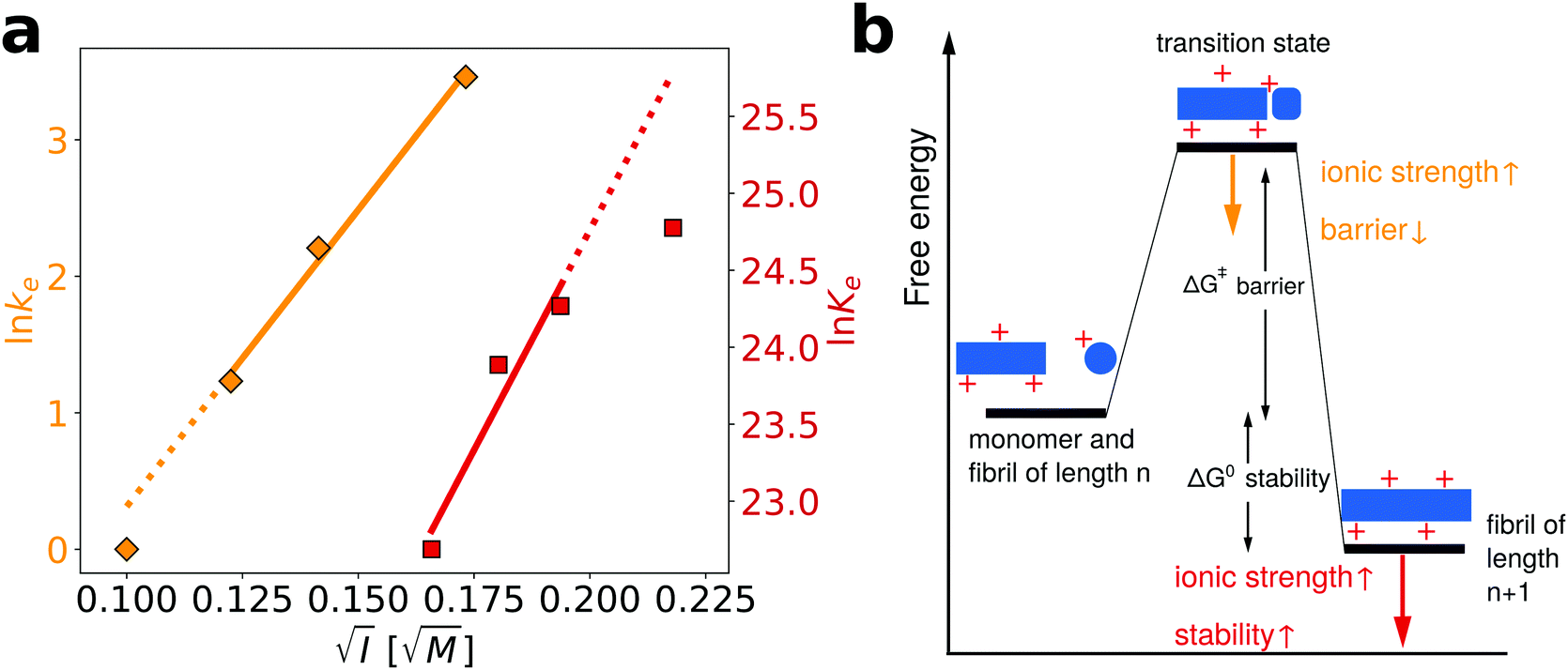 | ||
| Fig. 5 Comparison of the influence of salt on the kinetics and thermodynamics of the elongation of PI3K-SH3 amyloid fibrils. (a) In orange, the data from ref. 27, concerning the dependence of the elongation kinetics of PI3K-SH3 on the concentration of NaCl, while in red the thermodynamic data from this study. The linear fits have the following slopes: kinetics = 43.5; thermodynamics = 57.2 (b) schematic energy landscape illustrating the stabilizing effect that an increase in the ionic strength of the solution has on both the transition state and the final state of the elongation reaction. | ||
In Fig. 5b, we illustrate with a schematic energy landscape of fibril elongation how screening of the electrostatic charges by salt ions in solution can lower the free energy barrier,27 as well as lower the energy of the fibril and hence stabilize it (see Section S3 for details, ESI†).
4 Discussion
4.1 Intrinsic fluorescence as a probe of amyloid fibril depolymerization
The determination of amyloid fibril stability from chemical depolymerization experiments requires a measurement of the relative populations of fibrillar and monomeric protein. This can be achieved through physical separation of aggregated from non-aggregated protein by centrifugation of the sample and measurement of the protein concentration in the supernatant.22,33 However, spectroscopic approaches, based on either circular dichroism21,32 or intrinsic protein fluorescence32 have also been proposed, that do not require a physical separation of aggregates from monomeric protein. This feature is useful, as the separation of aggregated from soluble protein can depend on the size distribution of the aggregated species in conjunction with the applied centrifugal force. Fibrillar and monomeric protein can in most cases be expected to have a different spectroscopic signature, due to differences in secondary structure and chemical environment of the aromatic amino acid residues. Therefore the change in relative populations can be monitored by analyzing the ratio of the fluorescence emission at two different wavelengths. We use this method here, based on Trp fluorescence, and show that it is equivalent to the direct concentration determination in the supernatant after centrifugation.4.2 Nature of the denaturant
The equilibrium between the soluble and fibrillar conformation can be altered by the addition of chemical denaturant, similar to the denaturation of folded proteins. Interestingly, we find here that urea can be a more powerful denaturant for amyloid fibrils than GndHCl, which is in contrast to the general finding that GndHCl is a more powerful denaturant for protein unfolding than urea.35 This inversion in the denaturant strength between monomeric proteins and amyloid fibrils can be explained through the differential importance of electrostatic effects. For the homo-molecular polymerization process of amyloid fibril formation, electrostatic interactions are generally unfavorable,42 and the addition of a denaturant salt, such as GndHCl, will stabilise amyloid fibrils electrostatically at the same time as destabilizing them through competition for the hydrogen bonding network.43 At comparably low denaturant concentrations, the stabilizing effect can be stronger than the destabilizing one, leading to a net stabilization. This stabilizing effect is virtually absent for non-ionic denaturants, such as urea, therefore explaining the depolymerization of some amyloid fibrils at lower urea than GndHCl concentrations. It can be expected that this effect is particularly pronounced under solution conditions where the proteins carry a high net charge, such as the low pH values used here.4.3 Appropriate model for the analysis of chemical depolymerization of amyloid fibrils
The theoretical framework for the description of the thermodynamics of linear polymers has been developed by Oosawa, initially for actin polymerization. There it was found that the system was best described by so-called helical polymers, whereby each monomer interacts with several other monomers in the fibril, not only with the two next neighbors, as expected for a perfectly one-dimensional polymer. This structural feature leads to the fact that the initial nucleus is difficult to form, as a monomer will not be able to form the same number of favorable interactions with the nucleus compared to when it adds onto a complete fibril. Amyloid fibril thermodynamics has so far exclusively been analyzed with the simplest form of the linear polymerization model,21,22,33 whereby a single equilibrium constant for monomer addition is postulated. As we show in this work, individual chemical depolymerization curves can be fitted very well with this simple model, but as soon as the total concentration of the protein is varied, it becomes obvious in some cases that this model cannot accurately describe the equilibrium behavior of amyloid fibrils. Inspired by the field of supramolecular polymerization, we find that a model that allows for a less favorable equilibrium constant of monomer attachment to species below a threshold size n provides a consistent theoretical description that allows to quantitatively account for the data. Interestingly, we find that the best fits are achieved for small values of n (approximately between 2 and 5) and for values of σ that are one to two orders of magnitude smaller than unity. We would like to stress here that this model is likely to still represent an oversimplification, given the real complexity of amyloid fibril formation, whereby in most cases different steps of monomer addition and structural rearrangement44–46 lead to the formation of a minimal fibril. Nevertheless, it is probably the simplest extension to the basic linear polymerization model and the fact that our data can be well-fitted suggests that at least for the thermodynamic behaviour of amyloid fibrils, a description in terms of an effective equilibrium constant for the addition of monomers to small, pre-fibrillar species captures the essence of the process. It is interesting to note that the numerical values of the free energy of monomer addition and σ together suggest that also the addition of monomers to the smaller structures is favorable, albeit less so than the addition of monomer to a fully formed fibril. These results can be compared to the kinetics of amyloid fibril nucleation, which is generally found to be very slow and therefore the formation of the nucleus is viewed as being highly unfavorable. Indeed, the finding that in many amyloid systems, fibrils are found that consist of thousands of individual monomers suggests that the rate of nucleation of a fibril is at least 3–4 orders of magnitude slower than the rate of its growth. These observations can be reconciled with our present analysis by considering that for the kinetic behavior, the height of the free energy barriers, as well as the reaction order of nucleation and growth processes, have to be taken into account. A detailed analysis of these parameters in the case of the amyloid β peptide has recently revealed that the free energy barrier for primary nucleation is indeed several times higher than that for fibril elongation.47 In our experiments, however, we probe the thermodynamic behavior of fibril formation. While the energy barrier for the formation of a dimer, trimer or tetramer might be much higher than for monomer addition to a fibril,47 our results suggest that once such a small oligomer is formed, it can be similarly stable, per monomer, as a fully grown fibril.It is also interesting to note that the increasing number of available high resolution structures of amyloid fibrils34,48,49 paints the consistent picture of a minimal fibril unit, consisting of between 2 and 8 monomers, which displays the full range of interactions of a fully grown fibril. It is plausible that structures smaller than this minimal unit are thermodynamically somewhat less stable than bigger structures, in contrast to classical nucleation theory, where structures smaller than the critical nucleus are considered unstable with respect to individual monomers.
4.4 Electrostatic effects to probe the nature of the transition state for amyloid fibril elongation
Our experimental and conceptual framework allows us to probe the effect of changes in solution ionic strength on the thermodynamic stability of amyloid fibrils. Here we study two proteins that form fibrils at acidic pH conditions, where the individual protein molecules carry a positive net charge that opposes homomolecular polymerization.27,42,52,53 The electrostatic repulsion of the individual monomers within the fibril can be expected to be screened by salt ions in solution, similar to what has been reported for the formation of surfactant micelles54 or virus capsids.55 The use of urea as a denaturant allowed us to probe this effect and we represent the resulting data by plotting the logarithm of the equilibrium constant against the square root of the total ionic strength (Fig. 4a). The slope of such a plot depends on the effective charge of the molecular interaction in question (see Section S3, ESI† for a detailed discussion of the underlying model). In agreement with the higher formal net charge of PI3K-SH3 compared to glucagon at acidic pH, we find that the stability of PI3K-SH3 amyloid fibrils is more strongly affected by solution ionic strength than that of glucagon fibrils. It is insightful to compare the dependence of both PI3K-SH3 amyloid formation thermodynamics and kinetics27 on NaCl concentration. For technical reasons (see above), the range of ionic strengths exploited in both studies overlaps only slightly, but if we extrapolate the respective slopes we are nevertheless able to compare kinetic and thermodynamic ionic strength dependencies. We find that the slopes of the kinetic and thermodynamic ionic strength dependency plots are very similar (Fig. 5a), with the thermodynamic slope being slightly higher. This result suggests that the monomer adopting the transition state for fibril elongation experiences very similar, albeit slightly less, electrostatic repulsion compared to the monomer fully incorporated into the fibril, thereby implying a high degree of similarity between the transition state and the final state of the fibril elongation reaction. Under conditions of acidic pH, where each monomer carries only positive charges and therefore a substantial net charge, electrostatic repulsion is probably mainly defined by the principal reaction coordinate,50 the center of mass distance between the monomer and the fibril end. Therefore, our results suggest that the transition state of PI3K-SH3 amyloid fibril elongation corresponds to a monomer in very close proximity of the fibril end, only marginally removed from its final position when incorporated into the fibril. This conclusion is in close agreement with the finding that the hydrophobic effect plays a major role in stabilizing the transition state of fibril elongation, as revealed through a strongly favorable entropy of activation.47,56 The picture that therefore emerges from this analysis is that the rate-limiting step of amyloid fibril elongation consists of a structural rearrangement of the monomer while being in close contact with the fibril end. Therefore, the defining energy barrier for fibril elongation is of an inter-molecular, rather than intra-molecular nature (see Fig. 6 for a comparison of these two distinct scenarios). This result is an important contribution to the ongoing discussion about the intra- vs. intermolecular nature of the protein misfolding events that lead to amyloid fibril formation. It has been proposed for several amyloid systems, such as poly-glutamine57 and tau58 that the crucial and rate-limiting event along the pathway of amyloid fibril formation is a purely intra-molecular misfolding event to form an 'aggregation competent state' that can add onto a fibril without any significant barrier crossing. Our results for PI3K-SH3, on the other hand, are more compatible with the picture whereby the misfolding transition is a highly cooperative event between monomer and fibril end.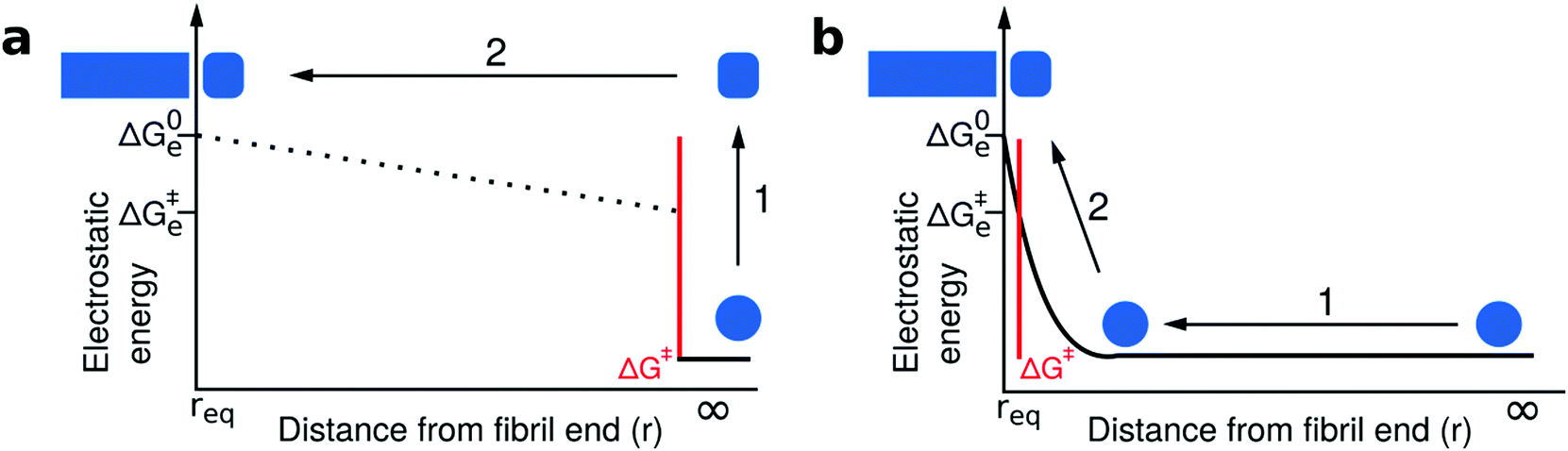 | ||
| Fig. 6 Comparison of two distinct scenarios of the transition state of amyloid fibril growth. The free energy barriers that define the kinetics of fibril growth are marked in red in both cases; however, we plot here only the electrostatic component of the free energy as a function of the center of mass separation between the approaching monomer and the fibril end. (a) The transition state of fibril elongation corresponds to an isolated misfolding event of the free monomer (1), which is electrostatically unfavorable followed by a diffusive search of the fibril end (2). The electrostatic signature of the latter cannot be elucidated from kinetic experiments, because only the highest free energy barrier is probed.50,51 The final state (monomer incorporated) is electrostatically less favorable than the initial state and the transition state, but has the lowest free energy. (b) The transition state of fibril elongation corresponds to a situation after a diffusional search (1), where the monomer is in close contact with the fibril end. The misfolding reaction is aided by the presence of the fibril end (2, templating effect). The transition state and the final state are similarly electrostatically unfavorable, because these two states have a very similar center-of-mass distance that dominates the electrostatic interactions. Therefore, we argue that our data on the ionic strength dependence of both kinetics and thermodynamics of fibril elongation is best compatible with a late, product-like transition state (scenario b). | ||
5 Conclusions
In summary, we have been able to show for the first time that the thermodynamics of amyloid fibril formation is best described by cooperative polymerization rather than by simple isodesmic linear polymerization. This result is in excellent agreement with mechanistic insight into amyloid fibril formation, as well as with the emerging high resolution structural information on amyloid fibrils. Furthermore, we have been able to accurately probe the role of electrostatic effects in amyloid fibril stability. In combination with previously available data on the influence of solution ionic strength on the kinetics of amyloid fibril growth, we are able to reveal the product-like nature of the transition state-ensemble for amyloid fibril elongation by PI3K-SH3 monomers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AKB and NV thank the Deutsche Forschungsgemeinschaft (DFG) for financial support. AKB thanks the Novo Nordisk Foundation for support through a Novo Nordisk Foundation professorship. The authors thank Sabine Metzger for help with the mass spectrometry, Florian Platten for helpful discussions and Novo Nordisk for a kind gift of glucagon.References
- F. Oosawa, S. Asakura, K. Hotta, N. Imai and T. Ooi, J. Polym. Sci., 1959, 37, 323–336 CrossRef CAS.
- G. G. Borisy and J. B. Olmsted, Science, 1972, 177, 1196–1197 CrossRef CAS.
- J. Hofrichter, P. D. Ross and W. A. Eaton, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4864–4868 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- F. Oosawa, Thermodynamics of the Polymerization of Protein, Academic Press Inc, 1975 Search PubMed.
- F. Oosawa and M. Kasai, J. Mol. Biol., 1962, 4, 10–21 CrossRef CAS.
- T. Oda, M. Iwasa, T. Aihara, Y. Maéda and A. Narita, Nature, 2009, 457, 441–445 CrossRef CAS PubMed.
- B. Magdoff-Fairchild and C. C. Chiu, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 223–226 CrossRef CAS PubMed.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, Structure, 2010, 18, 1244–1260 CrossRef CAS PubMed.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K. P. R. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS PubMed.
- K. A. Conway, J. D. Harper and P. T. Lansbury, Biochemistry, 2000, 39, 2552–2563 CrossRef CAS.
- W. S. Gosal, A. H. Clark and S. B. Ross-Murphy, Biomacromolecules, 2004, 5, 2408–2419 CrossRef CAS.
- A. Lomakin, D. B. Teplow, D. A. Kirschner and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7942–7947 CrossRef CAS PubMed.
- T. P. J. Knowles, C. A. Waudby, G. L. Devlin, S. I. A. Cohen, A. Aguzzi, M. Vendruscolo, E. M. Terentjev, M. E. Welland and C. M. Dobson, Science, 2009, 326, 1533–1537 CrossRef CAS PubMed.
- S. I. A. Cohen, M. Vendruscolo, M. E. Welland, C. M. Dobson, E. M. Terentjev and T. P. J. Knowles, J. Chem. Phys., 2011, 135, 065105 CrossRef PubMed.
- A. K. Buell, C. Galvagnion, R. Gaspar, E. Sparr, M. Vendruscolo, T. P. J. Knowles, S. Linse and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(21), 7671–7676 CrossRef CAS PubMed.
- E. P. O'Brien, Y. Okamoto, J. E. Straub, B. R. Brooks and D. Thirumalai, J. Phys. Chem. B, 2009, 113, 14421–14430 CrossRef PubMed.
- F. Bard, C. Cannon, R. Barbour, R. Burke, D. Games, H. Grajeda, T. Guido, K. Hu, J. Huang, K. Johnson-Wood, K. Khan, D. Kholodenko, M. Lee, I. Lieberburg, R. Motter, M. Nguyen, F. Soriano, N. Vasquez, K. Weiss, B. Welch, P. Seubert, D. Schenk and T. Yednock, Nat. Med., 2000, 6, 916–919 CrossRef CAS PubMed.
- X. Gao, M. Carroni, C. Nussbaum-Krammer, A. Mogk, N. B. Nillegoda, A. Szlachcic, D. L. Guilbride, H. R. Saibil, M. P. Mayer and B. Bukau, Mol. Cell, 2015, 59, 781–793 CrossRef CAS PubMed.
- T. Narimoto, K. Sakurai, A. Okamoto, E. Chatani, M. Hoshino, K. Hasegawa, H. Naiki and Y. Goto, FEBS Lett., 2004, 576, 313–319 CrossRef CAS PubMed.
- A. J. Baldwin, T. P. J. Knowles, G. G. Tartaglia, A. W. Fitzpatrick, G. L. Devlin, S. L. Shammas, C. A. Waudby, M. F. Mossuto, S. Meehan, S. L. Gras, J. Christodoulou, S. J. Anthony-Cahill, P. D. Barker, M. Vendruscolo and C. M. Dobson, J. Am. Chem. Soc., 2011, 133, 14160–14163 CrossRef CAS PubMed.
- E. Hellstrand, B. Boland, D. M. Walsh and S. Linse, ACS Chem. Neurosc., 2010, 1, 13–18 CrossRef CAS PubMed.
- C. N. P. J. K. Myers and J. M. Scholtz, Protein Sci., 1995, 4, 2138–2148 CrossRef PubMed.
- D. Zhao and J. S. Moore, Org. Biomol. Chem., 2003, 1, 3471–3491 RSC.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. de Greef, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- A. K. Buell, P. Hung, X. Salvatella, M. E. Welland, C. M. Dobson and T. P. J. Knowles, Biophys. J., 2013, 104, 1116–1126 CrossRef CAS.
- J. Zurdo, J. I. Guijarro, J. L. Jimenez, H. R. Saibil and C. M. Dobson, J. Mol. Biol., 2001, 311, 325–340 CrossRef CAS PubMed.
- G. H. Beaven, W. B. Gratzer and H. G. Davies, Eur. J. Biochem., 1969, 11, 37–42 CrossRef CAS PubMed.
- J. S. Pedersen, J. Diabetes Sci. Technol., 2010, 4, 1357–1367 CrossRef PubMed.
- J. I. n. Guijarro, M. Sunde, J. A. Jones, I. D. Campbell and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4224–4228 CrossRef CAS PubMed.
- J. S. Pedersen, D. Dikov, J. L. Flink, H. A. Hjuler, G. Christiansen and D. E. Otzen, J. Mol. Biol., 2006, 355, 501–523 CrossRef CAS PubMed.
- R. Porcari, C. Proukakis, C. A. Waudby, B. Bolognesi, P. P. Mangione, J. F. S. Paton, S. Mullin, L. D. Cabrita, A. Penco, A. Relini, G. Verona, M. Vendruscolo, M. Stoppini, G. G. Tartaglia, C. Camilloni, J. Christodoulou, A. H. V. Schapira and V. Bellotti, J. Biol. Chem., 2015, 290, 2395–2404 CrossRef CAS PubMed.
- C. Roeder, N. Vettore, L. N. Mangels, L. Gremer, R. B. Ravelli, D. Willbold, W. Hoyer, A. K. Buell and G. F. Schroeder, Nat. Commun., 2019, 10, 3754 CrossRef PubMed.
- J. S. Martin, J. Parker and A. R. Clarke, J. Mol. Biol., 1995, 253, 771–786 CrossRef PubMed.
- L. Kollipara and R. P. Zahedi, Proteomics, 2013, 13, 941–944 CrossRef CAS PubMed.
- A. Fersht, Structure and Mechanism in Protein Science, W.H. Freeman, New York, 1999 Search PubMed.
- A. K. Buell, Int. Rev. Cell Mol. Biol., 2017, 329, 187–226 Search PubMed.
- P. A. Korevaar, C. Schaefer, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 13482–13491 CrossRef CAS PubMed.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. Smulders, P. A. Hilbers, A. P. Schenning, T. F. De Greef and E. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed.
- C. Boesch, A. Bundi, M. Oppliger and K. Wüthrich, Eur. J. Biochem., 1978, 91, 209–214 CrossRef CAS PubMed.
- S. L. Shammas, T. P. J. Knowles, A. J. Baldwin, C. E. Macphee, M. E. Welland, C. M. Dobson and G. L. Devlin, Biophys. J., 2011, 100, 2783–2791 CrossRef CAS PubMed.
- E. P. O'Brien, R. I. Dima, B. Brooks and D. Thirumalai, J. Am. Chem. Soc., 2007, 129, 7346–7353 CrossRef PubMed.
- T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317–1321 CrossRef CAS PubMed.
- N. Cremades, S. I. A. Cohen, E. Deas, A. Y. Abramov, A. Y. Chen, A. Orte, M. Sandal, R. W. Clarke, P. Dunne, F. A. Aprile, C. W. Bertoncini, N. W. Wood, T. P. J. Knowles, C. M. Dobson and D. Klenerman, Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- G. A. Garcia, S. I. A. Cohen, C. M. Dobson and T. P. J. Knowles, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2014, 89, 032712 CrossRef PubMed.
- S. I. Cohen, R. Cukalevski, T. C. Michaels, A. Šarić, M. Törnquist, M. Vendruscolo, C. M. Dobson, A. K. Buell, T. P. Knowles and S. Linse, Nat. Chem., 2018, 10, 523 CrossRef CAS PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116–119 CrossRef CAS PubMed.
- A. W. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov, H. J. Garringer, R. A. Crowther, B. Ghetti, M. Goedert and S. H. Scheres, Nature, 2017, 547, 185 CrossRef CAS PubMed.
- A. K. Buell, J. R. Blundell, C. M. Dobson, M. E. Welland, E. M. Terentjev and T. P. J. Knowles, Phys. Rev. Lett., 2010, 104, 228101 CrossRef PubMed.
- T. C. Michaels, L. X. Liu, S. Curk, P. G. Bolhuis, A. Šarić and T. P. Knowles, Mol. Phys., 2018, 116, 3055–3065 CrossRef CAS.
- F. Massi, D. Klimov, D. Thirumalai and J. E. Straub, Protein Sci., 2002, 11, 1639–1647 CrossRef CAS PubMed.
- D. K. Klimov and D. Thirumalai, Structure, 2003, 11, 295–307 CrossRef CAS PubMed.
- G. Gunnarsson, B. Joensson and H. Wennerstroem, J. Phys. Chem., 1980, 84, 3114–3121 CrossRef CAS.
- W. K. Kegel and P. van der Schoot, Biophys. J., 2004, 86, 3905–3913 CrossRef CAS PubMed.
- A. K. Buell, A. Dhulesia, D. A. White, T. P. J. Knowles, C. M. Dobson and M. E. Welland, Angew. Chem., Int. Ed., 2012, 51, 5247–5251 CrossRef CAS PubMed.
- K. Kar, M. Jayaraman, B. Sahoo, R. Kodali and R. Wetzel, Nat. Struct. Mol. Biol., 2011, 18, 328–336 CrossRef CAS PubMed.
- H. Mirbaha, D. Chen, O. A. Morazova, K. M. Ruff, A. M. Sharma, X. Liu, M. Goodarzi, R. V. Pappu, D. W. Colby, H. Mirzaei, L. A. Joachimiak and M. I. Diamond, eLife, 2018, e36584 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp04524d |
| This journal is © the Owner Societies 2019 |