
Identification of active sites in CO oxidation over a Pd/Al2O3 catalyst†
Kazumasa
Murata
 a,
Eleen
Eleeda
a,
Junya
Ohyama
bc,
Yuta
Yamamoto
d,
Shigeo
Arai
d and
Atsushi
Satsuma
*ac
a,
Eleen
Eleeda
a,
Junya
Ohyama
bc,
Yuta
Yamamoto
d,
Shigeo
Arai
d and
Atsushi
Satsuma
*ac
aGraduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: satsuma@chembio.nagoya-u.ac.jp
bFaculty of Advanced Science and Technology, Kumamoto University, Kumamoto 860-8555, Japan
cElements Strategy Initiative for Catalysts and Batteries (ESICB), Kyoto University, Katsura, Kyoto 615-8520, Japan
dInstitute of Materials and Systems for Sustainability, Nagoya University, Nagoya, Japan
First published on 8th August 2019
Abstract
The active sites of Pd/Al2O3 catalysts for CO oxidations were identified by investigating the dependence of CO oxidation activities on the surface structure and morphology of Pd nanoparticles. The maximum catalytic activity was obtained for Pd particles approximately 2 nm in particle size. We performed structural analyses on the Pd surface through infrared (IR) spectroscopy of the adsorbed CO molecules. A positive correlation was obtained between catalytic activity and the fraction of linear CO adsorbed on Pd corner sites and Pd(111) facets, indicating that these sites are highly active for CO oxidation. X-ray absorption fine structure (XAFS) and spherical aberration-corrected scanning transmission electron microscopy (Cs-STEM) measurements demonstrated that Pd nanoparticles less than 2 nm in particle size with amorphous-like structures and Pd particles with large, well-ordered structures favor the formation of a high fraction of corner sites and Pd(111) facets, respectively.
Introduction
Supported metal nanoparticle catalysts have been widely used in a variety of industrially relevant catalytic processes such as purification of exhaust gas from automobiles. Thus, improving the performance of these heterogeneous catalysts has attracted considerable attention. One effective approach has been to control the size of the supported metal particles as the proportion of surface sites (e.g., corners or edges) on the nanoparticles changes dramatically depending on the particle size.1 For example, the abundance of low-coordination atoms on Au clusters improves the activities of CO oxidations,2 water gas shift reactions,3,4 and aldehyde hydrogenations.5 Additionally, it has been reported that the interfacial sites between Ni, Pd, or Pt nanoparticles and CeO2 supports, which increase with decreasing metal particle size, promote CO oxidations.6The crystal and surface structures of metal nanoparticles that are different from those of bulk metals occasionally improve the catalytic performance.7–12 For example, the controlled synthesis of 3 nm Ru nanoparticles improved the catalytic activity for hydrogen oxidations owing to the generation of an amorphous-like surface structure.11 Metal-support interaction (MSI) has also played an important role in determining the structure and catalytic activity of metal nanoparticles on supports.10,13,14 Recently, we investigated that effect of Pd particle size on methane combustion for Pd/Al2O3 with various alumina crystalline structures (γ-, θ-, and α-Al2O3).15 The interaction of γ-Al2O3 with Pd species is stronger than those of θ- and α-Al2O3. The morphology and surface structure of the Pd particles that varied based on Pd particle size and MSI had a profound effect on methane oxidation activity. The effects of size and morphology metal species have been well investigated using nanoparticle and single atom, especially CO oxidation and adsorption on Pt.16–19 In the case of extremely low metal loadings, metal species were present as an isolated metal atom on supports. It has been demonstrated that the isolated metal species (e.g., Pt,13,20 Pd,21 Rh,22,23 and Ru24) are generally more active than the corresponding nanoparticles in various catalytic reactions. Therefore, it is necessary to understand the influence of their structure on catalytic activity to improve their performance further and inform the design of new and efficient catalytic systems.
Pd-Based systems have been the most widely-used and effective catalysts in CO oxidations.25–29 Structure sensitivity studies probing the effect of Pd particle size on CO oxidation activity have also been of great interest, with the first report dating back almost 40 years ago.30 Despite the widespread use and interest in Pd-based CO oxidation catalysts, concerns on the sustainability and practicality of harsh reaction conditions have placed the emphasis on developing catalytic systems that can operate at lower temperatures (<200 °C). One of the important parameters influencing the reaction rate of CO oxidations is the size of the Pd particles. Wang and co-workers demonstrated that the activity of a Pd/Al2O3 CO oxidation catalyst increased with decreasing particle size from 50–70 nm to 4–8 nm,31 and Osaki observed a similar trend for particle sizes between 5 and 14 nm.32 In contrast, Haneda25 and Phan27 have reported that the catalytic activity of Pd/Al2O3 catalysts in CO oxidations is not affected by the Pd particle size for 4–14 nm and 5–12 nm structures, respectively. Moreover, Peterson et al. have reported that Pd dispersed atomically on Al2O3 is more effective for CO oxidations than Pd nanoparticles at low temperatures.29 Microcalorimetric measurements and density functional theory (DFT) calculations have demonstrated that the adsorption properties of CO on Pd nanoparticles (<1000 atoms) are considerably different from bulk Pd.33–36 Taken together, these studies suggest there is great variability on the relationship between Pd particle size and CO oxidation activity. We were therefore interested in probing the effects of non-reported particle sizes (<4 nm) to discern whether greater CO oxidation activity could be achieved.
Herein, we prepared Pd/Al2O3 catalysts of various particle sizes and alumina crystalline phases and evaluated their CO oxidation activities. We hypothesized that controlling the particle size and increasing the strength of the MSI would favorably change the shape and surface structures of the metal nanoparticles. We combined X-ray absorption fine structure (XAFS) spectroscopy, spherical aberration-corrected scanning transmission electron microscopy (Cs-STEM), and infrared (IR) spectroscopy using CO as a molecular probe to identify the size, shape, and surface structure of the Pd particles on Al2O3. A comparison between the relative fraction of various Pd surface sites and the catalytic activities revealed the most active sites for CO oxidations.
Experimental methods
Catalyst preparations
γ-Al2O3 was obtained by thermal decomposition of boehmite (Sasol, PURAL alumina) at 500 °C for 1 h. θ-Al2O3 (AKP-G07) was supplied from Sumitomo Chemical Co. Ltd and calcined at 500 °C for 1 h. Pd (Pd loading: 0.1–2 wt%) was deposited onto alumina supports with various crystalline phases via an impregnation method using a 4.5 wt% Pd(NO3)2 solution. The alumina supports were impregnated with an aqueous 4.5 wt% Pd(NO3)2 solution, and the suspension was stirred for 1 h. Excess water was removed by a rotary evaporator at 60 °C, and then the catalysts were dried at 80 °C for 8 h and calcined at 300 °C or 500 °C for 3 h. Some of the samples were further treated at 800 °C, 850 °C, or 900 °C under air for 10 h to obtain Pd/Al2O3 catalysts with various Pd particle sizes. The X-ray diffraction patterns, which were reported in our previous paper (Fig. S2 in ref. 15), indicated that the γ-Al2O3 phase is stable after calcination at temperatures at or below 900 °C.CO and H2 pulse chemisorption measurements
CO and H2 pulse chemisorption measurements were performed using the BEL-CAT-B instrument (MicrotracBEL). Approximately 50–200 mg of the sample was placed into a sample tube and pretreated with 100% O2 at 300 °C for 10 min and then 100% H2 at 300 °C for 10 min. After the sample was cooled to 50 °C under a He atmosphere, CO pulse chemisorption measurements were taken at 5% CO/He while monitoring the effluent with a thermal conductivity detector. For the H2 pulse chemisorption studies, the pretreated sample was first exposed to an Ar atmosphere at 300 °C for 10 min to eliminate adsorbed H atoms on Pd. The sample was then cooled to 0 °C, and the pulse chemisorption measurements were performed under a gaseous mixture of 5% H2/Ar. The Pd dispersion was calculated based on the total volume of adsorption gases by assuming that CO and H2 were adsorbed on the Pd surface at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Pd:CO) and 1:2 (Pd:H2) stoichiometries, respectively.
:1 (Pd:CO) and 1:2 (Pd:H2) stoichiometries, respectively.
Aberration-corrected scanning transmission electron microscopy (Cs-STEM)
The structure and size of the Pd nanoparticles supported on Al2O3 were observed using the Cs-STEM, JEM-ARM200F (JEOL Ltd), operating at 200 kV. Scanning transmission electron microscope (STEM) images were recorded by a high angle annular dark field (HAADF) detector. Pd/Al2O3 catalysts were pretreated under a flowing mixture of 10% O2/N2 for 10 min at 300 °C followed by 10% H2/N2 for 10 min at 300 °C. The samples were prepared by spreading a drop of the pretreated Pd/Al2O3 catalysts in a methanol suspension onto the microgrid carbon polymer supported on cupper. The Pd particle size distribution was also estimated after CO oxidation. Samples in Fig. S10 (ESI†) were prepared by an initial oxidation in 10% O2/N2 at 300 °C for 10 min and subsequent reduction in 10% H2/N2 at 300 °C for 10 min. These samples were then subjected to a mixture of 0.4% CO, 10% O2, and N2 balance at 130 °C for 30 min.Fourier transform infrared (FT-IR) spectroscopy studies
FT-IR measurements were performed using a quartz in situ IR cell and a JASCO FT/IR-6100 instrument (JASCO Co.) with a liquid-nitrogen-cooled HgCdTe (MCT) detector. IR spectra were obtained by averaging 128 scans at a resolution of 4 cm−1. The samples were pressed into approximately 50–100 mg of the self-supporting disk and mounted into the IR cell with a CaF2 window. The samples were pretreated under oxidation conditions in 10% O2/Ar at 300 °C for 10 min and then reduced in 10% H2/Ar at 300 °C for 10 min. It should be noted that samples used in our previous study were oxidized at 400 °C and reduced at 200 °C.15 This difference in pretreatment temperature should not have affected the oxidation state of the Pd catalysts because the oxidation form of Pd (i.e., PdO) can be reduced to Pd metal below room temperature. The samples were then cooled to room temperature under Ar, and IR spectra were taken for reference. CO (0.4% CO/Ar at a rate of 100 mL min−1) was introduced into the quartz in situ IR cell for 10 min. The physisorbed CO on Pd/Al2O3 catalysts were removed under a flow of 100% Ar, and IR spectra of adsorbed CO on Pd/Al2O3 were obtained.X-ray absorption fine structure (XAFS) studies
Pd K-edge XAFS measurements were conducted on the BL14B2 beamline of the SPring-8 synchrotron radiation facility at the Japan Synchrotron Radiation Research Institute in Hyogo, Japan. The data analysis was performed using the Athena software included in the Demeter package. The samples were pretreated under 10% O2/N2 for 10 min at 300 °C and then under 10% H2/N2 for 10 min at 300 °C.CO oxidation reactions
The CO oxidation reactions were conducted using a conventional fixed-bed flow reactor at atmospheric pressure. Prior to performing an activity test, each sample (10 mg) inside a U-shaped quartz tube (inside diameter of 4 mm) was exposed to a flowing mixture of 10% O2/N2 for 10 min at 300 °C and then 3% H2/N2 for 10 min at 300 °C. The CO oxidation test was performed under a gaseous mixture of 0.4% CO, 10% O2, and N2 balance at a total flow rate of 100 mL min−1, corresponding to a gas hourly space velocity of 600000 mL g−1 h−1. The effluent gas was analyzed by a nondispersive infrared CO/CO2 analyzer (Horiba VIA510). The CO conversion was measured at steady-state from 100 °C to 200 °C. The turnover frequency (TOF) was defined as the reaction rate per molar amount of surface Pd, as determined by CO pulse measurements. Samples showing >20% CO conversion at 130 °C were diluted with inert quartz to reduce CO conversion and exclude thermal and gas diffusion problems that would affect activity tests conducted at this temperature. The TOF values in Fig. S6 (ESI†) were calculated using the amount of surface Pd determined by H2 pulse chemisorption.
Results
Catalyst preparations
Table 1 shows all Pd/Al2O3 catalysts examined in the present study. In Table 1, “catalyst” is denoted as X nm Pd/Al2O3, where X corresponds to the average Pd particle size of Pd/Al2O3 estimated by CO pulse chemisorption. To ensure accurate measurements for the smaller Pd sized particles (i.e., 1.5–9.0 nm Pd/γ-Al2O3), catalyst preparations and CO pulse chemisorption studies were performed at least three times and average values for Pd dispersions and Pd particle sizes are displayed in Table 1. Based on the revised experimental design, we now characterize 1.9, 3.7, 5.4, and 7.7 nm Pd/γ-Al2O3 in ref. 31 as 2.6, 4.1, 5.2, and 9.0 nm Pd/γ-Al2O3, respectively. The particle sizes and dispersions calculated by H2 pulse chemisorption were consistent with those estimated by CO pulse chemisorption, with the exception of 0.1 wt% Pd/Al2O3. As it may be difficult to occur a dissociative adsorption of H2 molecules on isolated Pd atom, the H2 pulse technique underestimated the dispersion of 0.1 wt% Pd/Al2O3 (Table 1).| Catalyst | Pd loading (wt%) | 1st calcination temperature (°C) | 2nd calcination temperature (°C) | CO pulse chemisorption | H2 pulse chemisorption | ||
|---|---|---|---|---|---|---|---|
| Pd dispersion (%) | Average Pd particle sizea (nm) | Pd dispersion (%) | Average Pd particle sizea (nm) | ||||
| a Calculated from Pd dispersion assuming spherical Pd particles. | |||||||
| 1.5 nm Pd/γ-Al2O3 | 0.1 | 300 | — | 71 ± 7 | 1.5 ± 0.1 | 51 | 2.2 |
| 1.6 nm Pd/γ-Al2O3 | 0.2 | 300 | — | 71 ± 1 | 1.6 ± 0.1 | 60 | 1.9 |
| 2.2 nm Pd/γ-Al2O3 | 0.5 | 500 | — | 54 ± 11 | 2.2 ± 0.1 | 48 | 2.3 |
| 2.6 nm Pd/γ-Al2O3 | 1 | 500 | — | 45 ± 9 | 2.6 ± 0.5 | 38 | 2.9 |
| 4.1 nm Pd/γ-Al2O3 | 2 | 500 | — | 27 ± 3 | 4.1 ± 0.4 | 34 | 3.3 |
| 5.2 nm Pd/γ-Al2O3 | 2 | 500 | 800 | 24 ± 9 | 5.2 ± 1.1 | 21 | 5.3 |
| 9.0 nm Pd/γ-Al2O3 | 2 | 500 | 850 | 13 ± 2 | 9.0 ± 1.1 | 11 | 9.9 |
| 19 nm Pd/γ-Al2O3 | 2 | 500 | 900 | 6 | 19.1 | 7 | 15.9 |
| 1.2 nm Pd/θ-Al2O3 | 0.1 | 300 | — | 97 | 1.2 | 66 | 1.7 |
| 1.4 nm Pd/θ-Al2O3 | 0.2 | 300 | — | 83 | 1.4 | 77 | 1.5 |
| 1.5 nm Pd/θ-Al2O3 | 0.5 | 500 | — | 73 | 1.5 | 37 | 3 |
| 3.8 nm Pd/θ-Al2O3 | 1 | 500 | — | 29 | 3.8 | 25 | 4.5 |
| 5.4 nm Pd/θ-Al2O3 | 2 | 500 | — | 21 | 5.4 | 17 | 6.7 |
| 7.3 nm Pd/θ-Al2O3 | 2 | 500 | 800 | 15 | 7.3 | 13 | 8.5 |
| 14 nm Pd/θ-Al2O3 | 2 | 500 | 850 | 8 | 14.4 | 8 | 14.4 |
| 19 nm Pd/θ-Al2O3 | 2 | 500 | 900 | 6 | 19.2 | 6 | 18.1 |
XAFS measurements
XAFS measurements were performed to gain insights on the oxidation state and coordination structure of the Pd/Al2O3 catalysts. Fig. 1a and b show the Pd K-edge X-ray absorption near edge structure (XANES) spectra of Pd/Al2O3 using Pd foil and PdO as reference samples. The XANES spectra of Pd/Al2O3 with smaller Pd particles were similar to that of the PdO reference. This result indicates the presence of Pd2+ species in Pd/Al2O3 with particle sizes between 1 and 2 nm. As the particle size of Pd/Al2O3 increased, the X-ray absorption intensity of Pd/Al2O3 at approximately 24357 eV decreased. This relationship suggests that metallic Pd nanoparticles tended to form on Al2O3 as the Pd particle size increased.
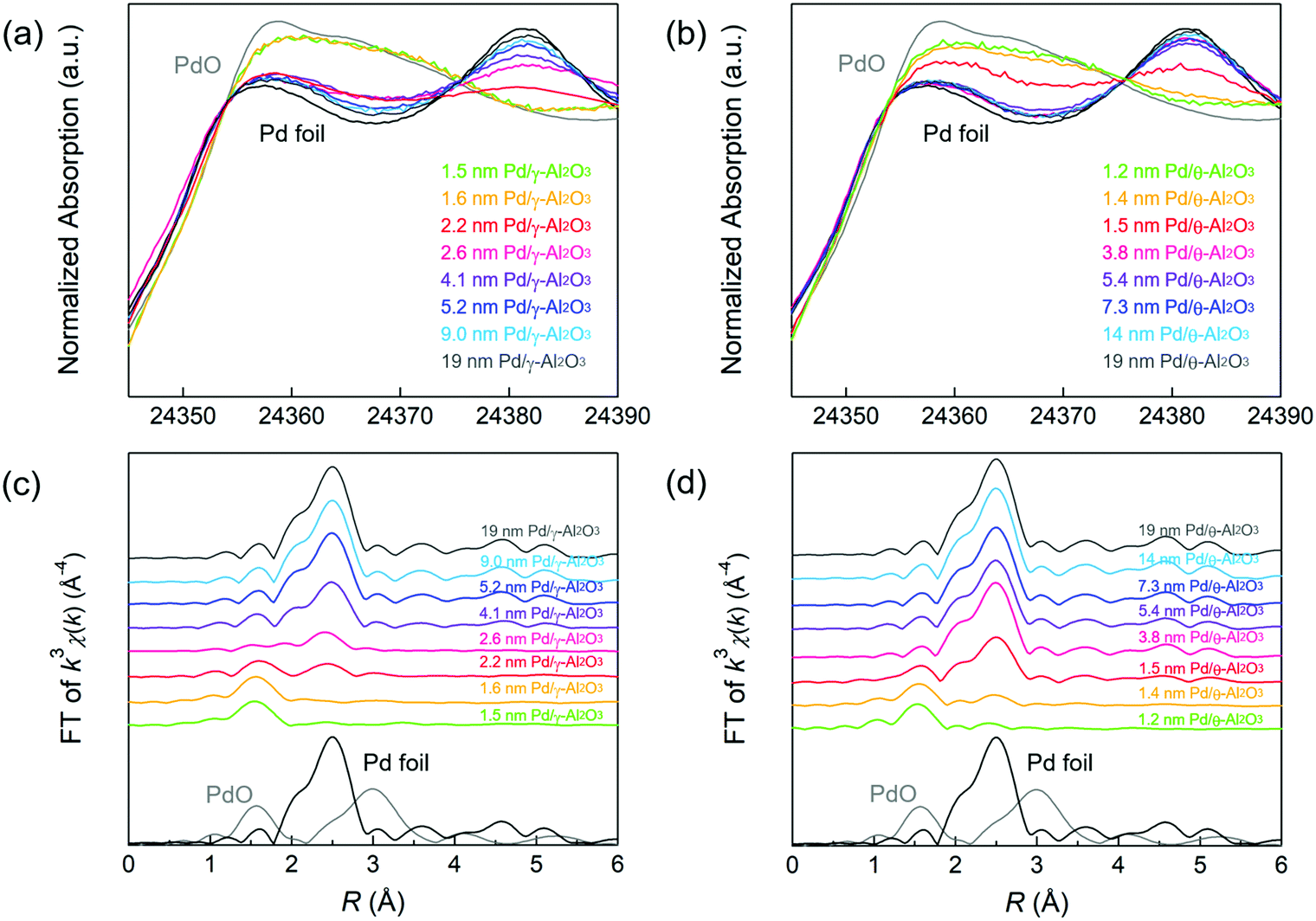 | ||
| Fig. 1 Pd K-edge (a and b) XANES and (c and d) FT-EXAFS spectra for Pd/Al2O3 with different average particle sizes after H2 reduction, together with those of Pd foil (black line) and PdO (gray line) as references. | ||
Fourier-transformed (FT)–extended X-ray absorption fine structure (EXAFS) spectra of Pd/γ-Al2O3 are displayed in Fig. 1c. Pd–O scattering in PdO and Pd–Pd scattering in the Pd foil samples were observed at 1.6 and 2.5 Å, respectively. In 1.5 and 1.6 nm Pd/γ-Al2O3, Pd–O scattering (1.6 Å) without Pd–Pd scattering (2.5 Å) was observed, suggesting that these samples are composed of isolated Pd atoms. In 2.2 and 2.6 nm Pd/γ-Al2O3, small peak due to Pd–Pd scattering was observed at 2.5 Å, indicating the formation of small amorphous-like Pd particles with low Pd–Pd coordination. The intensity of this Pd–Pd scattering peak was clearly detected for Pd particles larger than 2.6 nm while Pd–O scattering disappeared. These results indicate that Pd species are transformed from isolated atoms to small amorphous like particles and then to crystalline particles as the particle size increases on the Al2O3 support. A Pd–Pd scattering peak was clearly observed in the EXAFS spectrum of 1.5 nm Pd/θ-Al2O3 (Fig. 1d) but was not detected for 1–3 nm Pd/γ-Al2O3. The formation of crystalline Pd particles preferentially forms on θ-Al2O3 rather than γ-Al2O3 because isolated Pd atoms and amorphous Pd particles are less stable on θ-Al2O3, resulting in weak MSI.9
STEM images of Pd/Al2O3 catalysts
Cs-STEM was employed to visualize all Pd species (e.g., single atoms, small clusters) on Al2O3 at the atomic level. Fig. 2 shows STEM images of three typical Pd/γ-Al2O3 samples. The STEM images of other Pd/γ-Al2O3 catalysts are shown in Fig. S1 (ESI†). The STEM or transmission electron microscopy (TEM) images of 19 nm Pd/γ-Al2O3 and Pd/θ-Al2O3 of various Pd particle sizes are displayed in ref. 31. A STEM image of 1.5 nm Pd/γ-Al2O3 depicted isolated Pd atoms and amorphous-like Pd particles smaller than 2 nm on γ-Al2O3 (Fig. 2a). In 2.2 nm Pd/γ-Al2O3, amorphous-like Pd particles were mainly detected (Fig. 2b). When the particle size increased to 9.3 nm, the Pd particle was distorted in shape, and its surface had an amorphous-like structure (Fig. 2c). The Pd particles maintained a distorted shape even at particle sizes up to 19 nm. The size distributions of the Pd particles in Pd/γ-Al2O3 were obtained from STEM images (Table S1 and Fig. S2, ESI†). Pd particles less than or equal to 0.3 nm were denoted as an isolated Pd atom. The size distribution of 2.2 nm Pd/γ-Al2O3 exhibited a high fraction of Pd nanoparticles smaller than 2.1 nm excluding isolated Pd atoms. | ||
| Fig. 2 Typical STEM images of (a) 1.5 nm Pd/γ-Al2O3, (b) 2.2 nm Pd/Al2O3, and (c) 9.3 nm Pd/Al2O3. The red arrows indicate the positions of the single Pd atoms. | ||
Isolated Pd atoms and small amorphous Pd particles were detected in 1.5 nm Pd/θ-Al2O3, which displayed a similar STEM image as 2.2 nm Pd/γ-Al2O3. However, structural variations of the Pd particles on θ-Al2O3 with weaker MSI were notably different from those supported on γ-Al2O3. The 7.3 nm Pd/θ-Al2O3 were spherical in shape, and as the Pd particles increased in size to 19 nm, they transformed from spherical to well-faceted shapes. For particles ranging from 4–19 nm, the fraction of thermodynamically stable facets (e.g., Pd(111)) increased with increasing Pd particle size.
IR spectroscopy studies of adsorbed CO on Pd/Al2O3
IR spectroscopy using CO as a molecular probe was employed to identify surface sites on the Pd nanoparticles. Fig. 3a shows two representative IR spectra of adsorbed CO on Pd/Al2O3. The intensities of the IR bands were normalized because of differences in the Pd loadings and dispersions. IR spectra between 1750–2300 cm−1 were well-fitted with four stretching vibration bands corresponding to CO molecules adsorbed to Pd. The broad band centered at 2150 cm−1 is attributed to a linear CO molecule adsorbed on a cationic Pd species (Pd+–COlinear).37–40 The band at approximately 2075 cm−1 is attributed to linear CO adsorbed on the corner of Pd particles or Pd(111) (Pd0–COlinear);40–47 however, it is difficult to distinguish the corner sites from Pd(111) based on the-bands. The band at 1980 cm−1 is attributed to a bridged CO molecule adsorbed on Pd steps (Pd0step–CObridge).33,42,44,45,47 The broad bands at 1750–1960 cm−1 are derived mainly from bridged or three-fold CO molecules adsorbed on planes, which includes Pd(111) (Pd0plane–CObridge).33,37–47 Bands corresponding to linear and three-fold CO were observed when adsorbed CO reached saturation on Pd(111).41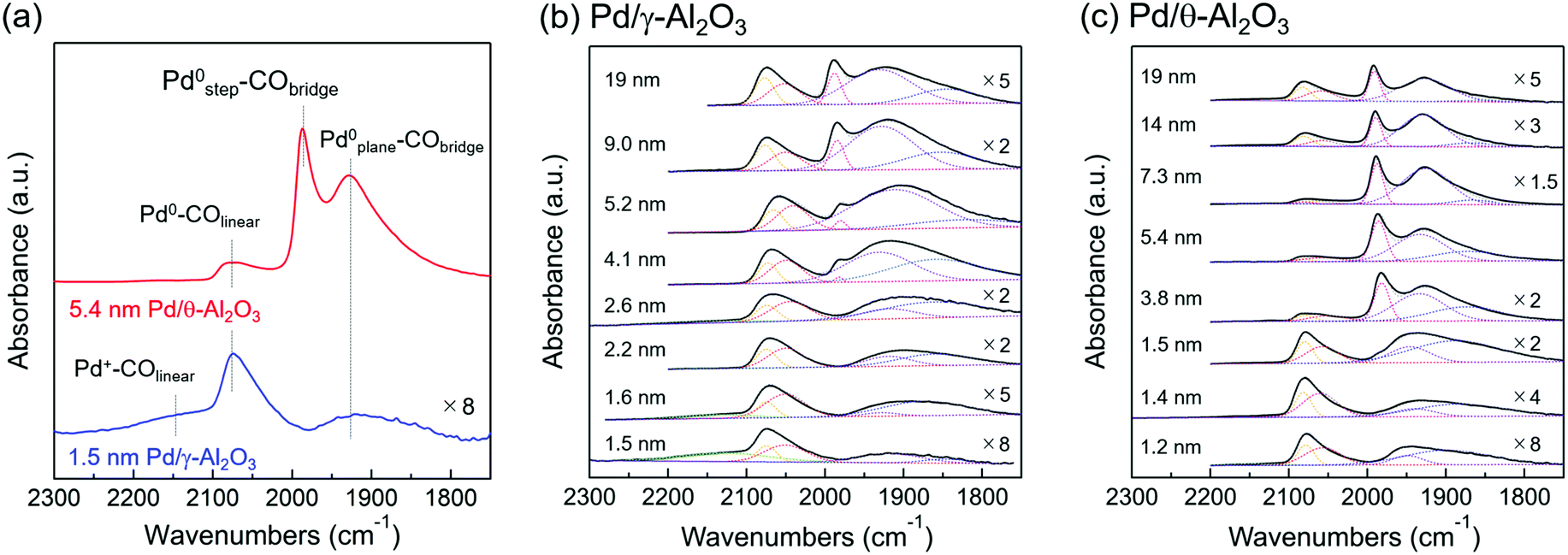 | ||
| Fig. 3 (a) IR spectra of CO chemisorbed to 1.5 nm Pd/γ-Al2O3 and 5.4 nm Pd/θ-Al2O3 at room temperature. The band centered at 2150 cm−1 is assigned to linear CO adsorbed on cationic Pd species (Pd+–COlinear). The band at around 2075 cm−1 is assigned to linear CO adsorbed on the corner of Pd particles or Pd(111) (Pd0–COlinear). The band at 1980 cm−1 is assigned to bridge adsorbed CO on the step (Pd0step–CObridge). The broad bands at 1750–1960 cm−1 are assigned mainly to bridged or three-fold adsorbed CO on planes, including Pd(111) (Pd0plane–CObridge). IR spectra of adsorbed CO at room temperature on (b) Pd/γ-Al2O3 and (c) Pd/θ-Al2O3 of various Pd particle sizes. IR spectra of 2.6–19 nm Pd/γ-Al2O3 and 1.5–19 nm Pd/θ-Al2O3 were previously reported in ref. 31, Fig. 2. Gaussian fittings of IR spectra were carried out to determine the band areas of 2100–2200, 2000–2100, 1980, and 1750–1960 cm−1. | ||
Fig. 3b and c (solid lines) show the IR spectra of CO adsorbed on Pd/Al2O3 for various Pd particle sizes. Fig. 3b and c (dotted lines) also show the IR bands of each adsorbed CO species fitted by a Gaussian function to quantify the Pd surface structures described in section of “Active Pd surface structure analyses”. Fig. 3b depicts the IR spectra of adsorbed CO on Pd/γ-Al2O3. For 1.5 nm Pd/γ-Al2O3, Pd+–COlinear and Pd0–COlinear bands were mainly observed. The cationic Pd species in this sample can be identified as an isolated Pd atom on Al2O3 based on the XAFS results and TEM images of 1.5 nm Pd/γ-Al2O3. As the Pd particle size of Pd/γ-Al2O3 increased from 1.5 to 2.2 nm, the relative intensity of the Pd+–COlinear band decreased, and the Pd0–COlinear and Pd0plane–CObridge bands increased in intensity. This variation corresponds to a decrease in the amount of isolated Pd atoms and the formation of small Pd nanoparticles with amorphous structures. Thus, the amorphous-like Pd particles likely play a role in the formation of Pd corner sites. The Pd0step–CObridge band appeared when the Pd particle reached 4 nm or greater in size. Fig. 3c shows the CO adsorption IR spectrum of Pd/θ-Al2O3. Spectra corresponding to catalysts composed of Pd particles smaller than 2 nm were similar to the spectra for 1–3 nm Pd/γ-Al2O3. However, increasing the particle size from 1.5 to 7.3 nm led to a sharp decrease in the relative intensity of the Pd0–COlinear band and subsequent increase of the Pd0step–CObridge band. This trend suggests that amorphous-like Pd particles become spherical in shape when there is a high fraction of step sites. Furthermore, as the particle size increased from 7.3 to 19 nm, the relative intensity of the Pd0–COlinear band increased due to transformation of the particles from spherical to well-faceted shapes (e.g., Pd(111)).
CO oxidation activities of Pd/Al2O3 catalysts
Fig. S3 and S4 (ESI†) show CO conversions against reaction temperature using 10 mg of Pd/Al2O3 catalysts with different particle sizes and alumina crystal phases. Since the Pd loading weights and dispersions are quite different between the Pd/Al2O3 catalysts, TOFs were calculated at <20% CO conversion where thermal and gas diffusion problems are negligible (Fig. S5, ESI†). The catalyst weights, molar amounts of surface Pd, CO conversions, and CO oxidation rates are summarized in Table S2 (ESI†). Fig. 4 shows the dependence of TOF on Pd particle size. As the particle size increased from 1.5 to 2.2 nm, the TOF of Pd/γ-Al2O3 in CO oxidations increased from 0.08 s−1 to a maximum value of 0.18 s−1. When the particle size increased to 4 nm, the TOF sharply decreased to 0.08 s−1 and then gradually increased again with increasing particle size up to 19 nm.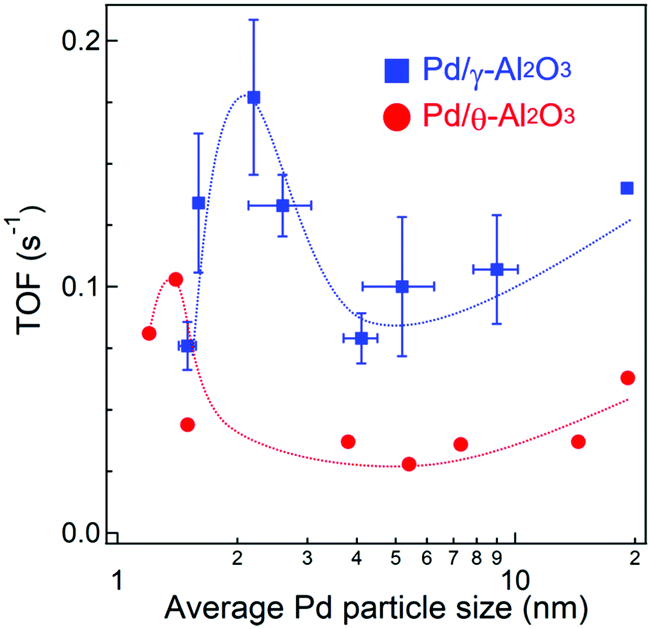 | ||
| Fig. 4 Dependence of TOFs at 130 °C on average Pd particle size. Experimental error is attributed to repeating the Pd/Al2O3 preparations, CO pulse measurements, and activity tests at least three times. | ||
The TOFs of Pd/θ-Al2O3 showed a similar trend to Pd/γ-Al2O3. However, the TOF of Pd/θ-Al2O3 was less than half of Pd/γ-Al2O3 for particles sized 4 nm or larger. The increase in TOF for the Pd/Al2O3 catalyst with particles sized between 4 and 14 nm was minimal, which is consistent with a previous study by Haneda and co-workers.25 When the TOFs were calculated using Pd dispersion based on H2 chemisorption measurements and plotted against Pd particle size (Fig. S6, ESI†), the trends were similar to those shown in Fig. 4.
Discussion
Active Pd surface structure analyses
To elucidate the active sites for CO oxidation, the surface structure of the Pd nanoparticles was quantified by the IR spectra depicted in Fig. 3. The Pd surface structure fraction was determined from the band area of adsorbed CO species obtained by Gaussian fitting (Fig. 3 and Table S3, ESI†). Also, we used six Gaussian functions to determine the band areas in the four regions, because the asymmetric bands at 2000–2100 and 1750–1960 cm−1 need two Gaussian functions for each band to be well fitted. The asymmetrical shape suggests two or more adsorption sites/manners as the band at 2000–2100 cm−1 is assignable to linearly adsorbed CO on corner or plane of Pd nanoparticles and as the band at 1750–1960 cm−1 is assignable to bridged or three-fold adsorbed CO on planes. Since the extinction coefficient of various adsorbed CO species is not known, it should be noted that the fraction of the Pd surface structure is a relative value and not an absolute value. The fraction of Pd0–COlinear was plotted against Pd particle size (Fig. 5a) and suggests a similar dependence of the Pd0–COlinear fraction on the particle size as the TOF. The highest faction of Pd0–COlinear was obtained for 2.2 nm Pd/γ-Al2O3, which also showed the highest catalytic activity. For particle sizes 8 nm or less, the high fraction of Pd0–COlinear could be attributed to the presence of amorphous Pd particles with a large amount of corner sites. In contrast, for particle sizes between 8 and 19 nm, an increase in the Pd0–COlinear fraction was observed, which is derived from formation of Pd(111) facets on the Pd particles. We estimated the particle size dependence of Pd site fractions on the surface of a model Pd cubo-octahedron (Fig. S7, ESI†).1,48 The particle size dependence of the fraction of Pd corner sites and Pd(111) facets on the Pd particle model was similar to that determined from IR spectroscopy (Fig. 5a). Therefore, the fraction of Pd0–COlinear for particle sizes <8 and >8 nm indicates the fraction of corner sites and Pd(111) facets, respectively (Fig. 5a). Catalysts with different crystalline alumina phases, i.e., γ-Al2O3, maintained their distorted Pd nanoparticles structure with a high fraction of corner sites due to stronger MSI. In contrast, Pd nanoparticles on θ-Al2O3 supports are characterized by weaker MSI and thus formed a higher fraction of step sites. TOF was plotted against the fraction of Pd0–COlinear (Fig. 5b) and showed that the fractions of Pd0–COlinear and TOF were proportional to one another. These studies suggest that corner sites on Pd particles and Pd(111) are highly active for CO oxidations. In contrast, a positive relationship was not observed between TOF and fractions of Pd+–COlinear, Pd0step–CObridge, or Pd0plane–CObridge (Fig. S8, ESI†).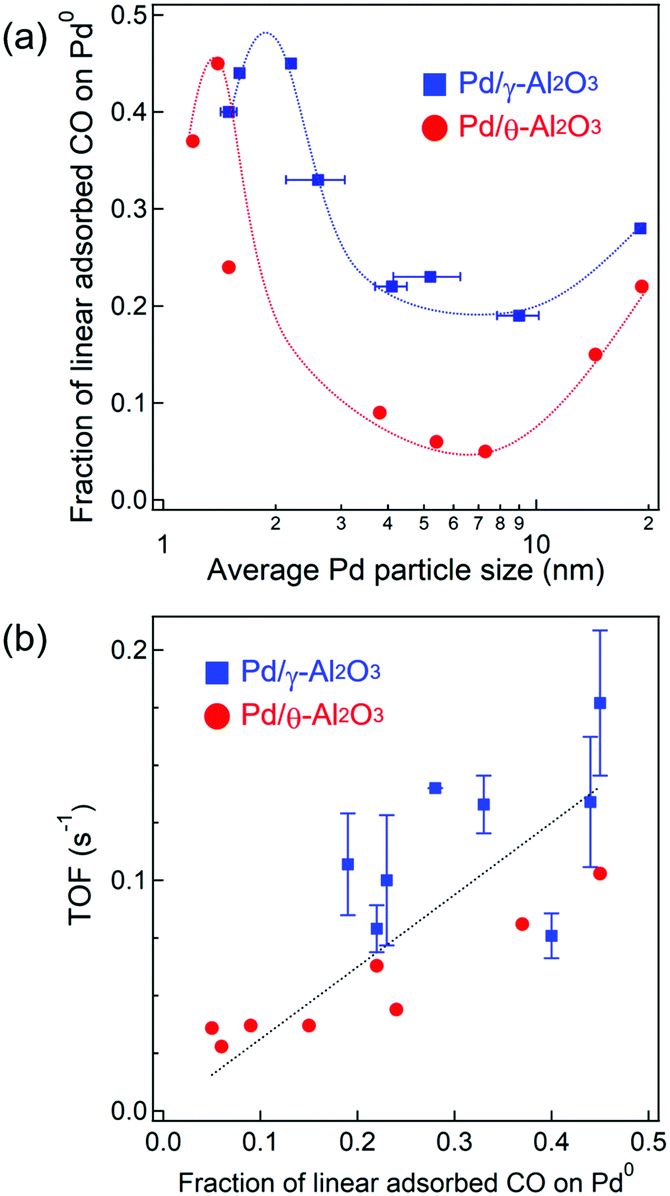 | ||
| Fig. 5 (a) Dependence of the fraction of linear CO adsorbed on Pd0 on the Pd particle size. (b) Plot of TOFs (at 130 °C) against the fraction of linear CO adsorbed on Pd0. | ||
The Pd/ZSM-5 catalyst (Pd loading: 0.26 wt%) was prepared via an ion exchange method to investigate the reactivity of isolated Pd atoms for CO oxidations. An IR spectrum of adsorbed CO on Pd/ZSM-5 showed that Pd was present as an isolated Pd species on ZSM-5 (Fig. S9a, ESI†). CO conversions for Pd/ZSM-5 were lower than all of the Pd/Al2O3 catalysts at 130 °C, and the TOF value for Pd/ZSM-5 was 0.009 s−1 (Fig. S9b, ESI†). Therefore, at this temperature, the isolated Pd atoms barely contribute to the CO oxidation activity, and the Pd metal clusters/nanoparticles are more active than the isolated Pd atoms.
Structure stability of Pd nanoparticles during CO oxidation
The stability of the isolated Pd atoms and Pd nanoparticles on Al2O3 during CO oxidation was confirmed by STEM. The Pd particle size distributions for 2.2 nm Pd/γ-Al2O3 before and after CO oxidation at 130 °C are shown in Fig. S10 (ESI†). Since the size distribution was barely affected by the reaction, we can conclude that the isolated Pd atoms and Pd nanoparticles were stable on Al2O3 under the studied reaction conditions.We hypothesized that CO-induced reconstruction of the Pd surface may have been occurring during CO oxidation at 130 °C. Avanesian reported surface reconstruction of Pt nanoparticles induced by CO adsorption at saturation for temperatures greater than 400 K.18 Pt(100) facets on Pt nanoparticles have also been reconstructed into step surfaces with high concentrations of under-coordinated Pt atoms as a result of CO adsorption. To determine whether the surfaces of the Pd nanoparticles were being reconstructed by CO exposure, we monitored the adsorbed CO species on Pd/Al2O3 before and after CO oxidation (Fig. S11, ESI†). The IR spectra of Pd/Al2O3 showed that the band intensity of linear CO adsorbed on the corner sites decreased after CO oxidation, whereas the band intensity of the bridged CO adsorbed on the facets (e.g., Pd(111) or Pd(100)) increased. These results indicated that CO-induced reconstruction of the Pd surface was occurring during CO oxidations at 130 °C because the bridged CO adsorbed on the Pd facet was more thermodynamically stable than the linearly bound CO adsorbed on the Pd corner. Variations in the measured fractions of linear CO on Pd0 was ∼10%. Changes in the Pd surface structure were more largely influenced by the Pd particle size and alumina crystalline phase than by the CO-induced reconstruction. Fig. S12 (ESI†) shows the plot of TOFs against the fraction of linear CO on Pd0 in Pd/Al2O3 after CO oxidation at 130 °C. The order of the Pd0–COlinear fraction among Pd/Al2O3 catalysts treated under CO oxidation conditions did not change. The fraction of Pd0–COlinear in Pd/Al2O3 after CO oxidation and the resulting TOFs were also directly correlated. Thus, we proposed that the Pd corner sites and Pd(111) facets worked as dominant active sites during CO oxidation.
Mechanistic studies
Kinetic analyses were conducted for Pd/Al2O3 catalysts of various Pd particle sizes. The apparent activation energies for CO oxidation by Pd/γ-Al2O3 with Pd particle sizes ranging from 1.5–9.0 nm were determined to be between 66–71 kJ mol−1 (Fig. S13 and Table S4, ESI†). Because there was little difference between their activation energy values, CO oxidation over Pd/Al2O3 catalysts of all studied sizes appeared to proceed via similar reaction mechanisms. We investigated the dependence of CO and O2 partial pressures on CO oxidation for Pd/γ-Al2O3 catalysts (Fig. S14 and Table S4, ESI†). Reaction orders with respect to CO and O2 were negative (between −0.58 and −0.89) and positive (between +0.66 and +1.08). Therefore, it was suggested that CO oxidation on the Pd metal surface proceeded via (i) a conventional Langmuir–Hinshelwood mechanism, which is reaction of adsorbed CO and adsorbed O atom49,50 or (ii) a mechanism involved O2 molecular adsorption and then an adsorbed CO-assisted O2 dissociation step.51 Although it is difficult to determine which mechanism is favored for CO oxidations over Pd particles at low temperatures, in both mechanisms, self-poisoning of the Pd metal surface by CO molecules would inhibit dissociative adsorption or molecular adsorption of O2. Thus, CO desorption from a Pd surface is likely the rate-limiting step for low temperature CO oxidations.26From the above kinetic analysis, it was inferred that desorption of CO from a Pd surface is an important step for CO oxidations. Given that CO desorption from the Pd surface is necessary for subsequent activation of O2, the desorption behavior of adsorbed CO from Pd/Al2O3 was next monitored by IR spectroscopy. CO was adsorbed onto pre-reduced 4.1 nm Pd/γ-Al2O3 at 40 °C and then the temperature of the IR cell was increased to 120 °C (in 10 °C intervals) under Ar flow. In Fig. 6a, the IR spectrum of 4.1 nm Pd/γ-Al2O3 at 40 °C exhibits three bands corresponding to Pd0–COlinear (2000–2100 cm−1), Pd0step–CObridge (1960–2000 cm−1), and Pd0plane–CObridge (1750–1960 cm−1), which is similar to the results shown in Fig. 3b. When the temperature was increased to 50–90 °C, the three CO stretching vibration bands decreased in intensity. The decrease in the band intensity of Pd0–COlinear was the most extreme and disappeared completely when the temperature was raised to 100 °C. To compare the adsorption strengths of CO on different Pd surface sites, the intensities of the CO absorbance bands for different surface sites were plotted against temperature (Fig. 6b). Assuming that CO coverage of each surface site at 40 °C and 120 °C is 100 and 0%, respectively, the CO coverage at various temperatures (between 50 °C and 110 °C) was evaluated based on normalized Δ absorbance values. A red shift of the Pd0plane–CObridge band was observed and attributed to a decrease in CO coverage. When the temperature increased from 40 °C to 80 °C, the normalized Δ absorbance of Pd0–COlinear decreased more intensely than Pd0step–CObridge and Pd0plane–CObridge. At 100 °C, bridged CO molecules adsorbed on the Pd plane were the only species observed. Thus, we concluded from these studies that the adsorption strength of CO on different Pd surface sites experienced the following trend: Pd0–COlinear < Pd0step–CObridge < Pd0plane–CObridge. This order indicated that linear CO on Pd corner sites and Pd(111) facets more readily desorbs than the bridged CO on Pd steps and planes. Using DFT calculations, Zeinalipour-Yazdi et al. reported that the order of CO adsorption energy follows the trend: linear < bridge < hollow-bound sites.52 To confirm that linear CO can easily desorb from Pd nanoparticles during CO oxidation, in situ IR spectroscopy measurements of adsorbed CO on Pd/Al2O3 during CO oxidations were conducted (Fig. S15, ESI†). The intensity of the Pd0–COlinear band (2000–2100 cm−1) decreased to half its original value, as temperature increased from 50 °C to 110 °C. In section of “Structure stability of Pd nanoparticles during CO oxidation”, we had determined that the fraction of Pd corner sites reconstructed to facets by adsorbed CO was approximately ∼10%. Therefore, the decrease of this Pd0–COlinear band was derived not only from CO-induced reconstruction but also from formation of Pd vacancy sites by CO desorption. In contrast, the intensities of the Pd0step–CObridge (1960–2000 cm−1) and Pd0plane–CObridge (1750–1960 cm−1) bands slightly increased with increasing temperature due to CO-induced reconstruction. At 130 °C, adsorbed CO on Pd particles completely disappeared as CO conversion nearly reached completion. The above in situ IR study suggests that at least a portion of the Pd corner sites and Pd(111) facets were present as vacancy sites to activate O2 molecules during CO oxidation.
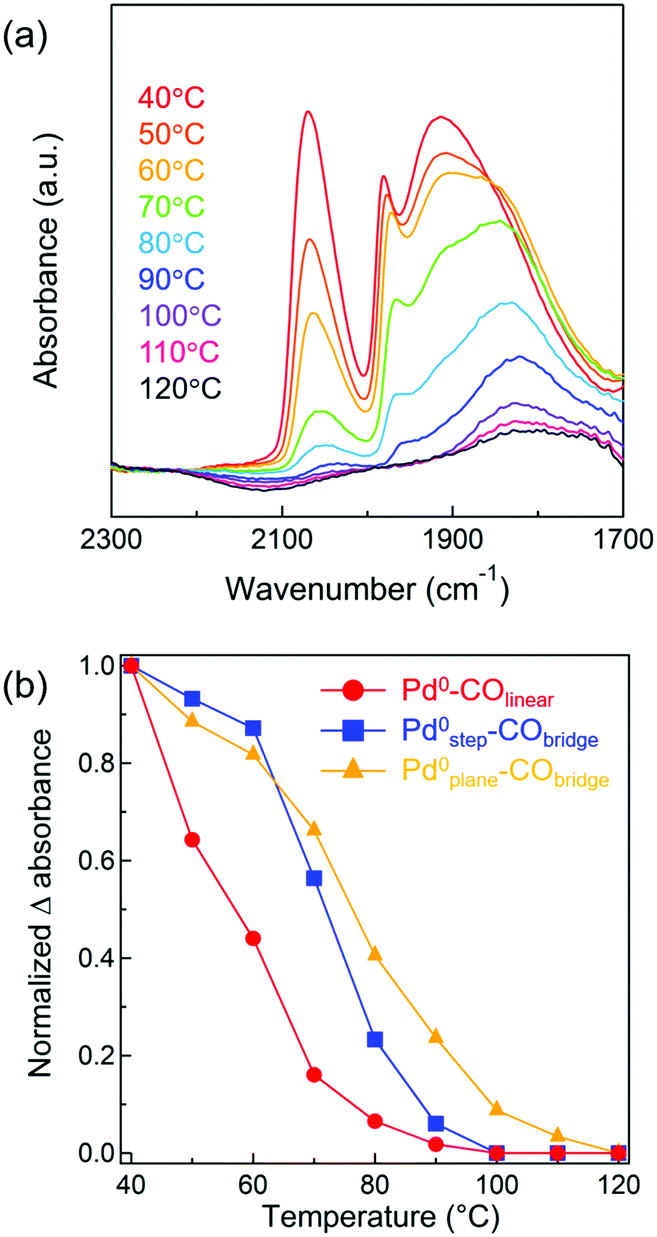 | ||
| Fig. 6 (a) FT-IR spectra of CO adsorbed on 4.1 nm Pd/γ-Al2O3 at various temperatures under Ar. (b) Plot of normalized Δ absorbance against temperature. Normalized Δ absorbance calculated from the IR band intensity (●: Pd0–COlinear (2000–2100 cm−1), ■: Pd0step–CObridge (1960–2000 cm−1), ▲: Pd0plane–CObridge (1750–1960 cm−1)). | ||
Conclusions
The effect of Pd particle size (1–19 nm) on CO oxidation for a range of Pd/Al2O3 catalysts was systematically studied, revealing complex behaviors and trends. Concretely, as the Pd particle size increased from approximately 1 to 2 nm, the catalytic activity increased; however, the activity sharply decreased with increasing particle size from 2 to 4 nm. Further growth of the Pd nanoparticles to 19 nm led to a slight increase in catalytic activity. The support structure also played a role in CO oxidation activity, as Pd/γ-Al2O3 showed higher activity than Pd/θ-Al2O3 for sizes ranging from 4 to 19 nm. These size effects could be rationalized based on the fraction of CO molecules adsorbed to Pd surface sites detected by IR spectroscopy. The positive correlation between the fraction of Pd0–COlinear and CO oxidation activity indicates that Pd corners and Pd(111) facets are highly active sites. The fraction of Pd0–COlinear also depended on the shape and surface structure of the Pd particles supported on Al2O3. For example, 2 nm Pd nanoparticles displayed an amorphous structure with a high fraction of corner sites. Increasing the size of the Pd particles to 19 nm promoted exposure of thermodynamically stable Pd(111) facets. When comparing Pd/γ-Al2O3 and Pd/θ-Al2O3, distorted Pd particles on γ-Al2O3 had a relatively higher fraction of highly active corner sites for CO oxidation than spherical or well-faceted Pd particles on θ-Al2O3. The structure sensitivity of CO oxidations over Pd nanoparticles was also apparent through CO adsorption strength studies, in which the adsorption strength was found to be dependent on the Pd surface site. Mechanistic studies clarified that corner sites and Pd(111) facets promote CO oxidation due to the ease at which CO can desorb from these surfaces.Our previous studies demonstrated that step sites on Pd nanoparticles are highly active for CH4 oxidation,15 whereas we identified corner sites and Pd(111) facets on Pd nanoparticles as being the most effective for high CO oxidation activity in the present work. Thus, the active surface of these heterogeneous catalysts strongly depends on the reaction substrate. To improve the catalytic activity of supported metal nanoparticles moving forward, it will be necessary to elucidate the shape and surface structure of metal nanoparticles that are most effective for the reaction substrate at the atomic level.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the JSPS KAKENHI Grant Number 16K14476 and 19J15440 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. A portion of this work was performed under management of the Elements Strategy Initiative for Catalysts & Batteries (ESICB), which is also supported by MEXT. XAFS measurement was conducted at BL14B2 of SPring-8 (Approval No. 2018B1756). The authors would like to thank Enago (http://www.enago.jp) for English language review.References
- R. V. A. N. Hardeveld and F. Hartog, Surf. Sci., 1969, 15, 189–230 CrossRef.
- X. M. Bu, C. H. Zhao, N. Zhang, S. Lin, F. Gao and X. W. Dai, Top. Catal., 2008, 16, 1074–1077 CAS.
- W. D. Williams, M. Shekhar, W. S. Lee, V. Kispersky, W. N. Delgass, F. H. Ribeiro, S. M. Kim, E. A. Stach, J. T. Miller and L. F. Allard, J. Am. Chem. Soc., 2010, 132, 14018–14020 CrossRef CAS PubMed.
- M. Shekhar, J. Wang, W. Lee, W. D. Williams, S. M. Kim, E. A. Stach, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Am. Chem. Soc., 2012, 134, 4700–4708 CrossRef CAS PubMed.
- J. Ohyama, A. Esaki, T. Koketsu, Y. Yamamoto, S. Arai and A. Satsuma, J. Catal., 2016, 335, 24–35 CrossRef CAS.
- M. Cargnello, V. V. T. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Science, 2013, 341, 771–773 CrossRef CAS PubMed.
- A. Carlsson, A. Puig-Molina and T. V. W. Janssens, J. Phys. Chem. B, 2006, 110, 5286–5293 CrossRef CAS PubMed.
- S. I. Sanchez, M. W. Small, E. S. Bozin, J. G. Wen, J. M. Zuo and R. G. Nuzzo, ACS Nano, 2013, 7, 1542–1557 CrossRef CAS PubMed.
- L. Li, L. L. Wang, D. D. Johnson, Z. Zhang, S. I. Sanchez, J. H. Kang, R. G. Nuzzo, Q. Wang, A. I. Frenkel, J. Li, J. Ciston, E. A. Stach and J. C. Yang, J. Am. Chem. Soc., 2013, 135, 13062–13072 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- J. Ohyama, T. Sato, Y. Yamamoto, S. Arai and A. Satsuma, J. Am. Chem. Soc., 2013, 135, 8016–8021 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- D. Mei, J. H. Kwak, J. Hu, S. J. Cho, J. Szanyi, L. F. Allard and C. H. F. Peden, J. Phys. Chem. Lett., 2010, 1, 2688–2691 CrossRef CAS.
- K. Murata, Y. Mahara, J. Ohyama, Y. Yamamoto, S. Arai and A. Satsuma, Angew. Chem., Int. Ed., 2017, 56, 15993–15997 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- L. Derita, S. Dai, K. Lopez-zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef CAS PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS PubMed.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- N. Tang, Y. Cong, Q. Shang, C. Wu, G. Xu and X. Wang, ACS Catal., 2017, 3–7 Search PubMed.
- M. Haneda, M. Todo, Y. Nakamura and M. Hattori, Catal. Today, 2017, 281, 447–453 CrossRef CAS.
- A. Satsuma, K. Osaki, M. Yanagihara, J. Ohyama and K. Shimizu, Appl. Catal., B, 2013, 132–133, 511–518 CrossRef CAS.
- D. Q. Phan and S. Kureti, Top. Catal., 2017, 60, 260–265 CrossRef CAS.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57–71 CrossRef CAS.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- C. R. Henry, Surf. Sci. Rep., 1998, 31, 231–325 CrossRef CAS.
- Z. Wang, B. Li, M. Chen, W. Weng and H. Wan, Sci. China: Chem., 2010, 53, 2047–2056 CrossRef CAS.
- T. Osaki, J. Porous Mater., 2012, 11, 697–711 Search PubMed.
- M. Peter, J. M. Florescamacho, S. Adamovski, L. K. Ono, K. H. Dostert, C. P. O’Brien, B. Roldancuenya, S. Schauermann and H. J. Freund, Angew. Chem., Int. Ed., 2013, 52, 5175–5179 CrossRef CAS PubMed.
- I. V. Yudanov, M. Metzner, A. Genest and N. Röseh, J. Phys. Chem. C, 2008, 112, 20269–20275 CrossRef CAS.
- S. M. Lang, I. Fleischer, T. M. Bernhardt, R. N. Barnett and U. Landman, ACS Catal., 2015, 5, 2275–2289 CrossRef CAS.
- J. H. Fischer-Wolfarth, J. A. Farmer, J. M. Flores-Camacho, A. Genest, I. V. Yudanov, N. Rösch, C. T. Campbell, S. Schauermann and H. J. Freund, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 8–11 CrossRef.
- W. Juszczyk, Z. Karpiński, I. Ratajczykowa, Z. Stanasiuk, J. Zieliński, L. L. Sheu and W. M. H. Sachtler, J. Catal., 1989, 120, 68–77 CrossRef CAS.
- D. Amalric-Popescu and F. Bozon-Verduraz, Catal. Lett., 2000, 64, 125–128 CrossRef CAS.
- M. Skotak, Z. Karpiński, W. Juszczyk, J. Pielaszek, L. Kępiński, D. V. Kazachkin, V. I. Kovalchuk and J. L. D’Itri, J. Catal., 2004, 227, 11–25 CrossRef CAS.
- J. Xu, L. Ouyang, W. Mao, X.-J. Yang, X. Xu, J.-J. Su, T.-Z. Zhuang, H. Li and Y.-F. Han, ACS Catal., 2012, 2, 261–269 CrossRef CAS.
- J. Szanyi, W. K. Kuhn and D. W. Goodman, J. Vac. Sci. Technol., A, 1993, 11, 1969 CrossRef CAS.
- K. Wolter, O. Seiferth, H. Kuhlenbeck, M. Bäumer and H.-J. Freund, Surf. Sci., 1998, 399, 190–198 CrossRef CAS.
- E. Ozensoy, D. C. Meier and D. W. Goodman, J. Phys. Chem. B, 2002, 106, 9367–9371 CrossRef CAS.
- H. Tiznado, S. Fuentes and F. Zaera, Langmuir, 2004, 20, 10490–10497 CrossRef CAS PubMed.
- T. Lear, R. Marshall, J. A. Lopez-Sanchez, S. D. Jackson, T. M. Klapötke, M. Bäumer, G. Rupprechter, H. J. Freund and D. Lennon, J. Chem. Phys., 2005, 123, 174706 CrossRef PubMed.
- J. Szanyi and J. H. Kwak, Phys. Chem. Chem. Phys., 2014, 16, 15117–15125 RSC.
- L. Ding, H. Yi, W. Zhang, R. You, T. Cao, J. Yang, J. Lu and W. Huang, ACS Catal., 2016, 6, 3700–3707 CrossRef CAS.
- J. Ohyama, T. Nishiyama and A. Satsuma, ChemCatChem, 2018, 10, 1651–1656 CrossRef CAS.
- T. Engel and G. Ertl, Chem. Phys. Lett., 1978, 54, 95–98 CrossRef CAS.
- M. M. Montemore, M. A. Van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS PubMed.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 4498–4517 CrossRef CAS PubMed.
- C. D. Zeinalipour-Yazdi, D. J. Willock, L. Thomas, K. Wilson and A. F. Lee, Surf. Sci., 2016, 646, 210–220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp03943k |
| This journal is © the Owner Societies 2019 |