
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon dioxide and water co-adsorption on the low-index surfaces of TiC, VC, ZrC and NbC: a DFT study†
Matthew G.
Quesne
 *,
Alberto
Roldan
,
Nora H.
de Leeuw
and
C. Richard A.
Catlow
*,
Alberto
Roldan
,
Nora H.
de Leeuw
and
C. Richard A.
Catlow
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: quesnem@cardiff.ac.uk
First published on 14th May 2019
Abstract
We present a theoretical DFT study into the activation of CO2 by TiC, VC, ZrC and NbC. Particular focus is given to the study of CO2/H2O co-adsorption and interaction on four carbide low-index surfaces: {001}, {011}, carbon-terminated {111} and metal-terminated {111}. The adsorption and activation of CO2 is shown to be most exothermic and indeed barrierless on the metal-terminated {111} surfaces, whilst adsorption on the {001} and {011} planes occurs via a small activation energy barrier. In contrast, the carbon-terminated {111} surface proves to be unstable in the presence of the adsorbates. Both water and carbon dioxide adsorb most strongly on TiC and most weakly on NbC, with the strongest co-adsorption interactions being seen in conformations that maximise hydrogen-bonding.
Introduction
As atmospheric carbon dioxide (CO2) concentrations continue to rise, policy makers and the general public are becoming increasingly aware of its unfolding negative consequences for climate change and ocean acidification.1,2 To slow the rate of this increase and realise the ambitious targets set out in the Paris Agreement,3 three main options appear viable: (i) reducing emissions through higher energy efficiency,4 (ii) developing sustainable resources in order to reduce reliance on fossil fuels5 and (iii) chemically trapping waste CO2 thereby reducing the volume of atmospheric emissions via an integrated carbon capture and storage (CCS) protocol.6 Carbon capture, utilisation and storage has become a major area of research7–9 with progress being made using both heterogeneous10–12 and homogeneous13–15 catalysts. However, the high stability of CO2 means that, for many catalysts, there is a high barrier for chemical interaction. This problem, combined with the need for a high enough adsorption energy for the material to retain the adsorbate, limits the number potential catalysts. Candidate materials must efficiently bind CO2 in the presence of many other compounds dispersed in, for example flue gas.16,17 Water in the form of steam is also of particular importance.18 There have been several previous theoretical studies that have confirmed the important role of H2O in both reducing the barrier and directing the pathways for CO2 reduction to methanol on solid state materials.19,20 Fortunately, carbon dioxide possesses a considerable quadrupole moment, which in the presence of the right catalyst can result in a relatively small barrier for chemical adsorption,21 due to the strong interaction between the quadrupole moment of carbon dioxide and specific binding sites on the catalyst. Thus, an increased strength of charge–charge, hydrogen-bonding and many other interactions decrease the relative activation barriers by increasing the exothermicity of chemisorbed species, when compared to non-quadrupolar adsorbates.22,23 Additionally, since this chemical adsorbed state requires electron transfer from the catalyst to produce an activated anionic CO2δ− species,24,25 there is potential for the development of chemical processes that further reduce CO2 by sustainably produced hydrogen.26–29 The search for the ideal catalyst to perform this reaction has been frustrated by low activity,30 or because of the need for expensive and scarce metals.31,32 However, it has become clear in the years following the seminal work of Levy and Boudart in 197333 that transition metal carbides (TMCs) have platinum-like activity and can catalyse a diverse array of reactions, including hydrogenation,34 water–gas shift,35,36 CO oxidation37,38 and importantly the hydrogenation of CO2.39–42 Recently, combined experimental and computational studies have confirmed that TMCs catalyse CO2 reduction to CO, as the first step in hydrogenation.43,44 Our previous work has used periodic Density Functional Theory (DFT) calculations to study a large range of bulk and surface properties of TMCs, which indicated that the low-index surfaces of TiC, VC, ZrC and NbC could be highly active in catalysing CO2 reduction.45 This work focuses on carbides with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometries and rock-salt structures, which are only consistently found with carbides that include metal from group 3–5 metals.46,47 Therefore, in this paper we present a detailed computational study of CO2 and H2O binding and activation on these surfaces.
:1 stoichiometries and rock-salt structures, which are only consistently found with carbides that include metal from group 3–5 metals.46,47 Therefore, in this paper we present a detailed computational study of CO2 and H2O binding and activation on these surfaces.
Methodology
All calculations were undertaken with the standard Perdew–Burke–Ernzerhof functional,48 as implemented in the Vienna Ab initio Simulation (VASP) package.49–51 This method has been benchmarked previously for the systems reported here, using periodic density functional calculation.45 The Perdew–Burke–Ernzerhof48 exchange functional (PBE) in combination with plane-wave basis sets were applied to the valence electrons, whilst the core electrons were described via the projected augmented wave method (PAW).52 Long range interactions were included using the D3 Grimme dispersion method.53,54 Bulk structures for TiC,55 VC,55 ZrC,56 and NbC57 were taken from the Inorganic Structure Database (ICSD).58 The lattice parameters and the internal coordinates of these bulk structures were then fully optimized and cut along the {001}, {011} and {111} planes using the METADISE code.59 Since there are alternating layers of metal and carbon along the {111} direction of TMCs,60–62 two surfaces were prepared that describe either the metal or carbon termination. Our previous work45 benchmarked these pristine surfaces with those that were reconstructed by migration of surface atoms and confirmed that the unreconstructed models were valid, which is also in agreement with experiment.63–65 Finally, each unit cell was replicated 2 × 2 × 3 to give slabs with six atomic layers and 96 atoms.45 The low-index surfaces were modelled using Monkhorst–Pack grids of k-points of 5 × 5 × 1. All energies were converged to within a cut-off of 520 eV and the threshold for ionic relaxation was set to 0.01 eV Å−1. The electronic threshold for the self-consistency cycles (SCF) was set at 10−5 eV and the convergence was determined via the Blöchl smearing method.66 All structures were fully optimised, with only the bottom two atomic layers of each slab constrained at their bulk-optimised positions to replicate the bulk. No other internal coordinate or symmetry constraints were added and spin polarization was enabled in all systems. Adsorption energies for H2O and CO2 are defined as:| Eads = E(slab/molec) − [E(slab) + E(molec)], | (1) |
Results
Carbon dioxide activation
The adsorption of CO2 onto each surface was studied by initially conducting full geometry optimisations with the CO2 placed high in the vacuum, which led, in most cases, to an initial physisorbed state, whereby the adsorbate retained an O–C–O bond angle of 180° and oriented in such a way as to lie parallel to the surface (see Fig. 1). The adsorbate position was then systematically scanned towards the surface, to locate both the chemisorbed global minimum and approximate transition state. During these scans, all internal coordinates except for that associated with the Z-position of the carbon atom of CO2 were allowed to relax fully (see ESI† Tables S1–S4). Additionally, chemically adsorbed structures were fully optimised and transition states located via NEB calculations.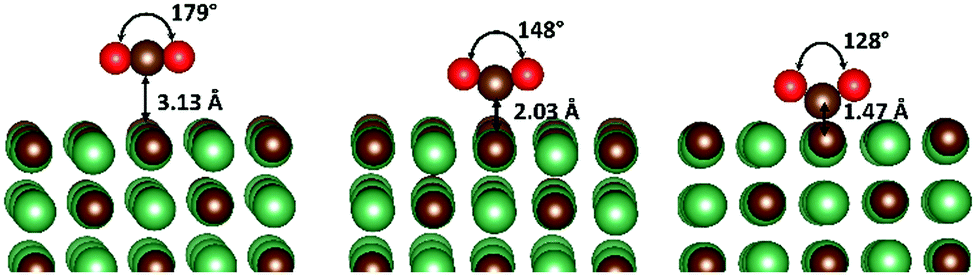 | ||
| Fig. 1 One electron reduction of CO2. The physisorbed (left) transition state (middle) and chemisorbed (right) structure, obtained with CO2 adsorbed on the NbC{001} surface. | ||
An illustration of the physisorbed, transition state and activated structures of the CO2 chemisorption process on the {001} surface of NbC is given in Fig. 1, although, it is important to note that this is only one of the two possible chemisorption pathways, which are discussed in detail below. Energies for each minimum and transition state are shown in Table 1. Recently, Illas et al.,21 studied chemically adsorbed CO2 on the {001} surface of the same series of TMCs and, whilst no other surfaces or states were considered, the available results are in excellent agreement with ours (see 2Itheo, column in Table 1). This agreement also helps to validate our approach of using geometry scans to locate global minima, since our results closely mirror their lowest energy species. The O–C–O bond angle of the adsorbate is transformed from 180° in the physically adsorbed structures, via a transition state bond angle of ∼150°, to <130° in the bent structure. These two forms of chemisorbed CO2 correspond to an increase of ∼1 to 2 electrons in the CO2 group according to the Bader charges (β) analysis, of atomic charges (see Table 2).
| 1I | TSCO2 | iFreq | 2I | 2Itheo | |
|---|---|---|---|---|---|
| Imaginary frequencies (iFreq) are given in cm−1. | |||||
| TiC{001} | −0.26 | 0.25 | 219.37 | −0.86 | −0.81 |
| TiC{011} | −0.45 | 0.00 | 203.43 | −3.45 | |
| TiC{111}M | — | — | −3.05 | ||
| VC{001} | −0.32 | 0.17 | 439.18 | −0.33 | −0.19 |
| VC{011} | −0.22 | 0.05 | 171.27 | −0.21 | |
| VC{111}M | — | — | −1.93 | ||
| ZrC{001} | −0.27 | −0.15 | 186.51 | −1.62 | −1.60 |
| ZrC{011} | −0.23 | −0.19 | 117.59 | −1.03 | |
| ZrC{111}M | — | — | −3.32 | ||
| NbC{001} | −0.20 | 0.06 | 320.54 | −0.76 | −0.87 |
| NbC{011} | −0.29 | −0.12 | 208.17 | −0.39 | |
| NbC{111}C | −0.21 | 0.61 | −0.43 | ||
| NbC{111}M | — | — | −1.98 | ||
| Φ r | Φ phys | Φ chem | β phys | β chem | R surf-C | OCO | r CO,ave | |
|---|---|---|---|---|---|---|---|---|
| TiC{001} | 3.93 | 3.73 | 4.58 | 0.06 | 0.92 | 1.47 | 128.1 | 1.29 |
| TiC{011} | 3.39 | — | 3.55 | — | 1.27 | 1.26 | 146.4 | 1.50 |
| TiC{111}C | 5.25 | 4.01 | — | 0.02 | — | — | — | — |
| TiC{111}M | 5.33 | — | 5.18 | 0.06 | 1.79 | 1.85 | 138.5 | 1.21 |
| VC{001} | 3.90 | 3.42 | 4.27 | 0.05 | 0.76 | 1.48 | 127.6 | 1.29 |
| VC{011} | 3.98 | 3.64 | 4.37 | 0.06 | 0.67 | 1.46 | 125.0 | 1.37 |
| VC{111}C | 5.51 | 5.13 | — | 0.02 | — | — | — | — |
| VC{111}M | 5.20 | — | 5.26 | — | 1.12 | 2.07 | 103.2 | 1.42 |
| ZrC{001} | 3.97 | 3.64 | 4.00 | 0.08 | 1.01 | 1.49 | 130.9 | 1.28 |
| ZrC{011} | 3.00 | 2.90 | 3.37 | 0.12 | 0.96 | 1.26 | 147.3 | 1.49 |
| ZrC{111}C | 5.10 | 4.62 | — | 0.03 | — | — | — | — |
| ZrC{111}M | 5.08 | — | 5.27 | — | 1.84 | 1.76 | 140.1 | 1.20 |
| NbC{001} | 3.21 | 2.75 | 3.58 | 0.07 | 0.93 | 1.47 | 124.5 | 1.30 |
| NbC{011} | 3.16 | 3.18 | 3.78 | 0.07 | 0.71 | 1.21 | 143.0 | 1.50 |
| NbC{111}C | 5.75 | 5.21 | — | 0.07 | — | — | — | — |
| NbC{111}M | 5.37 | — | 5.45 | — | 1.19 | 2.43 | 107.2 | 1.60 |
Results highlighted in Table 2 show that the workfunction (φ) of all carbide surfaces increases with the chemisorption of CO2, which is the result of a deeper d-band caused by electron transfer. The workfunction is calculated by subtracting the energy of the Fermi level from the vacuum energy, and is an important component linking the redox potential at the ground state to the valence bond curves (see Fig. 3). Unfortunately, except for the workfunction of the carbide surfaces, it is impossible to calculate the energetics of the other components of the excited wavefunction (ϕ*) and therefore this figure is intended as a powerful schematic representation of the valence bond diagrams for the two main reaction profiles. As mentioned above, the CO2 reduction was confirmed by the Bader charge analysis, which indicates that the adsorbate's valence electrons increased by at least ∼1 electron. We also observe that the variation in this charge transfer correlates very well with the exothermicity of the chemisorbed species (see Fig. 2). This clear correlation links a more exothermic adsorption energy with an increase in charge transfer, indicating that the more CO2 is reduced the greater the exothermicity of the adsorption. We note that, as there is no stable chemically adsorbed species on the {111}C surfaces, there is also a lack of electronic and structural data, for these states.
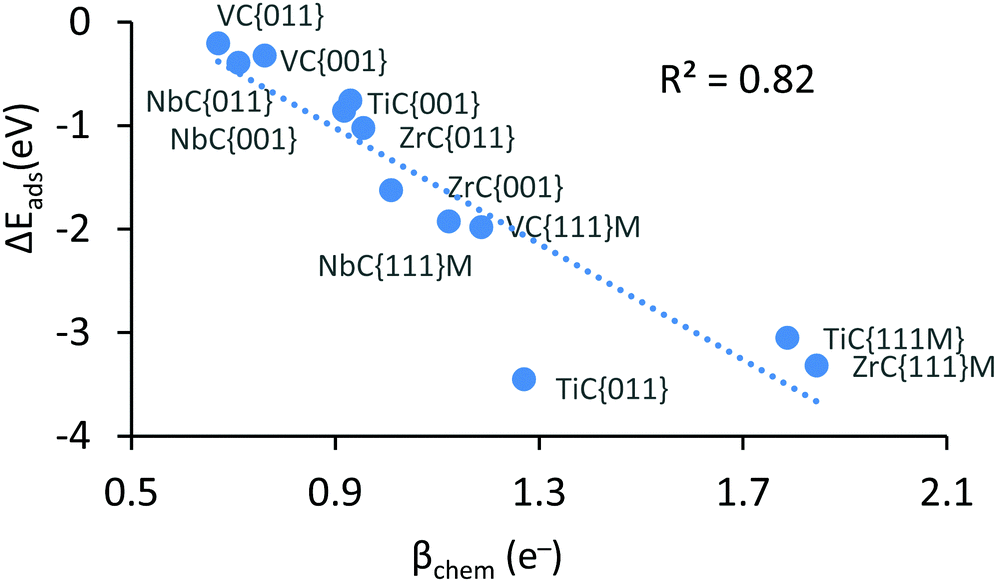 | ||
| Fig. 2 Negative correlation between chemical adsorption energy and amount of CO2 reduction, upon chemisorption. | ||
When the CO bond length in the activated adsorbate is plotted against adsorption energy, we observe two distinct trends, which also strongly correlate with the number of electrons transferred to the chemisorbed species (see Fig. 4). The first series has surface geometries of adsorption that promote activation of a double bond, with CO bond lengths that are between >1.20 Å and <1.45 Å. Additionally, adsorption on a surface of the second series promotes an activated single bond greater than 1.5 Å. Importantly, the latter also correlates to a two-electron reduction of the adsorbate, and represents the reaction pathway (b) in Fig. 3. These different trends are mostly caused by the different binding geometries, which are promoted by each surface. Variations in surface area and surface morphology change the most stable adsorption geometry for the carbon dioxide molecule. This adsorption geometry either leads to a stable P1 single electron transfer intermediate (a) or can promote a barrier-less pathway to a second electron transfer (P2), which in turn is the result of an elongated C–O bond length in the adsorbate of P1.
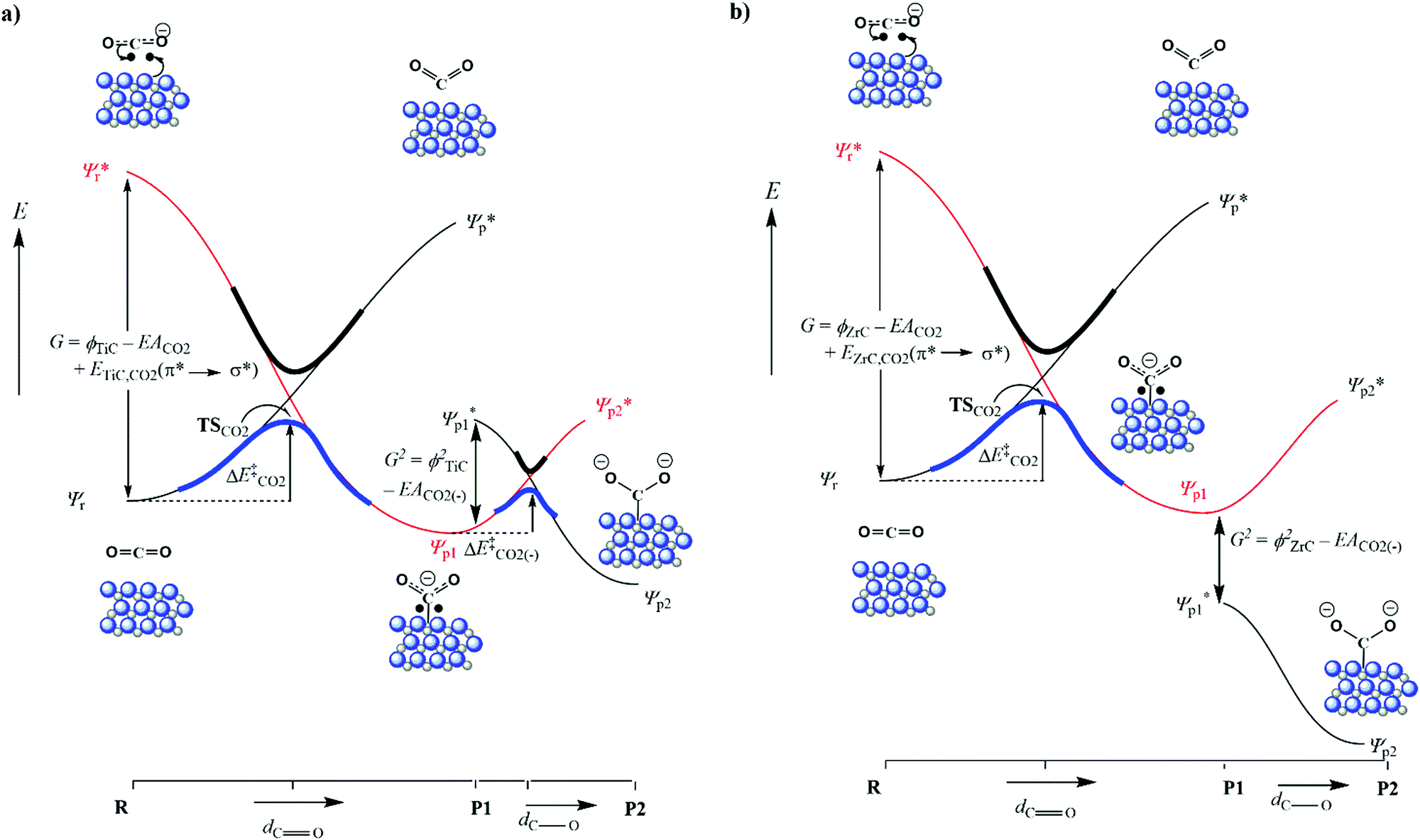 | ||
| Fig. 3 Schematic valence bond crossing diagrams for CO2 reduction on carbide surfaces. (a) Single electron reduction of CO2 leading to intermediate P1. (b) Two electron reduction of CO2 leading to intermediate P2. | ||
It has previously been reported that changes in the value of transition state imaginary frequencies are often closely linked to changes in reactivity,67,68 which seems also to apply to the current results, where the smallest imaginary frequency (186.51 cm−1) is associated with the most reactant-like transition state (i.e. occurring at the greatest distance from the surface). Importantly, the largest frequency was found for CO2 adsorption onto VC{001}, which was also described as the most product-like transition and indeed leads to the most exothermic adsorption, whilst CO2 adsorption on ZrC{001} was least exothermic. However, when the magnitude of the imaginary frequencies from each transition state was plotted against the adsorption energy of the chemically adsorbed state, a relatively poor correlation was observed (see Fig. S5 ESI†). It has frequently been reported that when different species catalyse the same redox process, there is often a correlation between the stability of the product ground state and the position along the potential energy surface at which the transition state occurs.69–71 This correlation is due to the position of the avoidance crossing (kinetic barrier) when plotted on a valence bond (VB) diagram (see Fig. 3). Transition states that happen earlier occur higher in the vacuum and have imaginary frequencies with smaller wavenumbers, and it should therefore be possible to directly correlate the imaginary frequencies of a transition state to the reaction thermodynamics. Fig. 3 graphically illustrates two distinct chemical adsorption pathways and helps to explain the lack of correlation, shown in Fig. S5, in the ESI.† The surfaces responsible for these two pathways will be discussed in more depth below. However, it is clear from the valence bond analysis that only the first adsorption type (a) will lead to a stable one electron absorbed intermediate (P1) that can be connected directly to a single conical intersection, since in the second reaction pathway (b) the adsorption geometry of the CO2, in P1, increases the electron affinity enough to overcome the surface workfunction and lower the ϕP1 below 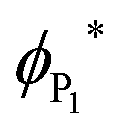 . In other words, the geometry of P1 promotes a barrier-less second electron transfer and the adsorption energy of the subsequent P2 intermediate can no-longer be directly linked to the avoidance crossing of the wavefunction linking the ground state product and the excited state reactant with the wavefunction linking the excited state reactant and the ground state product.
. In other words, the geometry of P1 promotes a barrier-less second electron transfer and the adsorption energy of the subsequent P2 intermediate can no-longer be directly linked to the avoidance crossing of the wavefunction linking the ground state product and the excited state reactant with the wavefunction linking the excited state reactant and the ground state product.
Fig. 4 shows, that of the two surfaces where transition states are observed, only CO2 adsorption on the {001} surfaces exclusively follow reaction pathway a, whereby a single electron transfer is coupled to limited C–O bond elongation. We therefore decided to evaluate the correlation between the position of the transition states and the sum of the energy of surface workfunctions and the chemical adsorption energies (see Fig. 5), which in practice means a subtraction of the energy gained by chemical adsorption (right hand side of VB curve) from the energy lost through the reduction of the carbide d-band (left hand side of VB curve). In this case, the lower the energy the earlier the transition state, which is to be expected since the energies are positive and therefore the higher the energy the later the transition state (conical intersection). This correlation is important, because it helps to confirm the similarities and trends of all potential energy surfaces and means that the heights of the respective barriers are comparable.
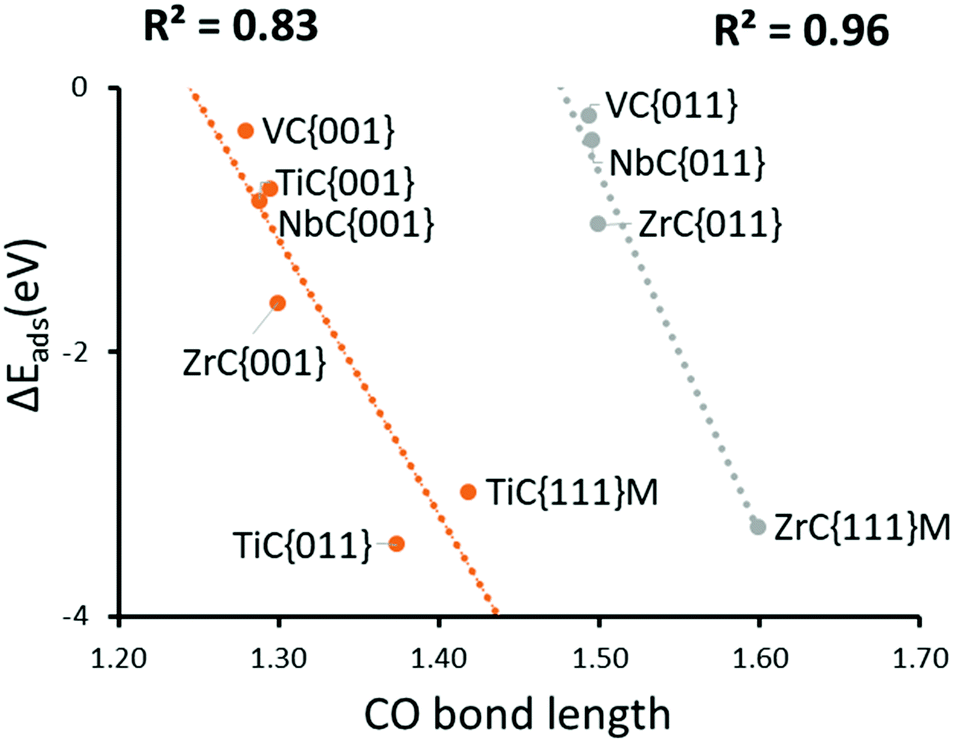 | ||
| Fig. 4 Correlation between chemical adsorption energy and the CO bond length of CO2 reduction transition state. (red) activated double bonds. (green) activate single bonds. | ||
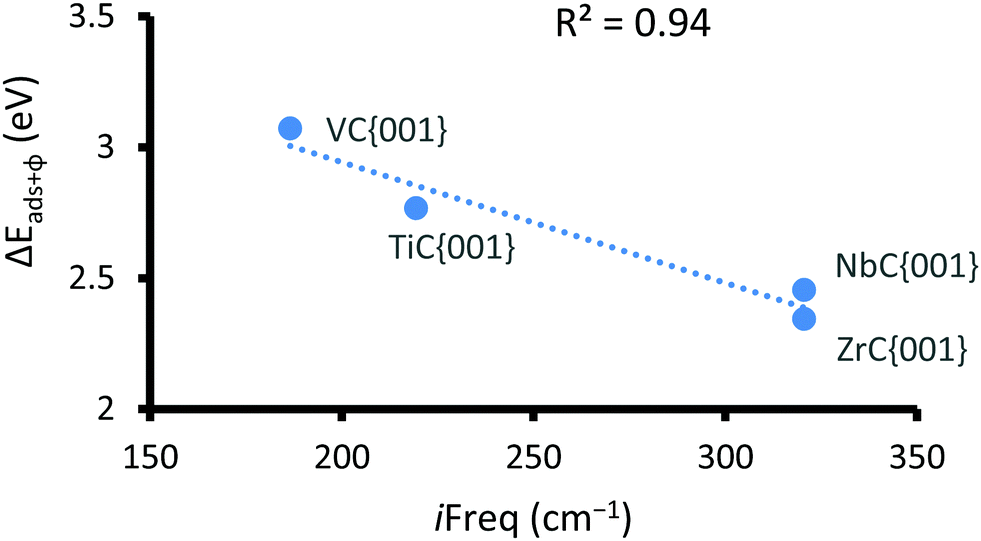 | ||
| Fig. 5 Negative correlation between the sum of the φ and chemical adsorption energy and the imaginary frequency for the CO2 reduction transition state. | ||
The {011} surface of transition metal carbides are also of significance, as previous work45 has shown that they are the second lowest energy surface in all the carbides reported here. Therefore, a Wulff construction72 of any TMC nanoparticles would probably indicate that the {011} surface would be the second most dominant, in conditions of thermodynamic equilibrium and in the absence of stabilizers and surfactants. Such a prediction has been supported by many studies that have used DFT-derived surface energies to predict such morphologies.73,74 Owing to the importance of these surfaces, a detailed study of their reactivity was also undertaken, which showed that chemical adsorption proceeded via small barriers. It is notable that for all carbides, the {011} surfaces greatly increase C–O bond activation above that of the comparative {001} surfaces (see Fig. 4). It is also noteworthy that carbides with parent metals of the fourth group appear to adsorb CO2 much more strongly than those of the fifth. Indeed, this is a general trend across all surfaces and is accompanied with generally stronger adsorption of period four metal carbides.
Unlike the behaviour on the {001} and {011} surfaces, no physically adsorbed structures were found on any of the metal terminated {111} surfaces, which instead catalysed direct chemical adsorption pathways. Despite an optimized adsorbate being placed high in the vacuum, the initial geometry optimisations lead inevitably and without an energy barrier to the formation of a carbide-bound, activated and bent carboxylate (CO2δ−) species.75 Attempts were made to locate any shallow physically adsorbed intermediate by scanning the adsorbates up into the vacuum and running stepwise optimisation with the only fixed degree of freedom associated position of the CO2 in the Z-direction. However, in all cases the results indicated a barrierless chemical adsorption process without stable physically adsorbed states (see ESI† Tables S1 and S2). It has previously been argued that the {111}M surfaces of early TMCs represent a polar metastable phase that can eventually reconstruct to produce open stepped and defect surfaces.76,77 However, there is no evidence of such a reconstruction and it appears that these pristine surfaces are stable.63–65 These surfaces are therefore considered to be important and their activity indicates that they represent examples of a small and privileged class of solid surfaces that are able to activate CO2 without any activation energy.78,79
Geometries and relevant bond lengths for these chemically adsorbed states are shown in Fig. 6. It is very noticeable that despite binding motifs that appear to position the bent CO2 molecule with the oxygen atoms pointing up into the vacuum, in all cases, the bond distances between the surface metals and all three adsorbate atoms are very similar, which is to be expected since all the structures reported represent the lowest energy binding configurations. These binding motifs are explained by the central carbon atom filling the hollow sites between metal atoms, on the {111}M surfaces, thereby allowing maximum overlap between the dangling orbitals of the surface atoms and atoms of the adsorbate. Very similar binding motifs are seen on the {001} and {011} surfaces, where the Contop position enables the maximum number of interactions between two adjacent metal atoms. As stated above, there are also two very pronounced trends between the CO2 adsorption energies and C–O bond lengths. For both activated modes, the two carbides with the most elongated C–O bonds (TiC and ZrC) were also the materials causing the greatest adsorption energies (see Fig. 4). It is also important to note that such elongated C–O bond lengths would imply lower barriers for further reduction,44,80 which in turn would suggest that surfaces from the elongated single bond series could be expected to have lower CO2 dissociation barriers, even if their adsorption is less exothermic than that seen in a comparable surface from the double bond elongation series.
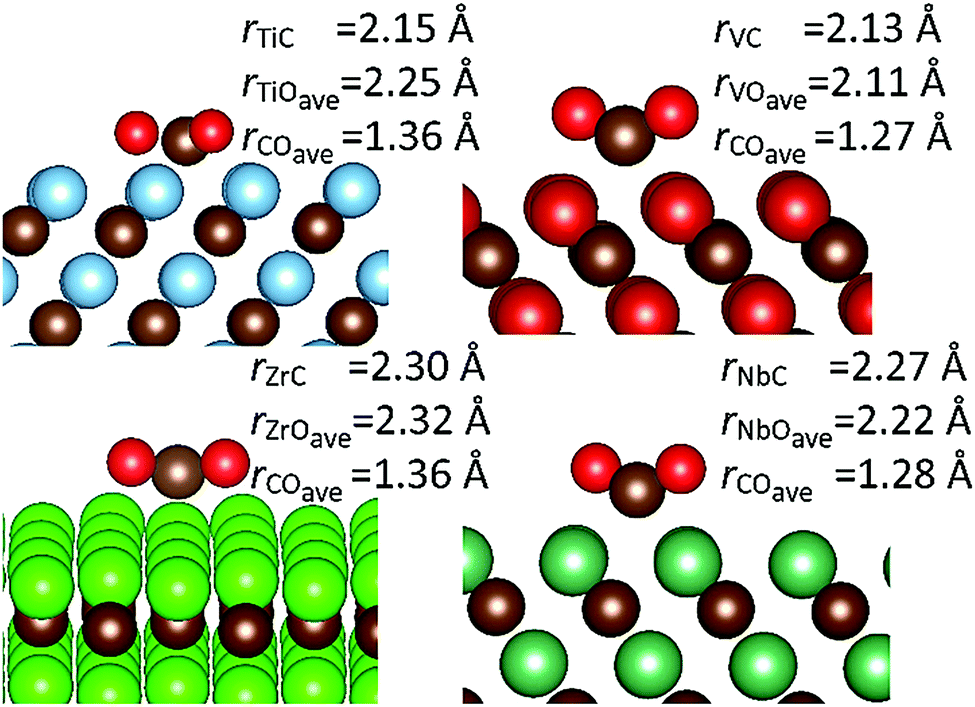 | ||
| Fig. 6 Chemically adsorbed CO2 on the {111}M carbide surfaces. Structural images and important bond distances are given for: TiC (top left), VC (top right), ZrC (bottom left) and NbC (bottom right). All distances are given in angstroms. | ||
In contrast to the metal-terminated {111} surfaces, those terminated with carbon proved to be very unstable in all cases except NbC. Although the NbC{111}C surface did remain stable during the chemical adsorption of CO2, the barrier for this reaction (at >0.8 eV) was comparatively high. This result is in excellent agreement with our previous computational work that predicted that the metal-terminated {111} surfaces would become progressively less favoured over the carbon-terminated {111} surfaces as the parent metal moves across the groups and down the periods.45 Other work focused on the hydrogenation of these same carbon-terminated {111} surface also found these termination to be unstable.81 Furthermore, these results are also in good agreement with experimental observations, which report that the {111} surface of TiC is exclusively terminated by titanium.60,82 However, since all these studies showed that a carbon-terminated {111} surface might be important for NbC, we have still included these surfaces for the sake of completeness.
Water activation
As already mentioned, H2O is an important component in flue gas that has the ability to direct the hydrogenation of CO2, as catalysed by solid materials.19,20 The H2O molecule prior to adsorption was initially placed high in the vacuum and sequentially scanned towards the surfaces, in a manner similar to that discussed in the previous section for CO2. Also as seen for the CO2 adsorption, this protocol led to lowest binding energy conformation and indeed it was not possible to locate more exothermic energies using alternative techniques. This protocol produced two clearly favoured binding configurations as shown in Fig. 7. In the two surfaces that maintained the surface stoichiometry of the bulk, namely the {001} and {011} surfaces, the lowest energy binding configuration was the on-top (OTS) metal site. The carbon-terminated {111} surface were all unstable in the presence of water, in a manner that is not only observed in the presence of CO2, but also seen on the hydrogenated surfaces, where in the presence of molecular hydrogen only the {111}C surface of NbC was stable.81 In contrast to the carbon terminated surface, which were only stable in the absence of an adsorbate, the metal terminated {111} surfaces were both stable and strongly adsorbed H2O in the hollow site (HS) configuration (see Fig. 7).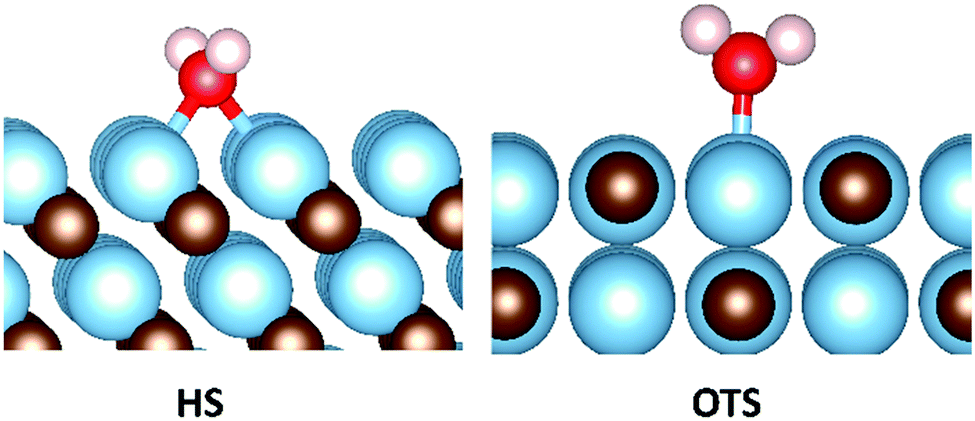 | ||
| Fig. 7 Depiction of the two lowest energy water binding configurations. The hollow site configuration (HS) is the most exothermic adsorption motif on the {111}M surface, whilst the metal on-top configuration (OTS) is most stable on the {001} and {011} surfaces. | ||
A strong trend in the binding energies is seen in the remaining three low-index surfaces, as shown in Fig. 8. In all cases the binding energy of H2O on the {111}M is far higher than that found for the {011} and {001} surfaces, which both show very comparable adsorption energies. The one exception to this observation is that the {011} surface of TiC binds water more exothermically than the TiC{111}M surface, which can be explained by the fact that the TiC{011} surface adsorbs H2O dissociatively. Geometry and Bader charge analysis confirms that adsorption on this surface produces bound hydrogen and hydroxyl groups, and this values is therefore not so much a measure of the binding energy of H2O as of the formation energy of hydroxyl and hydrogen species on the TiC{011} surface. Multiple attempts were made to locate a chemisorbed local minima that included an intact H2O, with all cases leading to either dissociative or physisorbed states upon full optimisation.
 | ||
| Fig. 8 Binding energies of water on three low-index surfaces of TiC, VC, ZrC and NbC. The orange diamond bar (left) represent the {001} surfaces; whilst, the brown square bar (middle) and the blue diagonally stripped bar (right) represent the {011} and {111}M surfaces respectively. Water adsorption is an exothermic process on the NbC{011} surface, so the physically adsorbed energy has been included instead and marked with an asterisk (*). | ||
In contrast to the adsorption of CO2, there appears to be no correlation between average O–H bond lengths and chemical adsorption energies of H2O (see ESI† Fig. S7). However, charge transfer is also extremely small in all but the three most exothermic cases. These three surfaces, namely ZrC{111}M, TiC{111}M and TiC{011}, see charge transfer to H2O of 0.11, 0.16 and 0.67 electrons respectively (see ESI,† Table S6). In every case, this additional electron density is transferred to the oxygen of the adsorbate. Once this centre is fully reduced a subsequent heterogeneous O–H cleavage will lead to the formation of a surface hydroxyl group and hydrogen atom. Importantly, ZrC{111}M and TiC{111}M are the surfaces that promote the second and third longest O–H bond lengths during chemical adsorption, with the longest O–H bond lengths reserved for the TiC{011} surface that promotes complete cleavage of this bond and consequently promotes the most electron transfer.
H2O decomposition on the {001} carbide surfaces
The occurrence of any adsorbate co-binding configuration depends strongly on whether the final product is more stable than the sum of CO2 and the water dissociation products. Therefore, we accessed the dissociative adsorption energies of H2O on the {001} surfaces of all the carbides studied. To achieve this, the distance between the hydrogen and hydroxyl groups were sequentially elongated before full geometry optimisations of the minima of the scans. The resulting bond distances, Bader charges and dissociative adsorption energies are shown in Fig. 9. Interestingly, although no chemical adsorption of water was observed on the {001} surface of NbC, dissociative adsorption is an exothermic process. Also of note is the finding that the changes in electronic structures of the dissociative adsorption states are almost identical, i.e. instead of protons we observe adsorbed hydrogen atoms that all retain one valence electron, which result is in excellent agreement with our work on molecular hydrogen adsorption.81 It appears that the surface d-bands donate approximately half of this electron density with the other half coming from the hydroxyl group.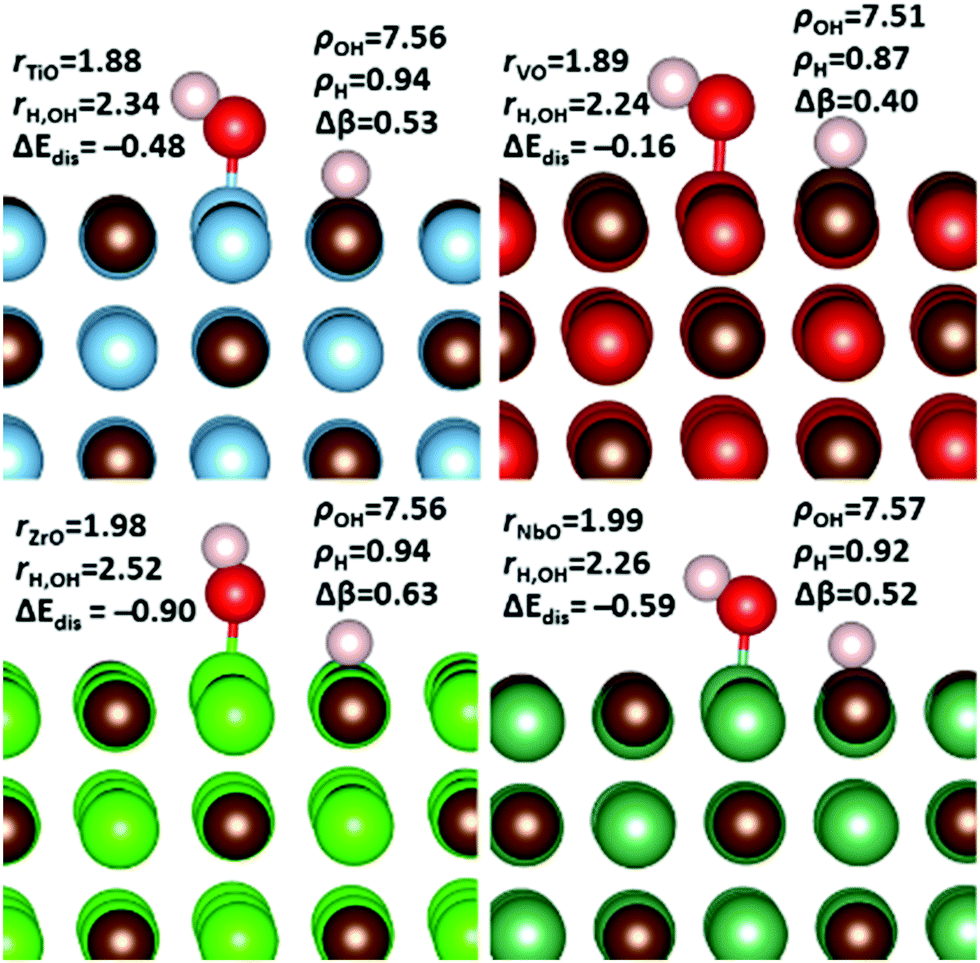 | ||
| Fig. 9 Bader charges, adsorption energies and important bond distances resulting from dissociative adsorption of H2O on the {001} surfaces of TIC (top left), VC (top right), ZrC (bottom left) and NbC (bottom right). The metal–oxygen (rXO) and hydrogen–hydroxyl (rH,OH) distances are included alongside the dissociative adsorption energy (ΔEdis). The total bader charges for the hydroxyl (βOH) and hydrogen (βH), as well as the total change in grouped bader charge (βOH). All distances are given in Å, whilst energy values are in eV. | ||
From the results shown in Fig. 9, it is clear that on the {001} surface of group four carbides, dissociative absorption of H2O is either energetically the same or less favourable than the alternative chemically adsorbed state. In practice, this result means that chemically adsorbed water would probably remain intact on the {001} surface of TiC and VC, where they could form strong hydrogen bonding networks with CO2. In contrast, dissociation of water on the {001} surfaces of NbC and ZrC is indeed an exothermic process. Indeed, as mentioned previously, dissociative adsorption is the only exothermic type of chemically adsorbed H2O on the {001} surface of NbC. However, this process would require a concerted surface binding and O–H bond breaking transition state that would probably make this binding motif a rare event. On the {001} surface of ZrC, dissociative adsorption is 0.31 eV more exothermic than the intact chemical adsorption of H2O, which could indicate that the dissociative adsorption of water could be important on this specific surface. Therefore, it is possible that chemical adsorption of molecular water could form a relatively stable intermediate state preceding the formation of surface-bound hydrogen and hydroxyl groups via a HO–H bond stretch. Such a process is expected to be heavily dependent on the presence of other adsorbate molecules in the local environment and the potential hydrogen-bonding network such species would initiate. In order to assess the importance of such interactions, a detailed study of the co-adsorption of both CO2 and H2O on the {001} surfaces of all four carbides is hence presented below.
CO2 and H2O co-adsorption on the {001} carbide surfaces
Competitive binding of water and carbon dioxide was assessed to examine the surface interactions between the two adsorbates. Of particular interest is the possibility of surface-mediated bicarbonate (HCO3−) formation via a carbonic acid (H2CO3) intermediate, which was examined on the lowest energy {001} surface only, since that is the dominant surface where these co-binding effects are believed to be most prevalent, as described previously.12 Sequestering carbon dioxide in the form of carbonic acid is becoming an increasingly important research area in the field of green chemistry.83–85 It has been hypothesised that surface mediated carbonic acid formation on a suitable catalyst would sequester CO2 at higher concentrations and at much faster rates than in the absence of such a material.86 Such a process has obvious application in CCS technologies and could also form an intermediate compound in the formation of calcium bicarbonate, whereby the natural process of rock weathering is replicated and accelerated, leading to affordable and long-term carbon sequestration.87To improve the probability of locating the global minimum for the co-binding of the adsorbates, a water molecule was placed high in the vacuum and sequentially scanned onto surfaces preloaded with CO2, see ESI† Tables S8 and S9. In all cases, this approach led to a co-bound configuration that includes a single OCO–H–OH hydrogen-bond. Transitions states were located using the same climbing NEB protocol mentioned previously and the resulting two-dimensional potential energy landscapes are shown in Fig. 10. All physisorption energies (1IX) are extremely similar, whilst there is much more divergence observed for the reaction barriers (1TS) and exothermicities (2IX). VC{001} showed both the smallest barriers as well as the most exothermic co-adsorption; whilst the reverse was true for TiC{001}. Importantly, none of the surfaces studied reported a co-adsorption barrier above 0.65 eV and all reported exothermic processes. Interestingly, whilst it was shown in the previous section that water decomposition is a likely occurrence on the {001} surfaces of ZrC and NbC, the energies of such processes are both remarkably similar to the co-binding energies of intact H2O on these surfaces, preloaded with CO2.
 | ||
| Fig. 10 Potential energy surface for the co-adsorption of H2O on the CO2 loaded {001} surfaces of TiC, VC NbC and ZrC. All values are given in eV relative to one molecule of H2O and the CO2 loaded surfaces. | ||
To assess the strength of the CO2 and H2O interactions on the different surfaces studied for the reaction outlined in eqn (2), the interaction energy (Einter) was calculated using eqn (3).88
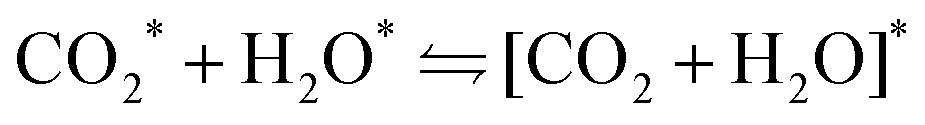 | (2) |
| Einter = [E(slab) + E(slab + CO2 + H2O)] − E(slab + CO2) − E(slab + H2O) | (3) |
Also, none of these binding conformations would be expected to promote efficient carbonic acid formation. Therefore, to locate the optimal binding confirmation to promote H2CO3 formation, it was decided to study the reverse reaction (from carbonic acid to separate adsorbates).
To investigate the possibility of the carbide-mediated formation of H2CO3, carbonic acid was adsorbed onto the {001} surfaces of each carbide, before following the reaction pathway that included both a hydrogen atom transfer and a C–O bond formation step. Fig. 11 shows the potential energy surface for the carbide-mediated formation of carbonic acid, on the {001} surfaces of TiC, VC, NbC and ZrC. Although the results were obtained in the reverse direction, the figure shows the potential energy surface along the reaction path, whereby, the co-bound adsorbates (2I*X) transform into the carbonic acid products (3IX) via a carbon–carbon bond cleavage transition state (2TSX), where (X = TiC/VC/NbC/ZrC). Very high barriers of between 0.76 eV and 1.20 eV are observed for the concerted hydrogen transfer and C–O bond formation step from the 2I*X intermediates. However, none of these intermediate states represents the lowest energy co-binding confirmations (2IX). These are the same binding confirmations shown in Fig. 10, whereby the dominant (low energy) binding geometries are very far from optimal for carbonic acid formation. Therefore, the surface mediated formation of carbonic acid is probably a rare event and would have to proceed via an activation barrier of >1 eV, with the end products being endothermic. Optimised structures of carbonic acid adsorbed on TiC{001} and VC{001} surfaces are shown in Fig. 12. Although the adsorption geometries of both species are very similar, there is a slightly longer average metal–oxygen distance for carbonic acid adsorbed on VC, which is caused by the absence of one formal V–C bond and helps to explain why adsorption on the {001} surface of TiC is more exothermic. This is shown clearly in Fig. 12, where the distal bond of the hydroxyl group of carbonic acid on VC{001} points up into the vacuum.
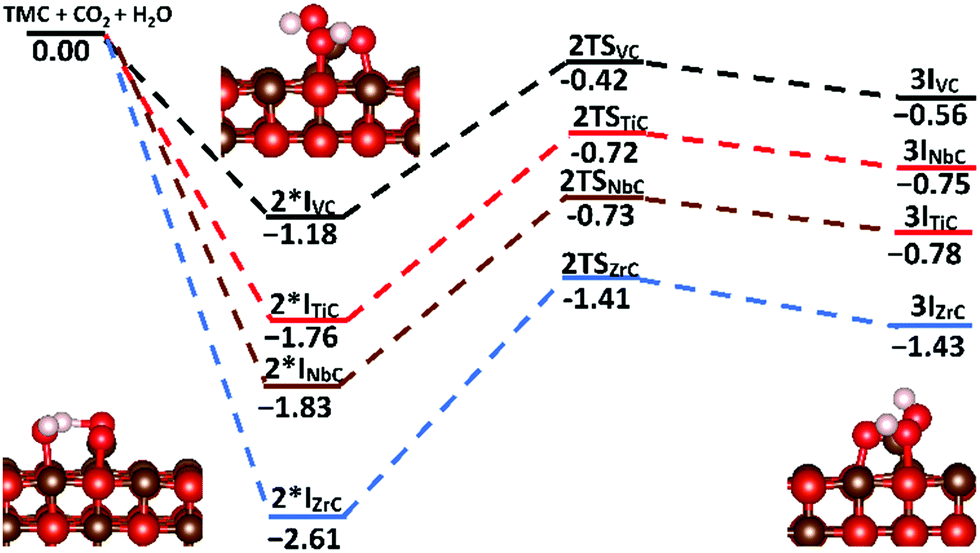 | ||
| Fig. 11 Potential energy surface for the surface mediated formation of carbonic acid, on the {001} surfaces of TiC, VC, NbC and ZrC. All values are given in eV relative to separate surface and adsorbate energies. | ||
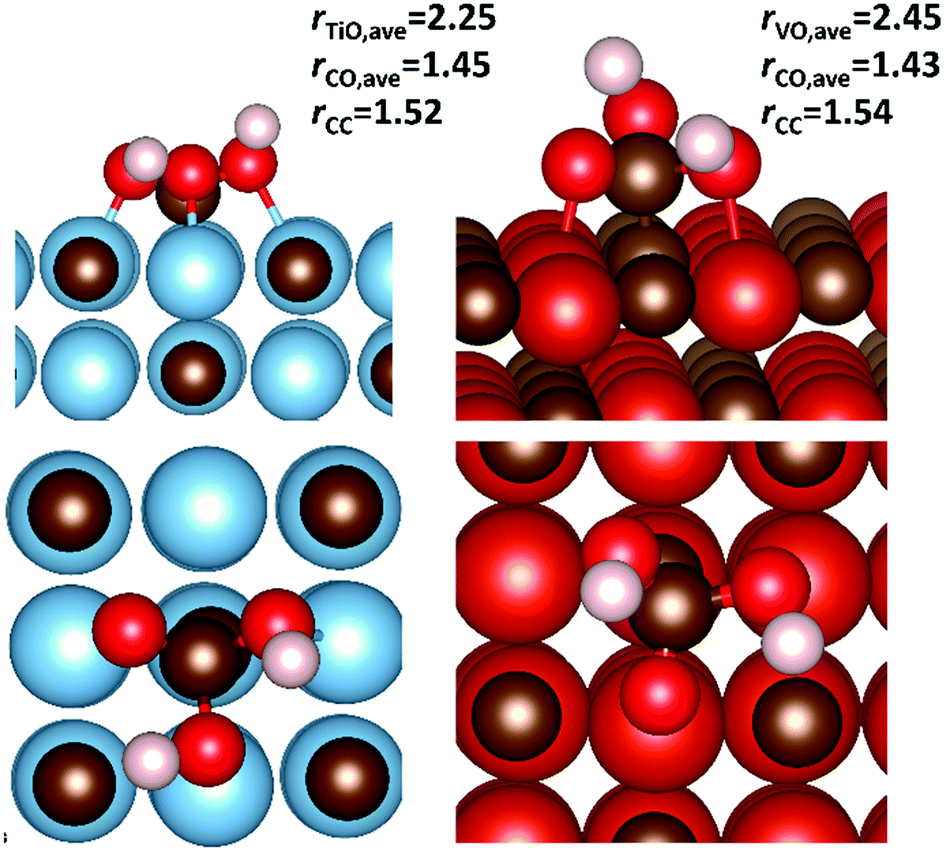 | ||
| Fig. 12 Side and top adsorption geometries of carbonic acid on the {001} surfaces of TIC (left) and VC (right). The average metal–oxygen (rXO,ave) and carbon–oxygen (rCO,ave) distances are given alongside the length of the carbon–carbon bond (rCC), formed between the adsorbate and the surface. All values are given in Å. | ||
Summary and conclusions
We have used a DFT methodology to model the catalytic activity of the low-index surfaces of TiC, VC, ZrC and NbC towards water and carbon dioxide. Our results indicate that the metal terminated {111} surfaces of all the carbides will be the most reactive and that in general reactivity is greater in period four metal carbides. We also report two distinct CO2 bonding modes that correspond to differing amounts of electron transfer and C–O bond elongation. Water adsorption is mostly a very exothermic process, even though it results in very modest amounts of charge transfer. On NbC{001} and TiC{011} surfaces only dissociative H2O adsorption is found to be exothermic. This work predicts the formation of hydrogen bonds when the two adsorbates are co-adsorbed. These strongly hydrogen bonded species are formed via relatively accessible energy barriers on all the {001} surfaces studied. However, surface-mediated carbonic acid formation is unlikely to proceed due to relatively high activation energy barrier and endothermic reaction energies, that could be outcompeted by alternative pathways that are initiated by water dissociation on the {001} surfaces of ZrC and NbC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded as part of an EPSRC low carbon fuels grant (EP/N009533/1). Computing facilities for this work were provided by ARCCA at Cardiff University, HPC Wales, and through our membership of the UK's Materials Chemistry Consortium (MCC). The MCC is funded by EPSRC (EP/F067496). All data created as part of this study are openly available at DOI: 10.17035/d.2019.0070602444.Notes and references
- Intergovermental Panel on Climate Change, Climate Change 2014–Impacts, Adaptation and Vulnerability: Regional Aspects, Cambridge University Press, 2014 Search PubMed.
- International Energy Agency, An energy system under stress, 2014.
- Report FCCC/CP/2015/L.9Rev.1, Adoption of the Paris Agreement, UNFCCC, Paris, France, 2015.
- J. Morrissey and R. E. Horne, Life cycle cost implications of energy efficiency measures in new residential buildings, Energy Build., 2011, 43, 915–924 CrossRef.
- P. A. Owusu and S. Asumadu-Sarkodie, A review of renewable energy sources, sustainability issues and climate change mitigation, Cogent Eng., 2016, 3, 1167990 Search PubMed.
- D. Y. C. Leung, G. Caramanna and M. M. Maroto-Valer, An overview of current status of carbon dioxide capture and storage technologies, Renew. Sustain. Energy Rev., 2014, 39, 426–443 CrossRef CAS.
- A. Sayari, Y. Belmabkhout and R. Serna-Guerrero, Flue gas treatment via CO2 adsorption, Chem. Eng. J., 2011, 171, 760–774 CrossRef CAS.
- L. Espinal, D. L. Poster, W. Wong-Ng, A. J. Allen and M. L. Green, Measurement, Standards, and Data Needs for CO2 Capture Materials: A Critical Review, Environ. Sci. Technol., 2013, 47, 11960–11975 CrossRef CAS PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- B. Wang, H. Jin and D. Zheng, Recovery of CO2 with MEA and K2CO3 absorption in the IGCC system, Int. J. Energy Res., 2004, 28, 521–535 CrossRef CAS.
- H. Gao, S. Pishney and M. J. Janik, First principles study on the adsorption of CO2 and H2O on the K2CO3(001) surface, Surf. Sci., 2013, 609, 140–146 CrossRef CAS.
- M. Mercy, S. F. R. Taylor, J. Jacquemin, C. Hardacre, R. G. Bell and N. H. De Leeuw, The addition of CO2 to four superbase ionic liquids: a DFT study, Phys. Chem. Chem. Phys., 2015, 17, 28674–28682 RSC.
- S. F. R. Taylor, C. McCrellis, C. McStay, J. Jacquemin, C. Hardacre, M. Mercy, R. G. Bell and N. H. de Leeuw, CO2 Capture in Wet and Dry Superbase Ionic Liquids, J. Solution Chem., 2015, 44, 511–527 CrossRef CAS.
- J. P. H. Fee, J. M. Murray and S. R. Luney, Molecular sieves: an alternative method of carbon dioxide removal which does not generate compound A during simulated low-flow sevoflurane anaesthesia, Anaesthesia, 1995, 50, 841–845 CrossRef CAS PubMed.
- A. Taheri Najafabadi, CO2 chemical conversion to useful products: an engineering insight to the latest advances toward sustainability, Int. J. Energy Res., 2013, 37, 485–499 CrossRef CAS.
- K. Yu, K. Kiesling and J. R. Schmidt, Trace Flue Gas Contaminants Poison Coordinatively Unsaturated Metal–Organic Frameworks: Implications for CO2 Adsorption and Separation, J. Phys. Chem. C, 2012, 116, 20480–20488 CrossRef CAS.
- S. M. Klara and R. D. Srivastava, U.S. DOE integrated collaborative technology development program for CO2 separation and capture, Environ. Prog., 2002, 21, 247–253 CrossRef CAS.
- Q.-L. Tang, Q.-J. Hong and Z.-P. Liu, CO2 fixation into methanol at Cu/ZrO2 interface from first principles kinetic Monte Carlo, J. Catal., 2009, 263, 114–122 CrossRef CAS.
- Y.-F. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, Insight into methanol synthesis from CO2 hydrogenation on Cu(1 1 1): complex reaction network and the effects of H2O, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- C. Kunkel, F. Vin and F. Illas, Biogas Upgrading by Transition Metal Carbides, ACS Appl. Energy Mater., 2018, 1, 43–47 CrossRef CAS.
- Z. Bao, L. Yu, Q. Ren, X. Lu and S. Deng, Adsorption of CO2 and CH4 on a magnesium-based metal organic framework, J. Colloid Interface Sci., 2011, 353, 549–556 CrossRef CAS PubMed.
- S. Bourrelly, P. L. Llewellyn, C. Serre, F. Millange, T. Loiseau and G. rard Fé rey, Different Adsorption Behaviors of Methane and Carbon Dioxide in the Isotypic Nanoporous Metal Terephthalates MIL-53 and MIL-47, J. Am. Chem. Soc., 2018, 15, 3 Search PubMed.
- A. Morales-Gar, A. An Fer Andez-Fer Andez, F. V. Nes and F. Illas, CO2 abatement using two-dimensional MXene carbides, J. Mater. Chem. A, 2018, 6, 3381 RSC.
- H.-J. Freund and M. W. Roberts, Surface chemistry of carbon dioxide, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- M. E. Boot-Handford, J. C. Abanades, E. J. Anthony, M. J. Blunt, S. Brandani, N. Mac Dowell, J. R. Fernández, M. C. Ferrari, R. Gross, J. P. Hallett, R. S. Haszeldine, P. Heptonstall, A. Lyngfelt, Z. Makuch, E. Mangano, R. T. J. Porter, M. Pourkashanian, G. T. Rochelle, N. Shah, J. G. Yao and P. S. Fennell, Energy Environ. Sci., 2014, 7, 130–189 RSC.
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: opportunities and challenges, Dalton Trans., 2007, 2975 RSC.
- G. Centi and S. Perathoner, Catalysis: Role and Challenges for a Sustainable Energy, Top. Catal., 2009, 52, 948–961 CrossRef CAS.
- T. Xiao, A. P. E. York, K. S. Coleman, J. B. Claridge, J. Sloan, J. Charnock and M. L. H. Green, Effect of carburising agent on the structure of molybdenum carbides, J. Mater. Chem., 2001, 11, 3094–3098 RSC.
- A. Roldan, N. Hollingsworth, A. Roffey, H.-U. Islam, J. B. M. Goodall, C. R. A. Catlow, J. A. Darr, W. Bras, G. Sankar, K. B. Holt, G. Hogarth and N. H. De Leeuw, Bio-inspired CO2 conversion by iron sulfide catalysts under sustainable conditions, Chem. Commun., 2015, 51, 7501–7504 RSC.
- S. Posada-Pérez, F. Viñes, J. A. Rodriguez and F. Illas, Fundamentals of Methanol Synthesis on Metal Carbide Based Catalysts: Activation of CO2 and H2, Top. Catal., 2015, 58, 159–173 CrossRef.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes, Energy Environ. Sci., 2013, 6, 3112 RSC.
- R. B. Levy and M. Boudart, Platinum-like behavior of tungsten carbide in surface catalysis, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- P. M. Patterson, T. K. Das and B. H. Davis, Carbon monoxide hydrogenation over molybdenum and tungsten carbides, Appl. Catal., A, 2003, 251, 449–455 CrossRef CAS.
- P. Liu and J. A. Rodriguez, Water-Gas-Shift Reaction on Molybdenum Carbide Surfaces: Essential Role of the Oxycarbide, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- N. M. Schweitzer, J. A. Schaidle, O. K. Ezekoye, X. Pan, S. Linic and L. T. Thompson, High Activity Carbide Supported Catalysts for Water Gas Shift, J. Am. Chem. Soc., 2011, 133, 2378–2381 CrossRef CAS PubMed.
- L. K. Ono, D. Sudfeld and B. Roldan Cuenya, In situ gas-phase catalytic properties of TiC-supported size-selected gold nanoparticles synthesized by diblock copolymer encapsulation, Surf. Sci., 2006, 600, 5041–5050 CrossRef CAS.
- K.-Z. Qi, G.-C. Wang and W.-J. Zheng, A first-principles study of CO hydrogenation into methane on molybdenum carbides catalysts, Surf. Sci., 2013, 614, 53–63 CrossRef CAS.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides, Chem. Commun., 2015, 51, 6988–6991 RSC.
- C. Kunkel, F. Viñes and F. Illas, Transition metal carbides as novel materials for CO2 capture, storage, and activation, Energy Environ. Sci., 2016, 9, 141–144 RSC.
- S. Posada-Pérez, F. Viñes, P. J. Ramirez, A. B. Vidal, J. A. Rodriguez and F. Illas, The bending machine: CO2 activation and hydrogenation on δ-MoC(001) and β-Mo2C(001) surfaces, Phys. Chem. Chem. Phys., 2014, 16, 14912–14921 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Molybdenum Carbide as Alternative Catalysts to Precious Metals for Highly Selective Reduction of CO2 to CO, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 Search PubMed.
- M. D. Porosoff, S. Kattel, W. Li, P. Liu and J. G. Chen, Identifying trends and descriptors for selective CO2 conversion to CO over transition metal carbides, Chem. Commun., 2015, 51, 6988–6991 RSC.
- X. Liu, C. Kunkel, P. Ramírez de la Piscina, N. Homs, F. Viñes and F. Illas, Effective and Highly Selective CO Generation from CO2 Using a Polycrystalline α-Mo2 C Catalyst, ACS Catal., 2017, 7, 4323–4335 CrossRef CAS.
- M. G. Quesne, A. Roldan, N. H. de Leeuw and C. R. A. Catlow, Bulk and surface properties of metal carbides: implications for catalysis, Phys. Chem. Chem. Phys., 2018, 20, 6905–6916 RSC.
- L. Johansson, Electronic and structural properties of transition-metal carbide and nitride surfaces, Surf. Sci. Rep., 1995, 21, 177–250 CrossRef CAS.
- S. T. Oyama, Preparation and catalytic properties of transition metal carbides and nitrides, Catal. Today, 1992, 15, 179–200 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- K. Nakamura and M. Yashima, Crystal structure of NaCl-type transition metal monocarbides MC (M = V, Ti, Nb, Ta, Hf, Zr), a neutron powder diffraction study, Mater. Sci. Eng., B, 2008, 148, 69–72 CrossRef CAS.
- A. N. Christensen, A neutron-diffraction investigation on single-crystals of titanium-oxide, zirconium carbide and hafnium nitride, Acta Chem. Scand., 1990, 44, 851–852 CrossRef CAS.
- G. Will and R. Platzbecker, Crystal Structure and Electron Density Distribution in Niobium Carbide, Z. Anorg. Allg. Chem., 2001, 627, 2207 CrossRef CAS.
- M. Hellenbrandt, The Inorganic Crystal Structure Database (ICSD)—Present and Future, Crystallogr. Rev., 2014, 10, 17–22 CrossRef.
- G. W. Watson, E. T. Kelsey, N. H. de Leeuw, D. J. Harris and S. C. Parker, Atomistic simulation of dislocations, surfaces and interfaces in MgO, J. Chem. Soc., Faraday Trans., 1996, 92, 433 RSC.
- H. H. Hwu and J. G. Chen, Surface Chemistry of Transition Metal Carbides, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- S. Zaima, Y. Shibata, H. Adachi, C. Oshima, S. Otani, M. Aono and Y. Ishizawa, Atomic chemical composition and reactivity of the TiC(111) surface, Surf. Sci., 1985, 157, 380–392 CrossRef CAS.
- A. L. Stottlemyer, T. G. Kelly, Q. Meng and J. G. Chen, Reactions of oxygen-containing molecules on transition metal carbides: surface science insight into potential applications in catalysis and electrocatalysis, Surf. Sci. Rep., 2012, 67, 201–232 CrossRef CAS.
- R. Souda, T. Aizawa, S. Otani and Y. Ishizawa, Effects of chemical bonding on the electronic transition in low energy He+ scattering, Surf. Sci., 1990, 232, 219–227 CrossRef CAS.
- W. Hayami, R. Souda, T. Aizawa, S. Otani and Y. Ishizawa, Analysis of the NbC(111)-(√3 × √3)R30°-Al surface structure by impact-collision ion-scattering spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 13752–13758 CrossRef CAS.
- T. Aizawa, W. Hayami, R. Souda, S. Otani and Y. Ishizawa, Hydrogen adsorption on transition-metal carbide (111) surfaces, Surf. Sci., 1997, 381, 157–164 CrossRef CAS.
- P. E. Blöchl, O. Jepsen and O. K. Andersen, Improved tetrahedron method for Brillouin-zone integrations, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 16223–16233 CrossRef.
- H.-G. Yu, J. T. Muckerman and T. J. Sears, A theoretical study of the potential energy surface for the reaction OH + CO → H + CO2, Chem. Phys. Lett., 2001, 349, 547–554 CrossRef CAS.
- M. G. Quesne and S. P. De Visser, Regioselectivity of substrate hydroxylation versus halogenation by a nonheme iron(IV)-oxo complex: possibility of rearrangement pathways, J. Biol. Inorg. Chem., 2012, 17, 841–852 CrossRef CAS PubMed.
- M. G. Quesne and S. P. de Visser, Regioselectivity of substrate hydroxylation versus halogenation by a nonheme iron(IV)–oxo complex: possibility of rearrangement pathways, JBIC, J. Biol. Inorg. Chem., 2012, 17, 841–852 CrossRef CAS PubMed.
- A. Timmins, M. G. Quesne, T. Borowski and S. P. de Visser, Group Transfer to an Aliphatic Bond: A Biomimetic Study Inspired by Nonheme Iron Halogenases, ACS Catal., 2018, 8685–8698 CrossRef CAS.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. De Visser, Origin of the Enhanced Reactivity of μ-Nitrido-Bridged Diiron(IV)-Oxo Porphyrinoid Complexes over Cytochrome P450 Compound i, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- G. Wulff, XXV. Zur Frage der Geschwindigkeit des Wachsthums und der Auflösung der Krystallflächen, Z. Krist. – Cryst. Mater., 1901, 34, 449–530 CAS.
- D. Santos-Carballal, A. Roldan, R. Grau-Crespo and N. H. de Leeuw, A DFT study of the structures, stabilities and redox behaviour of the major surfaces of magnetite Fe3O4, Phys. Chem. Chem. Phys., 2014, 16, 21082–21097 RSC.
- A. Roldán, J. M. Ricart and F. Illas, Origin of the size dependence of Au nanoparticles toward molecular oxygen dissociation, Theor. Chem. Acc., 2011, 128, 675–681 Search PubMed.
- S. F. Viñes, J. A. Rodriguez, S. Posada-Pérez, P. J. Ramírez, R. A. Gutiérrez, D. J. Stacchiola, F. Viñes, P. Liu and F. Illas, The conversion of CO2 to methanol on orthorhombic β-Mo2C and Cu/β-Mo2C catalysts: mechanism for admetal induced change in the selectivity and activity, Catal. Sci. Technol., 2014, 6, 6766–6777 Search PubMed.
- A. Vojvodic, A. Hellman, C. Ruberto and B. I. Lundqvist, From electronic structure to catalytic activity: A single descriptor for adsorption and reactivity on transition-metal carbides, Phys. Rev. Lett., 2009, 103, 146103 CrossRef CAS PubMed.
- A. Vojvodic, A. Hellman, C. Ruberto and B. I. Lundqvist, From electronic structure to catalytic activity: A single descriptor for adsorption and reactivity on transition-metal carbides, 2009 Search PubMed.
- L. Zhang, X. Wang, M. Chen and Q.-Z. Qin, Activation of CO2 by Zr atom. Matrix-isolation FTIR spectroscopy and density functional studies, Chem. Phys., 2000, 254, 231–238 CrossRef CAS.
- S. Di Tommaso, T. Marino, F. Rondinelli, N. Russo and M. Toscano, CO2 Activation by Nb+ and NbO+ in the Gas Phase. A Case of Two-State Reactivity Process, J. Chem. Theory Comput., 2007, 3, 811–815 CrossRef CAS PubMed.
- J. A. Rodriguez, J. Evans, L. Feria, A. B. Vidal, P. Liu, K. Nakamura and F. Illas, CO2 hydrogenation on Au/TiC, Cu/TiC, and Ni/TiC catalysts: production of CO, methanol, and methane, J. Catal., 2013, 307, 162–169 CrossRef CAS.
- A. Roldan, N. H. de Leeuw, C. R. A. Catlow, M. G. Quesne and F. Silveri, Hydrogen adsorption on transition metal carbides: a DFT study, Phys. Chem. Chem. Phys. 10.1039/c8cp05975f.
- K. E. Tan, M. W. Finnis, A. P. Horsfield and A. P. Sutton, Why TiC(111) is observed to be Ti terminated, Surf. Sci., 1996, 348, 49–54 CrossRef CAS.
- X. Zhang, S. Yan, R. D. Tyagi, R. Y. Surampalli and T. C. Zhang, Carbon Capture and Storage, American Society of Civil Engineers, Reston, VA, 2015, pp. 401–419 Search PubMed.
- Y. Soong, D. L. Fauth, B. H. Howard, J. R. Jones, D. K. Harrison, A. L. Goodman, M. L. Gray and E. A. Frommell, CO2 sequestration with brine solution and fly ashes, Energy Convers. Manage., 2006, 47, 1676–1685 CrossRef CAS.
- H. Wijnja and C. P. Schulthess, Effect of Carbonate on the Adsorption of Selenate and Sulfate on Goethite, Soil Sci. Soc. Am. J., 2002, 66, 1190 CrossRef CAS.
- W. N. R. Wan Isahak, Z. A. Che Ramli, M. W. Mohamed Hisham and M. A. Yarmo, The formation of a series of carbonates from carbon dioxide: capturing and utilisation, Renewable Sustainable Energy Rev., 2015, 47, 93–106 CrossRef CAS.
- G. H. Rau and K. Caldeira, Enhanced carbonate dissolution: a means of sequestering waste CO2 as ocean bicarbonate, Energy Convers. Manage., 1999, 40, 1803–1813 CrossRef CAS.
- L. Liu, M. Lin, Z. Liu, H. Sun and X. Zhao, Density functional theory study of CO2 and H2O adsorption on a monoclinic WO3(001) surface, Chem. Res. Chin. Univ., 2017, 33, 255–260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cp00924h |
| This journal is © the Owner Societies 2019 |