
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromaticity and photophysics of tetrasila- and tetragerma-annelated tetrathienylenes as new representatives of the hetero[8]circulene family†
G. V.
Baryshnikov
 *ab,
R. R.
Valiev
cd,
V. N.
Cherepanov
e,
N. N.
Karaush-Karmazin
b,
V. A.
Minaeva
b,
B. F.
Minaev
b and
H.
Ågren
ae
*ab,
R. R.
Valiev
cd,
V. N.
Cherepanov
e,
N. N.
Karaush-Karmazin
b,
V. A.
Minaeva
b,
B. F.
Minaev
b and
H.
Ågren
ae
aDivision of Theoretical Chemistry and Biology, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, 10691, Stockholm, Sweden. E-mail: glibar@kth.se
bDepartment of Chemistry and Nanomaterials Science, Bohdan Khmelnytsky National University, 18031, Cherkasy, Ukraine
cResearch School of Chemistry & Applied Biomedical Sciences, National Research Tomsk Polytechnic University, Lenin Avenue 30, Tomsk 634050, Russia
dDepartment of Chemistry, University of Helsinki, FIN-00014, Helsinki, Finland
eTomsk State University, 36 Lenin Avenue, Tomsk, Russia
First published on 11th April 2019
Abstract
The electronic structure, absorption and emission spectra, aromaticity and photophysical behavior of the recently synthesized tetrasilatetrathia[8]circulene and tetragermatetrathia[8]circulene compounds have been studied computationally. Both compounds demonstrate a specific bifacial aromaticity, which is unusual for hetero[8]circulenes; the inner eight-membered core sustains an expected strong paratropic magnetically-induced ring current, while the outer perimeter contains saturated Si(Et)2 and Ge(Et)2 moieties which break the conjugation between the thiophene rings. The overall magnetically-induced ring current for both studied circulenes is close to zero because of the strong local diatropic currents in each thiophene ring that compensate the paratropic counterpart. The electronic absorption and emission spectra of tetrasilatetrathia[8]circulene and tetragermatetrathia[8]circulene demonstrate a clear visible vibronic progression. The 0–0 band is the most active one in the absorption spectra, while in the fluorescence spectra the 0–1 band composed of several normal vibrations is more intense compared with the 0–0 band in excellent agreement with experiment. Accounting for spin–orbit coupling effects, an analysis of the photophysical constants for the two compounds demonstrates: (1) a clear manifestation of the internal heavy atom effect on the inter-system crossing efficiency; (2) one to two order domination of non-radiative rates over the fluorescence rate; and (3) that the S1–S0 internal conversion is extremely slow and can not compete with the fluorescence, while the S1–Tn inter-system crossing is a main deactivation channel of the S1 excited state. These results provide new insight into the electronic structure and photophysics of tetrasilatetrathia[8]circulene and tetragermatetrathia[8]circulene as novel standalone representatives of hetero[8]circulenes – tetraannelated derivatives of tetrathienylene.
1. Introduction
Among the numerous representatives of hetero[8]circulenes the heteroannelated derivatives of tetraphenylene constitute the main and most studied ones.1–3 Some of them, like azaoxa[8]circulenes and tetraoxa[8]circulenes, demonstrate an intense blue fluorescence4,5 that is useful for fabrication of blue fluorescent organic light-emitting diodes (OLEDs) and extremely stable white exciplex-based OLEDs.6 At the same time, tetraoxa[8]circulenes and other heteroannelated tetraphenylenes (like tetraaza[8]circulenes and tetrathia[8]circulenes) demonstrate an ability to form continuous infinite one- and two-dimensional ribbons and sheets.7–11 These graphene-like materials sustain semiconductive properties and high dynamic stability, which makes them promising organic materials for use as semiconductors for modern organic electronics applications.7,11 The electronic structure, spectra and aromaticity of heteroannelated derivatives of tetraphenylene have been well studied both experimentally and theoretically1–3,12 and even some novel hypothetical circulenes have been predicted as upcoming targets of synthesis surveys.5Heteroannelated derivatives of tetrathienylene (cycloocta[1,2-c:3,4-c′:5,6-c′′:7,8-c′′′]tetrathiophene) constitute another much smaller class of hetero[8]circulenes. Among them only octathia[8]circulene (8S)13,14 and tetrathiatetraselena[8]circulene (4S4Se)15 have been prepared and characterized. Because of the high molecular symmetry and poor solubility of the 8S and 4S4Se circulenes their electronic absorption and emission spectra have only been sparsely studied (mainly by quantum-chemical methods),16,17 while the vibrational IR and Raman spectra have been well characterized and investigated in great detail, accounting for crystal packing effects and anharmonicity corrections.16,18,19 Both 8S and 4S4Se species have been used as promising organic semiconductors for fabrication of organic field-effect transistors with a maximum hole mobility of 9 × 10−3 cm2 V−1 s−1.15 Due to the planar structure and very high molecular symmetry, the 8S and 4S4Se species also demonstrate supramolecular assembly into 2D and 3D arrays, which is extremely important for modulation of the charge-transport properties in such systems.20,21
Very recently, Miyake et al. synthesized novel representatives of heteroannelated tetrathienylenes called tetrasilatetrathia[8]circulene22 (4S4SiEt2) and tetragermatetrathia[8]circulene23 (4S4GeEt2) by a palladium-catalyzed silylation/germylation process with subsequent rhodium-catalyzed intramolecular dehydrogenative cyclization. A similar technique has been also applied for the synthesis of tetrabenzotetrathia[8]circulenes24 (4S), which at the same time can be considered as tetrathiaannelated derivatives of tetraphenylene and tetrabenzoannelated derivatives of tetrathienylene (Fig. 1). Important to note is that the 4S compound was prepared for the first time by Wong et al. from octabromotetraphenylene by S-bridging,25 and one year later by Miyake et al. who independently prepared the 4S compound by a reverse strategy through (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) bridging of tetrathienylene.24
CH–) bridging of tetrathienylene.24
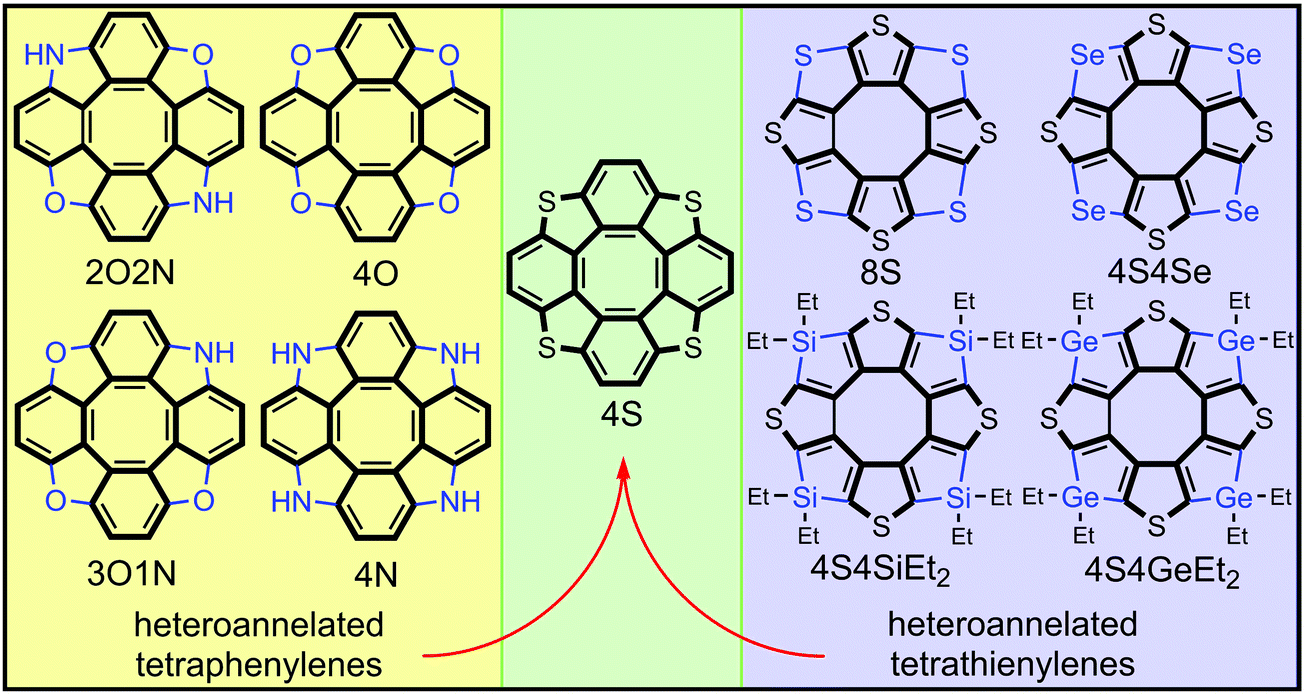 | ||
| Fig. 1 The structure of some representatives of heteroannelated derivatives of tetraphenylenes and tetrathienylenes. | ||
Circulene 4S has been comprehensively studied both theoretically and experimentally during the last ten years including the electronic and vibrational spectra, crystal structure, aromaticity etc.,26–29 while the novel 4S4SiEt2 and 4S4GeEt2 compounds22,23 still represent a challenge for computational chemistry. Especially important to note is that the photophysics of 4S4SiEt2 and 4S4GeEt2 has been studied experimentally by Miyake et al. in great detail22,23 including estimation of the rate constants for the internal conversion (IC), intersystem crossing (ISC), fluorescence and phosphorescence processes. That has motivated us to focus the present paper on high-level computations of the photophysical properties for the 4S4SiEt2 and 4S4GeEt2 compounds in order to explain and support the available experimental data.22,23 Moreover, the aromaticity of heteroannelated derivatives of tetrathienylene is still insufficiently studied and thus we have applied computations, in particular the gauge-including magnetically induced currents (GIMIC) methodology,30,31 to explain the distribution of diatropic and paratropic ring currents in the 4S4SiEt2 and 4S4GeEt2 molecules as novel representatives of fully annelated tetrathienylenes.
2. Computational details
The structure of the 4S4SiEt2 and 4S4SiEt2 molecules in the ground singlet electronic state (S0) and first excited state (S1) has been optimized at the DFT32 and TD DFT33 level of theory, respectively, in the gas phase using the B3LYP34,35 hybrid functional and the 6-31G(d,p)36 basis set within the Gaussian 16 program package.37 The vibronic absorption and emission spectra of the 4S4SiEt2 and 4S4GeEt2 molecules have been calculated within the Franck–Condon approximation through the time-domain formalism of Lax and Heller within the double harmonic parallel-mode approximation.38,39Magnetically induced current densities (current strengths) have been calculated using the GIMIC method.30,31 The NMR shielding calculations required for GIMIC computations have been performed at the B3LYP/def2-TZVP34,35,40 level of theory using the Turbomole program package.41 Ring-current strengths (I, nA T−1) and current strength pathways for the selected chemical bonds have been obtained by integrating the current density that flows through planes intersecting the chemical bonds. Additionally, the nucleus-independent chemical shift (NICS)42 descriptors have been calculated for the 4S4SiEt2 and 4S4GeEt2 molecules at the center (NICS(0)) and 1 Å above the center (NICS(1)) of each ring using the gauge-independent atomic orbital (GIAO)43 method at the B3LYP/6-311++G(d,p)34,35,44,45 level of theory. The Gaussian 16 program package37 was used for the NICS calculations.
The photophysical rate constants have been computed for the S1 state geometries based on extended multi-configuration quasidegenerate second order perturbation theory (XMC-QDPT2)46 calculations of the singlet and triplet excitation energies. The number of active electrons (e), the number of active orbitals (o), and the number of states (s) of the state-average (SA) complete active space self-consistent field (CASSCF) calculations have been assumed as 10e, 10o, and 5s. The XMC-QDPT2 calculations have been carried out using the Firefly program package.47
The spin–orbit coupling matrix elements 〈S1|HSO|Ti〉 between the S1 state and the energetically low-lying i-th triplet states Ti have been calculated at the ab initio CASSCF level of theory using the Gamess-US software.48 The IC rate constants have been computed by the original algorithm published in ref. 49 using the nonadiabatic coupling matrix elements calculated within the Turbomole package41 at the TD-DFT/B3LYP/def2-TZVP34,35,40 level of theory in the framework of perturbation theory. The spin–orbit coupling matrix elements required for the ISC rate calculations were taken from the previous CASSCF calculations. We assume that the vibrational frequencies and the Huang–Rhys factors are the same for the IC and ISC channels, something that was tested in our previous work.49 More details on the photophysical calculations can be found in the ESI.†
3. Results and discussion
3.1. Aromaticity (NICS and GIMIC criteria)
The aromaticity of hetero[8]circulenes is still a challenge for computational chemistry. The most acceptable concept for today is that hetero[8]circulenes are nonaromatic species in general because of the competitive diatropic (Idia) and paratropic (Ipara) currents in the outer (so-called rim) and inner (so-called hub) subsystems.28,30,50 Thus, the resulting magnetically-induced current (MIC) is close to zero (|Idia| ≈ |Ipara|) implying a non-aromatic character of most hetero[8]circulenes – derivatives of tetraphenylene and tetrathienylene. However, the studied 4S4SiEt2 and 4S4GeEt2 molecules contain sp3-hybrid Si and Ge atoms in the rim-subsystem that break conjugation in the outer perimeter. That is why the common double aromaticity concept can not be applied to the 4S4SiEt2 and 4S4GeEt2 molecules. Indeed, the current estimated along the Si(Ge)–C bonds is strictly equal to zero (Fig. 2).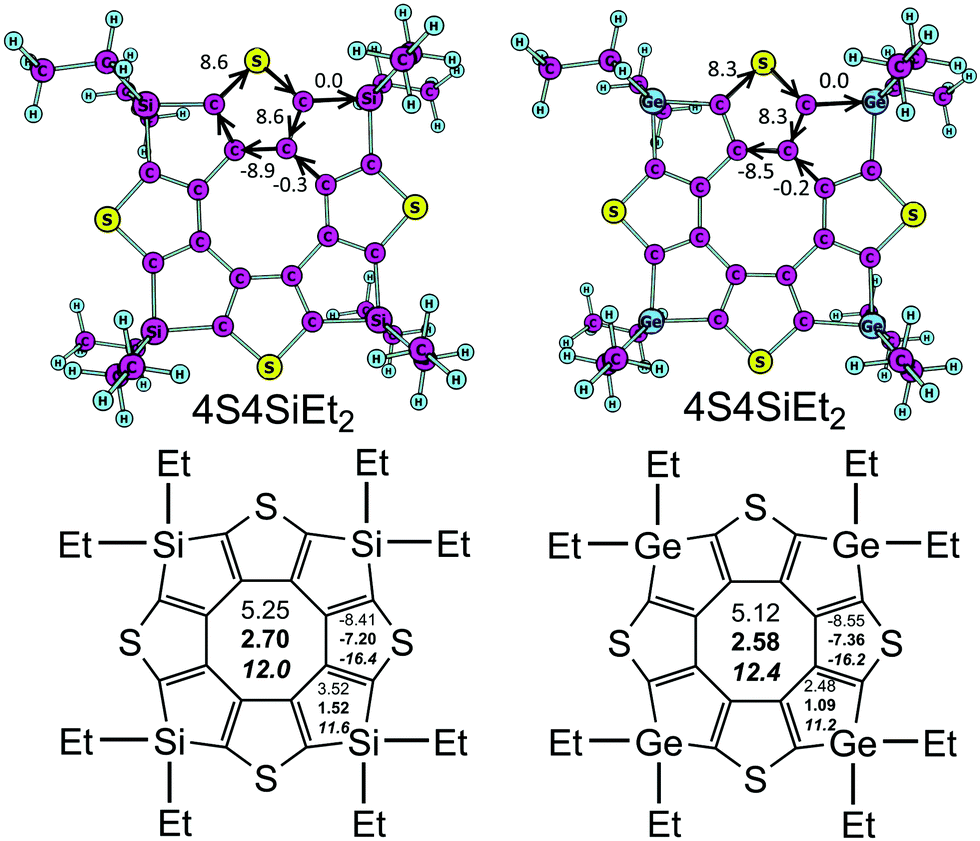 | ||
| Fig. 2 MIC strengths (top, numbers in nA T−1) and NICS(0), NICS(1) (in bold), and NICS(1)zz (italic) indices (bottom part, numbers in ppm) for the 4S4SiEt2 and 4S4GeEt2 molecules. | ||
At the same time each thiophene ring possesses its own strong local diatropic current (Idia = 8.6 and 8.3 nA T−1 for the 4S4SiEt2 and 4S4GeEt2 molecules, respectively) in contrast to the totally-conjugated circulenes like tetraoxa[8]circulene, octathia[8]circulene, etc.50 It is here rather surprising that the local diatropic MIC strength (Idia) is almost the same by absolute value as the paratropic MIC inside the planar eight-membered core (Ipara = −9.2 and −8.7 nA T−1 for the 4S4SiEt2 and 4S4GeEt2 molecules, respectively). This fact supports the same overall non-aromatic nature of the 4S4SiEt2 and 4S4GeEt2 molecules (Itot = −0.6 and −0.4 nA T−1, respectively) as for most of the hetero[8]circulenes.50 However, the topology of the MIC is principally different. Generally, the totally-conjugated hetero[8]circulenes do not sustain significant MIC along the radial C–C bonds, i.e. the diatropic and paratropic currents circulate in the rim and hub perimeters in opposite directions almost independently of each other (the rim and hub subsystems are separated by the C–C radial bonds). In the case of 4S4SiEt2 and 4S4GeEt2 circulenes the rim-system is broken by the sp3-Si(Ge) atoms, which induces MIC circulation along the radial C–C bonds (Fig. 3).
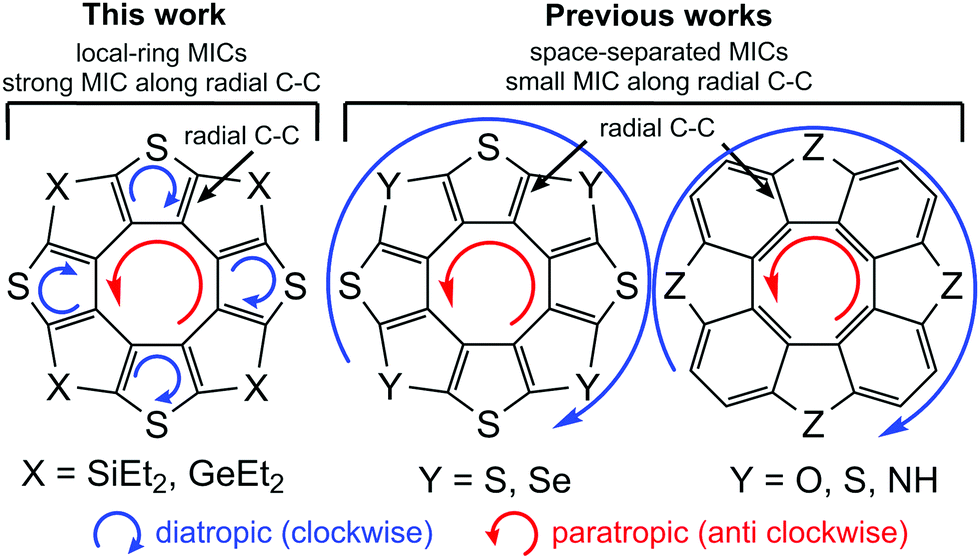 | ||
| Fig. 3 Principal difference between the ring-current topology in the totally conjugated heterocirculenes and the studied 4S4SiEt2 and 4S4GeEt2 compounds. | ||
The GIMIC results for the 4S4SiEt2 and 4S4GeEt2 molecules are in good qualitative agreement with the NICS calculations (Fig. 2). The inner cyclooctatetraene core sustains significantly positive NICS(0), NICS(1) and NICS(1)zz values for both studied molecules meaning an antiaromatic nature of this ring. At the same time, the NICS(0), NICS(1) and NICS(1)zz values for the thiophene rings are significantly negative (i.e. these rings are locally aromatic), but for the Si/Ge-containing rings the NICS(0) and NICS(1) descriptors are weakly positive (i.e. these rings are just weakly antiaromatic). Despite the fact that NICS(1)zz is significantly positive for the Si/Ge-containing rings, the two other xx and yy components of the magnetic shielding tensor are significantly negative (about −4 ppm) and compensate the zz one. Actually, Si/Ge-containing rings do not sustain significant π-electronic MICs and that is why NICS(1)zz indices are not really a good local aromaticity criterion for these rings. The overall balance between the NICS indices looks the same as obtained by the GIMIC method: local paratropic currents inside the cyclooctatetraene core are compensated by the local diatropic currents inside the thiophene rings, while Si/Ge-containing rings just weakly contribute with an antiaromatic component. Actually, the NICS data emphasize the local-ring topology of the MICs in the 4S4SiEt2 and 4S4GeEt2 molecules obtained by GIMIC and this model is principally different from the (C–C-radial)-separated topology of concentric MICs in most hetero[8]circulenes.28,30,50 An additional observation is that the outer ethyl substituents are not important in the GIMIC and NICS calculations; the deviations in Itot for model 4S4SiH2 and 4S4GeH2 are less than 0.1 nA T−1 and the differences in the NICS values are of the order of 10−2 ppm. This fact is expected because of the negligible effect of σ-type ethyl groups on the π-system of tetrathienylene.
3.2. Vibronic absorption and fluorescence spectra
4S4SiEt2 and 4S4GeEt2 circulenes are characterized by similar absorption and fluorescence spectra with clearly resolved vibronic structure.22,23 In order to explain these spectra and assign the vibronic bands we have initially optimized both the 4S4SiEt2 and 4S4GeEt2 circulenes using the TDDFT/B3LYP/6-31G(d,p) method in the first excited singlet state (S1), which is responsible for the fluorescence emission. The results are summarized in Table 1. By orbital symmetry type, the S1 state of 4S4SiEt2 and 4S4GeEt2 can be attributed to the ππ* nature corresponding to the HOMO–LUMO configuration (Fig. S1, ESI†).| State | 4S4SiEt2 | 4S4GeEt2 |
|---|---|---|
| S0 geometry | ||
| S1 | 3.302 (0.003); exp.: 3.23 | 3.485 (0.003); exp.: 3.40 |
| S2 | 3.34 (0.0001) | 3.52 (0.0001) |
| S1 geometry | ||
| S1 | 2.89 (0.001); exp.: 3.23 | 3.22; exp.: 3.40 |
The S1 state is structurally closely similar to the ground state – the main differences relate to an elongation of the radial C–C and C–S bonds, while the remaining skeletal bonds (C–C bonds in the inner COT core and Si/Ge–C bonds) become slightly shorter upon excitation into the S1 state (Table S1, ESI†). Such structural behavior is principally different from that for the totally-conjugated tetraoxa[8]circulene and mixed azaoxa[8]circulenes, which change bond alternation in the inner COT core upon excitation into the S1 state.51
Based on the calculated Hessians for the ground (S0) state and gradients of the excited (S1) states of real Et-substituted and model H-substituted compounds the absorption (S0 → S1) and fluorescence (S1 → S0) vibronic spectra were calculated within the Franck–Condon approximation using the Lax–Heller model.38,39 This model shows a good result for the cases of slightly-shifted potential energy surfaces (PES) of the S0 and S1 states. Indeed, the energy difference between the vertical and 0–0 electronic transitions is only 0.04 eV and 0.05 eV for the 4S4SiEt2 and 4S4GeEt2 molecules, respectively, something that allows us to use the simple Lax–Heller model.38,39
At the first stage, we have defined the vibronic activity of the outer ethyl groups. We have found that these groups do not significantly contribute to the main promotive skeletal vibrational modes both in absorption and fluorescence vibronic spectra and only some new low-frequency promotive modes appear in the spectra of the model 4S4SiH2 and 4S4GeH2 molecules. All of these vibrational modes are characterized by very small Huang–Rhys factors and actually are overlapped by the 0–0 transition. The calculated vs. experimental vibronic absorption and fluorescence spectra are presented in Fig. 4 and 5, respectively, while the shapes of the promotive modes are shown in the ESI† (Fig. S2 and S3 for the real 4S4SiEt2 and 4S4GeEt2 molecules and for the model H-substituted 4S4SiH2 and 4S4GeH2 species). As can be seen from Fig. 4, the 0–0 electronic transition takes place at 380 and 370 nm for the 4S4SiEt2 and 4S4GeEt2 molecules and it produces the first most intense band in the absorption spectra of both compounds (exp.: 384 and 365 nm, respectively). Just three vibrational modes promote the vibronic progression in the absorption spectra of the 4S4SiEt2 (4S4GeEt2) molecule in the region 320–360 nm (Table 2). The modes at 794 (763) and 1329 (1320) cm−1 correspond to the “breathing” symmetrical stretching vibrations of the inner COT core, while the mode 1329 (1320) cm−1 is additionally accompanied by an in-phase stretching of C–C radial bonds. These specific “breathing” vibrational modes are also active in the vibronic absorption spectra of other hetero[8]circulenes51 indicating that the rigid planar COT skeleton is mainly responsible for the experimentally observed vibronic structure in electronic spectra of hetero[8]circulenes as a unique class of heteroannelated cyclooctatetraenes. The third active mode at 1139 (1117) cm−1 corresponds to the in-plane out-of-phase bending vibrations of the radial C–C bonds but it is much less intense in the vibronic absorption spectrum compared with the COT “breathing” vibrations (Table 2).
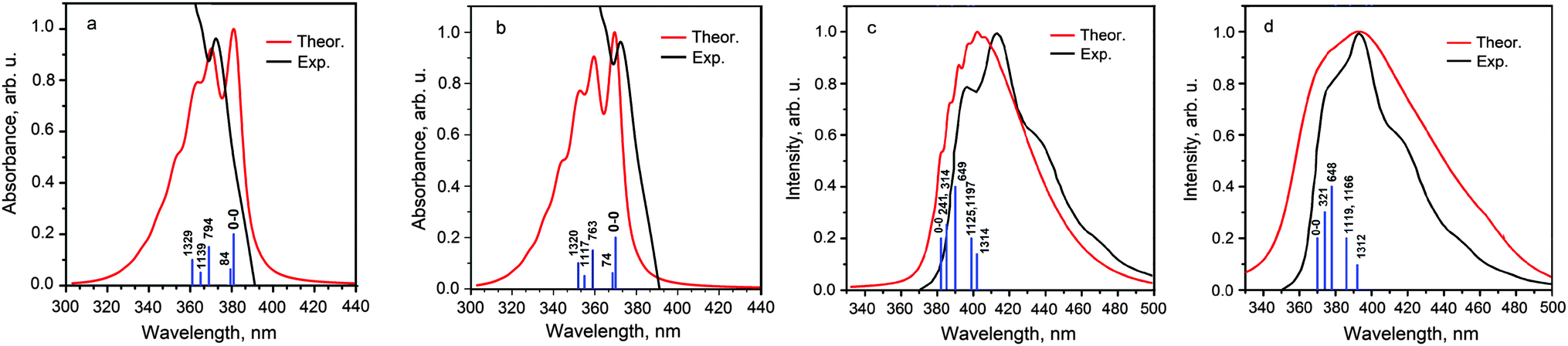 | ||
| Fig. 4 Theoretically calculated vs. experimental vibronic absorption (a and b) and fluorescence (c and d) spectra of the 4S4SiEt2 (a and c) and 4S4GeEt2 (b and d) molecules. Vertical sticks correspond to the energy of promotive vibrational modes (cm−1) relative to the 0–0 transition. Experimental curves are taken from ref. 22 and 23. | ||
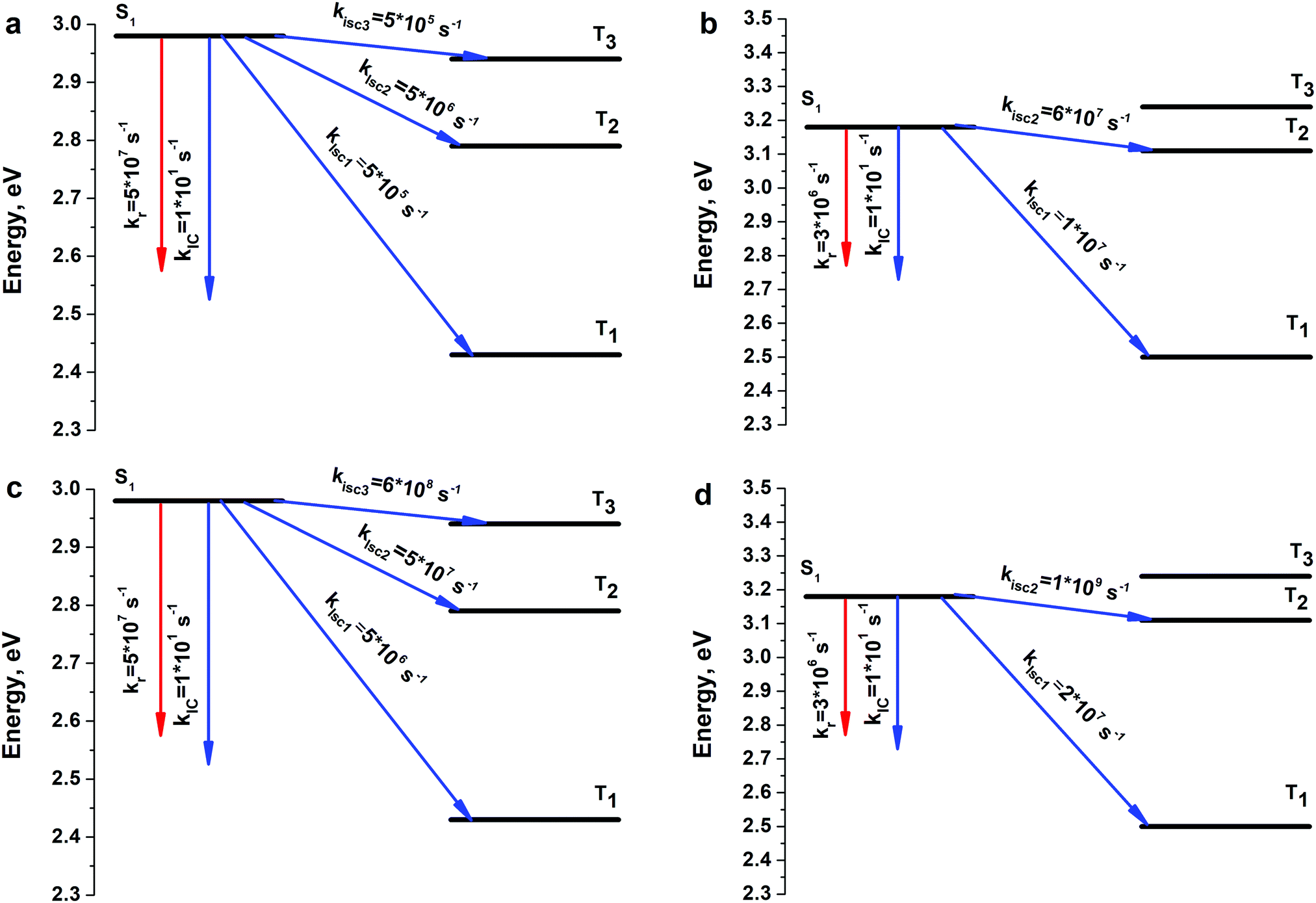 | ||
| Fig. 5 Calculated rate constants for the main photophysical processes of S1 state deactivation for the 4S4SiH2 (a and c) and 4S4GeH2 (b and d) molecules without (a and b) and with (c and d) accounting for spin–vibronic interactions. | ||
| Absorption (S0–S1) | Fluorescence (S1–S0) | |
|---|---|---|
| 4S4SiEt2 | 84(0.1); 794(0.5); 1139(0.06); 1329(0.31) | 241(0.3); 314(0.4); 369(0.4); 397(0.2); 649(0.6); 1125(0.1); 1197(0.20); 1314(0.2) |
| 4S4GeEt2 | 74(0.1); 763(0.65); 1117(0.08); 1320(0.31) | 223(0.36); 321(0.49); 343(0.43); 362(0.2); 648(0.75); 1119(0.14); 1166(0.30); 1312(0.2) |
| 4S4SiH2 | 784(0.75); 1152(0.07); 1326(0.29) | 250(0.36); 323(0.49); 373(0.43); 397(0.2); 666(0.75); 1137(0.14); 1193(0.30); 1312(0.2) |
| 4S4GeH2 | 754(0.65); 1138(0.1); 1317(0.29) | 180(0.43); 297(0.46); 321(0.25); 341(0.2); 628(0.33); 1127(0.18); 1185(0.30); 1331(0.2) |
In the vibronic fluorescence spectra the 0–0 transition is less intense than the 0–1 band promoted by the low-frequency mode at 666 (626) cm−1 for the 4S4SiEt2 (4S4GeEt2) molecule. Actually, both these 0–0 and 0–1 transitions form the first experimentally-observed band at 397 and 377 nm in the fluorescence spectra of the 4S4SiEt2 and 4S4GeEt2 compounds, respectively. The next most intense band in the experimental spectrum (412 and 392 nm, respectively) corresponds to the manifold of active modes in the region of 1100–1300 cm−1 combined with a double excitation of the low-frequency mode 666 (628) cm−1 (Table 2). All the low-frequency vibrational modes involve the bending vibrations of the CH bonds of the ethyl substituents (Fig. S2 and S4), but for the model 4S4SiH2 and 4S4GeH2 compounds these modes transform into the Si(Ge)–H bending vibrations of different symmetry. The active modes in the region 1100–1300 cm−1 mainly correspond to the asymmetrical stretching and bending vibrations of the radial C–C bonds (Fig. S3, ESI†).
One can stress that the ethyl groups play a more important role in vibronic fluorescence spectra compared with the absorption spectra because of the high vibronic activity of low-frequency bending vibrations of the CH groups in the ethyl moieties. But even the H-substituted models 4S4SiH2 and 4S4GeH2 adequately describe the experimentally observed fluorescence spectra because the Ge(Si)–H bending vibrations demonstrate a similar vibronic activity (Table 2).
3.3. Photophysical properties
In ref. 22 and 23 the main photophysical constants like the internal-conversion rate (kIC), intersystem crossing rate (kISC), fluorescence rate (kr) and fluorescence quantum yield (φfl) were experimentally estimated for both 4S4SiEt2 and 4S4GeEt2 circulenes. Actually, it was the first time a detailed experimental estimation was given for the photophysical properties of the hetero[8]circulenes. In previous sections we have stated that the 4S4SiH2 and 4S4GeH2 models adequately describe the electronic and spectroscopic parameters of the real 4S4SiEt2 and 4S4GeEt2 systems. Thus, in this section we are focusing on the photophysical properties of the model 4S4SiH2 and 4S4GeH2 compounds in comparison with experimentally measured constants.22,23 One can see from Fig. 5 that three (T1, T2 and T3) and two (T1 and T2) triplet excited states lie below the S1 level for the 4S4SiH2 and 4S4GeH2 molecules, respectively (the same pattern was also computed for the Et-substituted systems). All these triplet states participate in non-radiative quenching of the S1 state through multichannel ISC (kISC1, kISC2 and kISC3). Calculations of the corresponding spin–orbit coupling (SOC) matrix elements show that the S1–T1 radiationless transition is the main deactivation channel of the S1 state energy without accounting for spin–vibronic interactions (Table 3). One can also observe a clear internal heavy atom effect52 moving from the Si- to the Ge-containing circulene (〈ϕ(S1)|HSO|ϕ(Tn)〉 (4S4GeH2) > 〈ϕ(S1)|HSO|ϕ(Tn)〉 (4S4SiH2)). However, the total ISC rate (∑kISCi) estimated as a sum of partial kISC1,2,3 rates was found to be considerably underestimated compared with the experimental data (6.0 × 106 s−1vs. exp.: 4.6 × 108 for 4S4SiEt2 and 7.0 × 107 s−1vs. exp.: 1.3 × 108 s−1 for 4S4GeEt2, Table 3). In order to clarify the ∑kISCi rate, spin–vibronic interactions were taken into account through the distortion of geometries along the out-of-plane Eg normal modes (Fig. 6). As follows from the symmetry selection rules for the SOC operator,52–54 only the out-of-plane Eg vibrations affect the corresponding SOC matrix elements. As a result, all the 〈ϕ(S1)|HSO|ϕ(Tn)〉 matrix elements have been increased (especially 〈ϕ(S1)|HSO|ϕ(T3)〉), which produces an enhancement of ∑kISCi by one–two orders of magnitude, providing more reliable agreement with experiment (Table 3). Finally, taking account of spin–vibronic interactions provides excellent agreement with respect to the fluorescence quantum yield (0.07 vs. exp.: 0.012 for 4S4SiEt2, 0.003 vs. exp.: 0.0076 for 4S4GeEt2), which means a prevailing role of ISC quenching over the IC process.| Parameter | Without spin–vibronic interactions | With spin–vibronic interactions | ||
|---|---|---|---|---|
| 4S4SiH2 | 4S4GeH2 | 4S4SiH2 | 4S4GeH2 | |
| S1, eV (f) | 3.04(0.14) | 3.18(0.007) | — | — |
| S1 (exp.), eV | 3.23 | 3.40 | — | — |
| T1, eV | 2.43 | 2.5 | — | — |
| T2, eV | 2.79 | 3.11 | — | — |
| T3, eV | 2.94 | 3.24 | — | — |
| 〈ϕ(S1)|HSO|ϕ(T1)〉, cm−1 | 0.30 | 2.27 | 0.89 | 3.35 |
| 〈ϕ(S1)|HSO|ϕ(T2)〉, cm−1 | 0.08 | 0.58 | 0.21 | 0.13 |
| 〈ϕ(S1)|HSO|ϕ(T3)〉, cm−1 | 0.02 | 0.12 | 0.43 | 0.8 |
| k ISC1, s−1 | 5 × 105 | 1 × 107 s−1 | 5 × 106 | 2 × 107 |
| k ISC2, s−1 | 5 × 106 | 6 × 107 s−1 | 5 × 107 | 1 × 109 |
| k ISC3, s−1 | 5 × 105 | — | 6 × 108 | — |
| ∑kISCi, s−1 | 6 × 106 | 7 × 107 | 7 × 108 | 1 × 109 |
| ∑kISCi (exp.), s−1 | 4.6 × 108 | 1.3 × 108 | — | — |
| k IC, s−1 | 1 × 101 | 1 × 101 | 1 × 101 | 1 × 101 |
| k IC (exp.), s−1 | 6.3 × 108 | 2.7 × 108 | — | — |
| k r, s−1 | 5 × 107 | 3 × 106 | 5 × 107 | 3 × 106 |
| k r (exp.), s−1 | 1.3 × 107 | 3 × 106 | — | — |
| φ fl | 0.90 | 0.02 | 0.07 | 0.003 |
| φ fl (exp.) | 0.012 | 0.0076 | — | — |
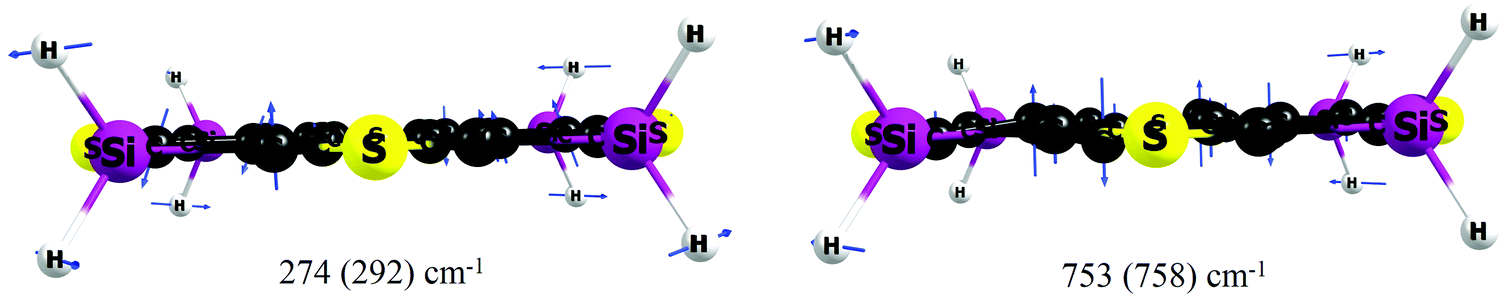 | ||
| Fig. 6 Scaled displacement vectors for the two out-of-plane Eg modes responsible for the enhancement of the SOC matrix elements upon geometry distortion of the 4S4SiH2 (4S4GeH2) molecule. Both modes are quite similar and their effects on the SOC matrix elements are almost identical (deviations are less than 0.02 cm−1). | ||
At the same time our calculations are considerably different (by seven orders of magnitude) from experimental data with respect to the internal conversion rates (kIC ∼ 101 s−1).22,23 Experimental estimations provide a 108 s−1 order of magnitude for the internal conversion process, which means that this channel is quite active in the S1 state deactivation. In our opinion, ref. 22 and 23 give an incorrect estimation of the kISC rate by measuring the decay of the T–T absorption during energy transfer from the 4S4SiH2 donor to the β-carotene acceptor.55,56 This method only means that the T1 state lies lower than the S1 state for the studied molecules and that only the T1 state of circulene is able to transfer energy to the T1 state of β-carotene. However, the ISC processes into the T2 and T3 states also quench the S1 state energy by a commensurate rate with the S1–T1 process. Most likely, both kISC2 and kISC3 rate constants of the order 107–108 s−1 were erroneously included in the IC channel by subtracting φfl and φISC (S1–T1) from unity. Indeed, the calculated total rate constant for the non-radiative quenching (kIC + ∑kISCi) is comparable with the experimental data for both compounds, while the ratio between the IC and ISC rates is strongly different compared with our calculations (theor.: ∑kISCi ≫ kIC, but exp.: ∑kISCi ≈ kIC). One more argument in favor of our interpretation that the experimental kISC rate is underestimated in ref. 22 and 23 is that the opposite internal heavy atom effect was obtained22,23 when comparing kISC values for 4S4SiEt2 (4.6 × 108 s−1) and 4S4GeEt2 (1.3 × 108 s−1). This contradicts the fundamental mass-dependent property of SOC effects (SOC increases with the rise of the atomic number of an element)52 and also the experimentally estimated radiative phosphorescence lifetimes (τphos(4S4SiEt2) = 235 ms > τphos(4S4GeEt2) = 98 ms), which correctly reproduce the internal heavy atom effect. We should stress here that the total phosphorescence lifetimes (τphos.tot.(4S4SiEt2) = 33 s, τphos.tot.(4S4GeEt2) = 1.2 s), estimated taking into account phosphorescence quantum yields (i.e. accounting for non-radiative quenching), are in excellent agreement with our calculations by the second-order approximate coupled-cluster (CC2) method57 (31 s and 2 s, respectively), indicating a high-accuracy of the theoretical calculations of the SOC effects. A more detailed computational study of the phosphorescence of 4S4SiEt2 and 4S4GeEt2 can be found in ref. 58.
Finally, we want to stress that despite the quantitative mismatch between the calculated and experimental kISC and kIC rates, the general conclusion obtained from theory and experiment is the same: ISC is the main deactivation channel of the S1 state energy compared with the fluorescence and internal conversion processes. The main reason lies in the quite large SOC matrix elements between the S1 and Tn (n = 1–3) states despite the similar ππ* nature of both the singlet and triplet counterparts. For common organic molecules the SOC matrix elements between S1 and Tn states are of the order of 0.1 cm−1.52,59 It means that 4S4SiEt2 and 4S4GeEt2 molecules possess quite specific photophysical behavior due to their unique circulenoid structure and due to the presence of heavy Si and Ge atoms in the structure of these molecules.
4. Conclusions
In the present work we have comprehensively studied the electronic structure, spectra, aromaticity and photophysical properties of two recently synthesized derivatives of tetrathienylene named tetrasilatetrathia[8]circulene (4S4SiEt2) and tetragermatetrathia[8]circulene (4S4GeEt2). Both studied molecules possess double aromaticity characterized by a local magnetically-induced current in contrast to the space-separated global current model for most hetero[8]circulenes. Generally, both 4S4SiEt2 and 4S4GeEt2 circulenes can be considered as globally non-aromatic species in terms of the magnetically-induced current criterion; the local diatropic currents inside the four thiophene rings are completely canceled by the paratropic currents inside the inner eight-membered core producing a close to zero net current strength typical for non-aromatic species. Both the 4S4SiEt2 and 4S4GeEt2 species demonstrate clearly visible vibronic structure in the absorption and emission spectra due to the activity of the stretching and bending C–C vibrations of the tetrathienylene skeleton. At the same time, the low-frequency CH bending vibrations play a crucial role for the fluorescence vibronic spectra. Accounting for spin–vibronic interactions through geometry distortion along the several out-of-plane Eg modes provides reliable agreement between the calculated and experimental photophysical constants. At the same time, neglecting the spin–vibronic interactions gives an underestimation of the spin–orbit coupling matrix element between the S1 and T1 states, which subsequently leads to a strong overestimation of the fluorescence rate and quantum yield. Moreover, the fact that several triplet excited states lie lower in energy than the S1 state plays a crucial role in the balance between the internal conversion and inter-system crossing rates. Accounting for this assumption we have concluded that the inter-system crossing channel constitutes a main deactivation process responsible for the deactivation of the S1 state energy. Actually, this paper contains for the first time a detailed photophysical investigation of representatives of the hetero[8]circulene family and provides a close comparison with experimental data. We hope that the current study will help to generalize and interconnect the aromaticity and spectral theory of hetero[8]circulenes as a unique class of totally-annelated cyclooctatetraenes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Ministry of Education and Science of Ukraine (projects no. 0117U003908 and 0118U003862), and by the Olle Engkvist Byggmästare foundation (contract no. 189-0223). The calculations were performed with computational resources provided by the High Performance Computing Center North (HPC2N) in Umeå, Sweden, through the project “Multiphysics Modeling of Molecular Materials” SNIC 2018-2-38. Also, this work has been supported by the Svenska Kulturfonden (grant number 136102) and Academy of Finland (grant number 1315600). R. V. is personally thankful to the Tomsk Polytechnic University Competitiveness Enhancement Program (VIU-RSCABS-142/2019). V. N. C. and H. Å. thank the Tomsk State University Competitiveness Improvement Program.References
- G. V. Baryshnikov, B. F. Minaev and V. A. Minaeva, Russ. Chem. Rev., 2015, 84, 455 CrossRef CAS.
- T. Hensel, N. N. Andersen, M. Plesner and M. Pittelkow, Synlett, 2016, 498 CAS.
- N. N. Karaush, G. V. Baryshnikov, V. A. Minaeva, H. Ågren and B. F. Minaev, Mol. Phys., 2017, 115, 2218 CrossRef CAS.
- C. B. Nielsen, T. Brock-Nannestad, T. K. Reenberg, P. Hammershoj, J. B. Christensen, J. W. Stouwdam and M. Pittelkow, Chem. – Eur. J., 2010, 16, 13030 CrossRef CAS PubMed.
- G. V. Baryshnikov, R. R. Valiev, N. N. Karaush, V. A. Minaeva, A. N. Sinelnikov, S. K. Pedersen, M. Pittelkow, B. F. Minaev and H. Ågren, Phys. Chem. Chem. Phys., 2016, 18, 28040 RSC.
- K. B. Ivaniuk, G. V. Baryshnikov, P. Y. Stakhira, S. K. Pedersen, M. Pittelkow, A. Lazauskas, D. Volyniuk, J. V. Grazulevicius, B. F. Minaev and H. Ågren, J. Mater. Chem. C, 2017, 5, 4123 RSC.
- A. V. Kuklin, G. V. Baryshnikov, B. F. Minaev, N. Ignatova and H. Ågren, J. Phys. Chem. C, 2018, 122, 22216 CrossRef CAS.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, RSC Adv., 2014, 4, 25843 RSC.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, Phys. Chem. Chem. Phys., 2014, 16, 6555 RSC.
- J. Yu, Q. Sun and P. Jena, J. Phys. Chem. C, 2016, 120, 27829 CrossRef CAS.
- J. Yu, Q. Sun, Y. Kawazoe and P. Jena, Nanoscale, 2014, 6, 14962 RSC.
- B. Minaev, N. Karaush-Karmazin, G. Baryshnikov and V. Minaeva, Electronic Structure and spectral properties of hetero[8]circulenes, Publishing dept. Chabanenko Yu. A., Cherkasy, Ukraine, 2018 Search PubMed.
- K. Yu. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367 CrossRef CAS PubMed.
- K. Yu. Chernichenko, E. S. Balenkova and V. G. Nenajdenko, Mendeleev Commun., 2008, 18, 171 CrossRef.
- A. Dadvand, F. Cicoira, K. Yu. Chernichenko, E. S. Balenkova, R. M. Osuna, F. Rosei, V. G. Nenajdenko and D. F. Perepichka, Chem. Commun., 2008, 5354 RSC.
- N. N. Karaush, B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Opt. Spectrosc., 2014, 116, 33 CrossRef CAS.
- G. V. Baryshnikov, B. F. Minaev, V. A. Minaeva and V. G. Nenajdenko, J. Mol. Model., 2013, 19, 4511 CrossRef CAS PubMed.
- V. Minaeva, N. Karaush-Karmazin, G. Baryshnikov and B. Minaev, Vib. Spectrosc., 2019, 100, 107 CrossRef CAS.
- S. S. Bukalov, L. A. Leites, K. A. Lyssenko, R. R. Aysin, A. A. Korlyukov, J. V. Zubavichus, K. Yu. Chernichenko, E. S. Balenkova, V. G. Nenajdenko and M. Yu. Antipin, J. Phys. Chem. A, 2008, 112, 10949 CrossRef CAS PubMed.
- O. Ivasenko, J. M. MacLeod, K. Yu. Chernichenko, E. S. Balenkova, R. V. Shpanchenko, V. G. Nenajdenko, F. Rosei and D. F. Perepichka, Chem. Commun., 2009, 1192 RSC.
- W. D. Xiao, Y. Y. Zhang, L. Tao, K. Aït-Mansour, K. Y. Chernichenko, V. G. Nenajdenko, P. Ruffieux, S. X. Du, H.-J. Gao and R. Fasel, Sci. Rep., 2014, 4, 5415 CrossRef CAS PubMed.
- Y. Serizawa, S. Akahori, S. Kato, H. Sakai, T. Hasobe, Y. Miyake and H. Shinokubo, Chem. – Eur. J., 2017, 23, 6948 CrossRef CAS PubMed.
- S. Akahori, H. Sakai, T. Hasobe, H. Shinokubo and Y. Miyake, Org. Lett., 2018, 20, 304 CrossRef CAS PubMed.
- S. Kato, Y. Serizawa, D. Sakamaki, S. Seki, Y. Miyake and H. Shinokubo, Chem. Commun., 2015, 51, 16944 RSC.
- X. Xiong, C.-L. Deng, B. F. Minaev, G. V. Baryshnikov, X.-S. Peng and H. N. C. Wong, Chem. – Asian J., 2015, 10, 969 CrossRef CAS PubMed.
- V. A. Minaeva, G. V. Baryshnikov, B. F. Minaev, N. N. Karaush, X.-D. Xiong, M.-D. Li, D. L. Phillips and H. N. C. Wong, Spectrochim. Acta, Part A, 2015, 151, 247 CrossRef CAS PubMed.
- V. A. Minaeva, G. V. Baryshnikov and B. F. Minaev, Opt. Spectrosc., 2015, 119, 620 CrossRef CAS.
- G. V. Baryshnikov, N. N. Karaush, R. R. Valiev and B. F. Minaev, J. Mol. Model., 2015, 21, 136 CrossRef PubMed.
- G. V. Baryshnikov, R. R. Valiev, N. N. Karaush, D. Sundholm and B. F. Minaev, Phys. Chem. Chem. Phys., 2016, 18, 8980 RSC.
- (a) H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500 RSC; (b) J. Jusélius and D. Sundholm, J. Chem. Phys., 2004, 121, 3952 CrossRef PubMed; (c) D. Sundholm, H. Fliegl and R. J. F. Berger, WIREs Comput. Mol. Sci., 2016, 6, 639 CrossRef CAS.
- Gauge-Including Magnetically Induced Currents (GIMIC) program is distributed on GitHub platform and can be downloaded through the link: https://github.com/qmcurrents/gimic.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople and M. S. Gordon, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. J. Heller, Acc. Chem. Res., 1981, 14, 368 CrossRef CAS.
- M. Lax, J. Chem. Phys., 1952, 20, 1752 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- TURBOMOLE V7.2 2017, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- P. v. R. Schleyer, C. Maerker, H. C. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS.
- A. A. Granovsky, J. Chem. Phys., 2011, 134, 214113 CrossRef PubMed.
- A. A. Granovsky, Firefly v. 8, http://classic.chem.msu.su/gran/firefly/index.html.
- M. S. Gordon and M. W. Schmidt, Advances in electronic structure theory: GAMESS a decade later, in Theory and Applications of Computational Chemistry: the first forty years, ed. C. E. Dykstra, G. Frenking, K. S. Kim and G. E. Scuseria, Elsevier, Amsterdam, 2005 Search PubMed.
- R. R. Valiev, V. N. Cherepanov, G. V. Baryshnikov and D. Sundholm, Phys. Chem. Chem. Phys., 2018, 20, 6121 RSC.
- G. V. Baryshnikov, R. R. Valiev, N. N. Karaush and B. F. Minaev, Phys. Chem. Chem. Phys., 2014, 16, 15367 RSC.
- N. N. Karaush, R. R. Valiev, G. V. Baryshnikov, B. F. Minaev and H. Ågren, Chem. Phys., 2015, 459, 65 CrossRef CAS.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500 CrossRef CAS PubMed.
- B. Minaev, Y.-H. Wang, C.-K. Wang, Y. Luo and H. Ågren, Spectrochim. Acta, Part A, 2006, 65, 308 CrossRef PubMed.
- B. Minaev, H. Ågren and O. Vahtras, Chem. Phys., 1993, 175, 245 CrossRef CAS.
- R. Bensasson, E. A. Dawe, D. A. Long and E. J. Land, J. Chem. Soc., Faraday Trans. 1, 1977, 1319 RSC.
- I. Carmichael and G. L. Hug, J. Phys. Chem. Ref. Data, 1986, 15, 1 CrossRef.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409 CrossRef CAS.
- R. R. Valiev, G. V. Baryshnikov, V. N. Cherepanov and D. Sundholm, Russ. Phys. J., 2019 Search PubMed , in press. The full text is available by e-mail to the corresponding author, glibar@kth.se.
- O. Loboda, I. Tunell, B. Minaev and H. Ågren, Chem. Phys., 2005, 312, 299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp01608b |
| This journal is © the Owner Societies 2019 |