
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular insight into carboxylic acid–alkali metal cations interactions: reversed affinities and ion-pair formation revealed by non-linear optics and simulations†
Adrien
Sthoer‡
 a,
Jana
Hladílková‡
b,
Mikael
Lund
b and
Eric
Tyrode
*a
a,
Jana
Hladílková‡
b,
Mikael
Lund
b and
Eric
Tyrode
*a
aDepartment of Chemistry, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden. E-mail: tyrode@kth.se
bDivision of Theoretical Chemistry, Lund University, P.O.B. 124, SE-22100 Lund, Sweden
First published on 10th May 2019
Abstract
Specific interactions between the carboxylic acid moiety and the monovalent salts CsCl, NaCl, and LiCl, have been investigated in Langmuir monolayers using vibrational sum frequency spectroscopy (VSFS) and complemented with coarse grained and all-atom molecular dynamics simulations. By exploiting VSFS's intrinsic surface specificity, an emphasis was made on targeting headgroup vibrations of both its charged and uncharged forms as well as water molecules in the interfacial layer. The degree of deprotonation of the monolayer as a function of cation concentration and pH was experimentally determined and theoretically rationalized. Starting from 100 mM, the surface charge was overestimated by the Gouy–Chapman model and varied depending on the identity of the cation, highlighting the appearance of ion specific effects. Agreement could be found using a modified Poisson–Boltzmann model that takes into account steric effects, with a fitted effective ion-size compatible with the hydrated ion diameters. The relative affinity of the cations to the carboxylic acid moiety was pH dependent: at pH 4.5 they arranged in the order Cs+ > Na+ > Li+, but fully reversed (Li+ > Na+ > Cs+) at pH 9. Simulations yielded microscopic insight into the origin of this behavior, with the cations showing contrasting interaction preferences for either the uncharged carboxylic acid or the charged carboxylate. Sum frequency spectra also provided evidence that all cations remained hydrated when interacting with the charged headgroup, forming solvent-separated or solvent-shared ion pairs. However, for the specific case of 1 M Li+ at pH 9, contact ion pairs were formed. Finally, the remarkable effect of trace metal multivalent cations in the interpretation of experiments is briefly discussed. The results provide exciting new insights into the complex interactions of alkali metal cations with the biophysically relevant carboxylic acid moiety.
1. Introduction
Specific ion effects play a key role in a wide range of physical phenomena, stretching from the systematic variation in the surface tension of simple salts solutions to the structure and function of complex biological surfaces.1–3 Despite being studied for over a century, starting with the work of Hofmeister4 who ranked the relative efficiency of different salts depending on their ability to precipitate proteins from solution, a comprehensive overarching molecular explanation remains elusive.5 Earlier explanations6 in terms of the ability of ions to affect the bulk structure of surrounding water molecules have been recently put into question.7,8 Instead, direct ion–surface interactions together with the associated and concurrent disruption of the interfacial water structure, are currently considered to be the underlying cause of ion-specific phenomena.1,5,9,10 To challenge and extend these theories, direct experimental observations of the molecular structure from ions and water in the interfacial region are required. Here, we use surface-sensitive experimental and simulations tools to address the specific interaction of alkali metal cations with the carboxylic acid moiety, an essential constituent of amino acids having an obvious biophysical relevance.The molecular interactions of alkali cations with the carboxylic acid groups, both in the bulk and at surfaces, have been the subject of a number of recent experimental studies. For instance, X-ray absorption spectroscopy (XAS),11,12 has been used to determine the relative interaction strength of the monovalent cations with the acetate in bulk solution. Cations were found to order in the sequence Li+ > Na+ ≥ K+ when considering the carbon K-edge,12 yet as Na+ > Li+ > K+ when targeting the oxygen K-edge,11 with no apparent explanation for the discrepancy.13 Polarization-resolved femtosecond mid-infrared spectroscopy,14,15 has also been used to classify cations in the bulk by measuring the dynamics of slow-down water molecules in formate salts, resulting in the sequence Na+ > Li+ > K+ > Cs+. On surfaces, vibrational sum frequency spectroscopy (VSFS) studies on fatty acid monolayers, concluded that K+ binds more strongly than Na+,16 while at relatively low concentrations (i.e., 1 mM salt) no discernible differences were reported between the monovalent cations with Li+ ≈ Na+ ≈ K+ ≈ Rb+.17 These apparent contradictions are not only limited to their relative ordering, but also on the type of interactions, in particular, the ion-pairs18 formed between the cations and the carboxylic acid moiety, which include solvent-separated (2SIP), solvent-shared (SIP), contact-ion pairs (CIP), and/or bound complexes.11,15–17,19 A potential explanation for this contrasting behavior, at least in terms of ion affinity, is hinted by simulations from Schwierz et al.,20 which suggest that the relative ordering of the cations depends on the concentration of ions as well as on solution pH. At certain conditions, even a complete reversal of the Hofmeister ordering would be expected (from Li+ > Na+ > Cs+ to Cs+ > Na+ > Li+).20,21 To our knowledge, however, there is currently no systematic experimental evidence that substantiates this claim.
Langmuir monolayers of fatty acid molecules offer practical advantages for the study of cations–carboxylic acid interactions, as they can readily be characterized using various experimental approaches. Firstly, surface pressure isotherms provide macroscopic 2D phase information of the monolayer, and in particular, a detailed account of the fatty acid's average molecular surface area.22–24 The molecular conformation and packing arrangement of the alkyl chains in the monolayer, has been determined, for example, using grazing incidence X-ray diffraction,25 complemented with polarization-modulation infrared reflection absorption spectroscopy (PM-IRRAS)26 and sum frequency vibrational spectroscopy.16,17,27 On the other hand, the specific carboxylic headgroup–cation interactions, have been preferentially examined using VSFS,16,17,28–31 where, in contrast to PM-IRRAS,26 background subtraction is not needed. With its intrinsic surface specificity, VSFS additionally provides structural information about the interfacial water near the fatty acid headgroup.17,28,32,33 However, the majority of these studies have exclusively focused on Na+ as monovalent cation and/or at low salt concentrations. Furthermore, the influence of trace amounts of polyvalent ions in the interpretation of the results, particularly when increasing the amount of salt in solution, has been largely neglected. From the computational side, the few studies on fatty acid Langmuir monolayers reported have been mainly concerned in understanding the 2D phase behavior and the packing of the alkyl chain,34 and also the interfacial water hydrating the headgroup.35,36 The interactions of selected monovalent cations with fatty acid Langmuir monolayers had only been explicitly considered in the work by Schwierz et al.20
In this paper, we employ VSFS to investigate the specific interactions of alkali metal chloride salts with the carboxylic acid group in an arachidic acid Langmuir monolayer. From the analysis of the carboxylate vibrational features, the absolute charge of the monolayer is explicitly determined, both as a function of salt concentration and pH. The concentration range chosen, starting from 100 mM and up to 1 M, covers the emergence of cation specific effects. We show that the quantitative determination of the surface charge allows for direct comparison, not only to theoretical models of charged interfaces, such as Gouy–Chapman or suitable extensions that take into account the finite size of the ions, but also to coarse grained and all-atom MD simulations, with the former capturing the dynamics of the fatty acid dissociation, while the latter provides a direct atomistic and molecular insight. VSFS reports on additional molecular aspects that we have contrasted with simulation results to validate, as well as to determine potential limitations of current computational approaches. We investigate the orientation of the uncharged carboxylic acid moiety upon charging of the monolayer as well as the type of ion pairs formed between the respective cations and the charged carboxylate at various salt concentrations and pH. Interestingly, we provide direct experimental evidence for the reversion of the relative interaction strength of the different cations when increasing the pH of the subphase. VSFS also provides information on interfacial water molecules, including those found in the diffused double layer and in closer proximity to the fatty acid monolayer. Finally, we briefly discussed the remarkable effect that trace amounts of polyvalent ions could have in the interpretation of the results if no active measures are taken to remove them.
2. Experimental section and simulation methods
2.1. Materials
NaCl (99.999% trace metals basis), CsCl (99.999% trace metals basis), LiCl·H2O (99.99% trace metals basis), NaOH (99.99% trace metals basis), ethylenediaminetetraacetic acid (EDTA 99.995% trace metals basis), eicosanoic-d39 acid (97%, dAA for deuterated arachidic acid), eicosanoic acid (99%, AA for arachidic acid), butanoic acid (>99%), and chloroform (anhydrous grade, stabilized with ethanol) were obtained from Sigma-Aldrich. NaCl and CsCl were baked at 500 °C for 1 h, and slowly cooled down to eliminate any traces of organic compounds. The remaining compounds were used as received. Nonetheless, in order to remove potential surface active contaminants, the surface of the salt solutions were systematically aspirated multiple times before use, with adsorption times varying up to 30 minutes before aspiration. Ultrapure water was obtained from an Integral 15 Millipore system featuring a constant conductivity (18.2 MΩ cm) and low total organic content (<3 ppb). Glassware was cleaned by a three-step sonication procedure: first in ethanol, followed by Deconex (commercial alkaline agent, Borer Chemie) and finally in ultrapure water (glassware was rinsed at least 10 times in ultrapure water between each step).2.2. Polyvalent ion trace contaminants
As elaborated in the results section below and ESI† (Section S1), trace amounts of polyvalent cations present in the high purity salts used, were sufficient to compete with the monovalent ions for adsorption sites at the fatty acid monolayer and radically change its behavior. This was particularly true for the concentrated salt solutions used in these studies. To remove the polyvalent ions, EDTA in a concentration ranging between 0.5 to 20.0 μM was added to the solutions. The pH was then adjusted when needed with a dilute solution of NaOH. We note that EDTA is not surface active in the concentrations used, as evidenced by the constant surface tension relative to water, of solutions of even 1 mM EDTA. Further, no spectral VSF features were detected in the CH and carbonyl stretching regions in neither pure water nor concentrated salt solutions, consistent with the absence of EDTA from the surface (see Section S1 in ESI†).2.3. Fatty acid Langmuir monolayers
The solutions of arachidic acid (AA and dAA) were prepared by diluting 10 mg of the fatty acid in 10 mL of chloroform. 20 μL of the solution were then spread on the surface of the subphase placed in a Langmuir trough from KSV NIMA (195 mm length, 50 mm width, and 4 mm in depth). The trough and compression barriers were made of Teflon and Delrin, respectively. Before compression, a waiting time of at least 10 minutes was observed to ensure full solvent evaporation. The monolayer compression rate was 10 mm per minute, and the surface pressure was measured using a paper Wilhelmy plate, 10 mm wide. All VSF spectroscopy measurements were carried out at a constant surface pressure and temperature of 20 mN m−1, and 22.0 ± 0.5 °C, respectively. As the trough surface was partly exposed to atmospheric CO2, the pH reported have an error of maximum ±0.2 pH units.2.4. VSF spectrometer
The femtosecond VSF spectrometer has been described in detail elsewhere.37 Briefly, it consists of a Ti:light oscillator and a Integra-HE amplifier (Amplitude, France) that generates 90 fs/1 kHz/805 nm pulses with a total output power of 6 W. The tuneable IR beam is produced by a traveling optical wave parametric amplifier (HE-TOPAS-C, Light Conversion, Lithuania), pumped by ∼4.5 W of the output power of the Integra amplifier. The remaining fraction of the output power of the Integra is converted in a home-built beam shaper to a bandwidth-tunable picosecond pulse. The 805 nm “visible” and tunable IR pulses are directed to the sample position in a co-propagating geometry, with the angles of incidence set to 70° and 55°, respectively. The laser beams were gently focused at the sample position using convex lenses with focal lengths of 1 m (visible) and 30 cm (IR). The SF signal collection optics display a high degree of automation that accommodate for changes in the angle of emission of the SF beam with IR frequency, allowing measurements in broad spectral regions. The SF signal is finally detected using a spectrometer (Shamrock SR202i-B, Andor, Ireland) and EM-CCD camera (Newton, Andor, Ireland). The power at the sample position for the IR and “visible” beam was typically set to 5 mW and ∼30 mW, respectively. Spectra were recorded in the polarization combinations SSP, SPS, and PPP, with a spectral resolution <3 cm−1, and normalized by the non-resonant SF response from a gold surface.37The spectra were fitted by convoluted Lorentzian and Gaussian line shapes of the form presented in eqn (1), which accounts for homogeneous (Lorentzian line shapes) and inhomogeneous broadening (Gaussian line shape), as well as the complex interference between neighboring bands.38
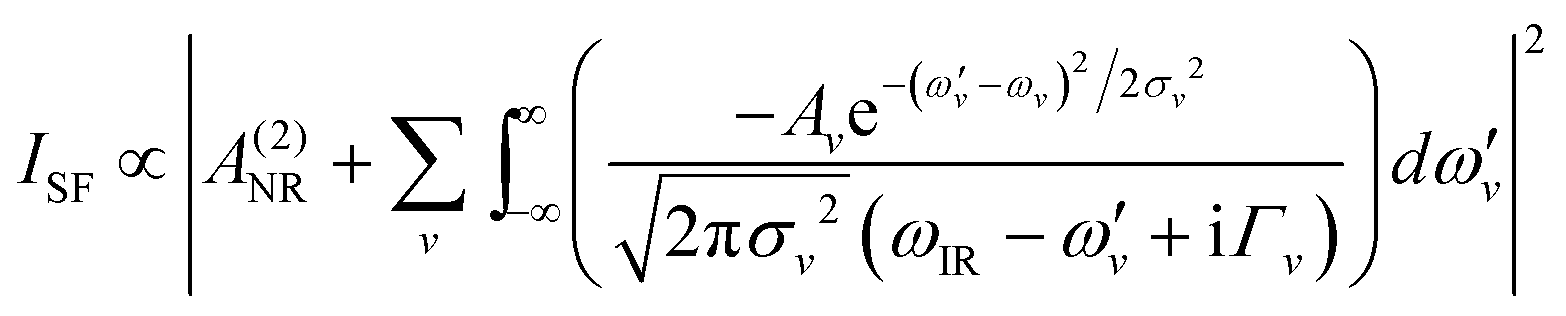 | (1) |
2.5. Gouy–Chapman and modified Poisson Boltzmann models
The dependence of the degree of deprotonation of the fatty acid monolayer on the bulk electrolyte concentration, can be estimated using the Gouy–Chapman model.23 The starting point is the acid–base equilibria of the acid at the surface (RCOOH ↔ RCOO− + H+), which relates the dissociation constant Ka, the degree of deprotonation α, the bulk hydronium concentration [H+]∞, and the surface potential, ψ0. | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolyte, in:
:1 electrolyte, in: | (3) |
 | (4) |
2.6. MD simulations
Classical all-atom molecular dynamics (MD) simulations were performed in a slab geometry with two carboxylic acid monolayers forming two water–vapour interfaces with 1 M salt solution of LiCl, NaCl, or CsCl in between (see Fig. 1). This setting allows for efficient configurational sampling as well as convergence checks. One monolayer consists of 116 arachidic acids (C20H41COOH) for a fully uncharged surface, or a mixture of arachidic acids and arachidates (C20H41COO−) for charged surfaces. 30 and 45% of arachidic acids were manually chosen and swapped to arachidates in order to have as starting point, an evenly distributed charge over the monolayer. The all-atom OPLS,42 as well as the charmm3643 force fields were used for arachidic acid and arachidate. The solution consists of 8350 SPC/E water molecules44 and 232 ions. Two sets of ion force fields were used for comparison. The first was developed to match activity coefficients of alkali chlorides and alkali acetates,45 while the latter is based on a charge scaling approach to match neutron scattering data.46,47 The simulation box has dimensions of 4.8 × 4.8 × 45 nm with periodic boundary conditions applied in all dimensions. This condition corresponds to the experimentally established value of 20 Å2 area per molecule (AM) for all salt solutions. We prepared the system by prolongation of carbon chains of palmitic acids of the previously published and converged structures.48 We followed the same simulation protocol with significantly prolonged productions runs; we equilibrated and propagated all systems for 36 and 500 ns, respectively, using the velocity rescale thermostat49 with a 2 fs time step. Electrostatic forces were calculated using the smooth Particle-Mesh Ewald method50 with a real-space cut-off of 1.0 nm for Coulomb and Lennard-Jonnes (LJ) interactions. Bonds containing hydrogens were constrained using the LINCS algorithm.51 All MD simulations were performed with the Gromacs simulation package (version 4.6.5).52 The standard Gromacs package tools,52 the VMD program package,53 as well as in-house scripts were used for data analysis. | ||
| Fig. 1 Left: Example of an all-atom molecular dynamics simulation box for a fully uncharged monolayer of arachidic acids. Arachidic acid (cyan – carbon, white – hydrogen, and red – oxygen atoms) with ions (sodium in green and chloride in orange) are depicted using van der Waals diameters. Water molecules are shown as red dots. Right: Example of a coarse grained, Grand Canonical Monte Carlo simulation box. Surfaces consist of a mixture of arachidic acid (grey) and arachidates (red). The amount of salt ions in the solution (sodium in green and chloride in orange) depends on pH and salt chemical potential. | ||
2.7. Coarse grained Monte Carlo simulations
Constant pH, coarse grained, grand canonical Monte Carlo (GCMC) simulations were performed using Faunus54 in a slab box 4.8 × 4.8 × 55 nm with two monolayers, similarly to the MD simulations (see Fig. 1). At low salt concentrations (1–50 mM), the box was prolonged in the z-direction (1550 nm) to keep at least one ion constantly present in the solution. Ions are treated explicitly while water is considered implicitly by a dielectric continuum with a relative dielectric constant, εr = 78. Ionic LJ parameters, shown in Table S3 (ESI†) were derived to reproduce experimental activity coefficients55 for all three salt solutions (LiCl, NaCl, CsCl) at 100 mM, 500 mM and 1 M concentrations. The monolayer is made of 115 COOH and COO− moieties, both coarse grained to a single bead, representing the arachidic acid in both protonation states (matching the experimental AM). The cation–COOH interactions follow the MD results (excess of cations at the surface) while the cation–COO− interactions result in the matching experimental values of activity coefficients of LiAc, NaAc, and CsAc solutions (all parameters used are summarized in Table S3 in ESI†). The MC Hamiltonian is composed of a COO−/COOH titration term,56 Coulomb and Lennard-Jones interactions. There were 1 million steps per simulation, each step can be (i) translation of ions/COOH/COO−, (ii) titration of the acid COOH ↔ COO− (pKa = 5.0), or (iii) salt insertion/deletion to/from the solution. As a result, an average protonation state of the monolayer is calculated according to the cation type, pH, and salt concentration. The obtained ensemble averages are directly comparable to the Sum Frequency experiments as developed below.3. Results and discussion
3.1. Description of the model system
Monomolecular insoluble films of arachidic acid were used as a framework for studying the interactions of the carboxylic acid moiety with selected monovalent cations. Besides 2D structural phase transition information, a key advantage of using Langmuir monolayers, is that they provide the average area per molecule, simplifying the interpretation of the VSF spectra. The compression isotherm at 22 °C of deuterated arachidic acid (dAA) on water is shown in the inset of Fig. 2a. The isotherm is characterized by the absence of a liquid expanded phase, with the direct coexistence of a gas phase with a tilted condensed phase at areas per molecules larger than ∼24 Å2. The tilted condensed phase extends up to ∼27 mN m−1, followed by an untilted condensed phase at higher surface pressures.17,57 Interestingly, the compression isotherms of dAA at the various salt and pH conditions used here, remained, within error, the same as that on water (see inset in Fig. 2a and Section S5 in ESI†). However, they could significantly vary depending on the nature, temperature, and pH of the subphase, as described in numerous studies.17,26,57–61 All experiments in this work were carried out at a constant surface pressure of 20 mN m−1, corresponding to an average area per AA molecule of ∼20 Å2 (tilted condensed phase). | ||
| Fig. 2 VSF spectra of a deuterated arachidic acid (dAA) Langmuir monolayer, on (a) LiCl subphase solution (b) NaCl subphase solution, and (c) CsCl subphase solution, collected under the SSP polarisation combination for different salt concentrations and in the presence of 20 μM of EDTA. The pH was adjusted to 6.0 with a dilute solution of NaOH to compensate for small change in pH caused by the added EDTA. (d) Deprotonation of the fatty acid is compared with the prediction of the Gouy–Chapman (G–C) model with a pKa of 5.0, and a modified Poisson–Boltzmann (MPB) model, accounting for steric effects (a = 7.5 Å). | ||
VSFS provides a molecular insight into the monolayer, in particular the conformational order of the alkyl chains, the degree of deprotonation of the polar head group, and structural information of the interfacial water molecules. The SF spectra of an AA (and dAA) monolayer on water, as well as its dependence on both the pH and the presence of monovalent ions at concentrations below 100 mM, have been thoroughly studied in a recent publication,17 thus only the main features relevant for higher salt concentrations will be highlighted here.
Consistent with the high packing density of the monolayer, the spectral signatures of the alkyl chains are dominated by the modes of the terminal methyl group. For deuterated chains these are the CD3 symmetric stretch (r+) and its Fermi resonance (rFR+), as well as the CD3 asymmetric stretch (r−) centered at ∼2075 cm−1, ∼2135 cm−1, ∼2217 cm−1, respectively (see Fig. 2a). The lack of discernible features linked to methylene modes, and in particular, the CD2 asymmetric stretch (d−) expected at ∼2200 cm−1 in the PPP and SPS polarization spectra (see Section S6 in ESI†), indicate that the alkyl chains have a high degree of conformational order, with methylene groups arranged mainly in an all-trans configuration.62 Deuterating the alkyl chain offers the advantage of shifting the methyl and methylene bending modes to lower frequencies which otherwise overlap with the carboxylate stretches.17 The latter, together with the carbonyl stretch (vC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at ∼1720 cm−1 are useful for determining the state of association of the head group. By varying the pH of the subphase, the apparent pKa of the arachidic acid monolayer was experimentally determined to be ∼10.8 (when only the pH determining ions are present in solution),17 that is, almost six orders of magnitude higher than the “thermodynamic” pKa, and interestingly, in agreement with the value predicted by the Gouy–Chapman theory.17,32 The high apparent pKa is a consequence of the monolayer's negative surface potential and resulting increase, relative to the bulk, of the surface hydronium ion concentration.17,63
O) at ∼1720 cm−1 are useful for determining the state of association of the head group. By varying the pH of the subphase, the apparent pKa of the arachidic acid monolayer was experimentally determined to be ∼10.8 (when only the pH determining ions are present in solution),17 that is, almost six orders of magnitude higher than the “thermodynamic” pKa, and interestingly, in agreement with the value predicted by the Gouy–Chapman theory.17,32 The high apparent pKa is a consequence of the monolayer's negative surface potential and resulting increase, relative to the bulk, of the surface hydronium ion concentration.17,63
3.2. Specific monovalent cation interactions: concentration dependence
The SF spectra of a dAA monolayer on an aqueous subphase containing LiCl, NaCl, and CsCl at three different concentrations and pH 6.0 are shown in Fig. 2. The constancy in the CD stretching modes, primarily the r+, rFR+, and the r− modes (>2000 cm−1), clearly indicates that for all cases the alkyl chains remain unperturbed with a high degree of conformational order. This scenario is supported by the lack of significant changes in the Langmuir compression isotherms (see Section S5 in ESI†). In Fig. 2a–c, the spectral signatures from the headgroup in both its charged and uncharged forms, specifically the symmetric carboxylate stretch (vsCOO−) at ∼1408 cm−1 and carbonyl (vCO) at 1720 cm−1, can be clearly observed.
Interestingly, the center wavelength of the carboxylate stretch remains constant for all salts and concentrations explored in Fig. 2. Following previous measurements performed at much lower ionic strengths and various pH conditions,16,17,32,33 the vsCOO− band at ∼1408 cm−1 is assigned to the hydrated carboxylate or following Marcus et al. classification,18 to the solvent-separated/solvent-shared ion-pair carboxylate group. The absence of a frequency shift indicates that even at a high bulk concentration of 1 M, no contact ion pairs or complexes are formed at pH 6 between the carboxylate and the monovalent cations at the surface. However, as elaborated in a later section, this is not necessarily the case at more alkaline conditions.
The intensity of the hydrated carboxylate band can be used to estimate the percentage of deprotonation. The procedure consists in using as reference the equivalent signal from a fully charged fatty acid monolayer with the same area per molecule.17 The SF intensity depends both on the mean orientation and the number squared of contributing oscillators.64 Nonetheless, since the orientation of carboxylate group in the Langmuir monolayer remains largely independent of the area per molecule,17 the band intensity thus simply reflects a square dependence with the number of charged species. The degree of deprotonation of the monolayers for the three alkali metal chloride salts at pH of 6.0 are summarized in Fig. 2d. The degree of deprotonation up to 50 mM of NaCl in the subphase has previously been shown to be consistent with the values predicted by Gouy–Chapman theory (open symbols in Fig. 2d), and independent of the nature of either the monovalent cation or anion for the specific case of 1 mM salt (see Experimental section for details of the model).17 However, already at 100 mM the degree of deprotonation clearly departs from the Gouy–Chapman prediction and differs for each cation, highlighting the emergence of ion specific effects. At 1 M the differences, although small, remain apparent. The degree of deprotonation of the monolayer at pH 6 follows the order: Cs+ ≥ Na+ > Li+, corresponding to ∼24% for Cs+ and only ∼19% for Li+. At high salt concentrations, G–C largely overestimates the experimentally determined surface charge, stemming partly from the neglect of finite ion-sizes. Fig. 2d shows that better agreement is obtained using a modified Poisson–Boltzmann (MPB) model that takes into account steric effects using a fitting parameter, a, linked to the effective ion size (a ∼ 7.5 Å for Cs+ and Na+ and ∼9 Å for Li+, which are roughly comparable to the hydrated ion diameters. See Experimental section).40 As developed in a later section, the MPB model also proves useful for estimating the surface potential at high ionic strengths. Nevertheless, the phenomenological parameter a remains a fitting parameter that likely encapsulate multiple competing effects beyond the effective ion size, including ion–ion correlations and other ion specific effects.40 We note that other MPB models have been proposed65 to specifically account for either the ion-dependent dielectric response,66 dispersion interactions,67 or water molecules as explicit point-like dipoles.68
Although the monoatomic cations, lacking covalent bonds, cannot directly be targeted with VSF spectroscopy, useful information can be extracted from their influence on the carboxylic acid headgroups, and also on interfacial water molecules as discussed later. Deprotonation with increasing salt concentration results from the presence of cations at the interface that screens electrostatic interactions and effectively reduce the surface potential. Consequently, the degree of deprotonation could be directly linked to the amount of ions in the interfacial region. This would indicate that at pH 6, Li+ ions interact less favorably with the carboxylic acid monolayer when compared to Cs+ and Na+. However, as will be shown in the pH dependent studies in combination with molecular simulations in Section 3.3, the interactions of ions with both the uncharged and charged headgroups influence the total number and relative amount of ions at the surface.
In contrast to the carboxylate band, a closer examination of the carbonyl stretch (vCO) at 1720 cm−1 shows no apparent intensity changes when increasing the concentration of salt (Fig. 2a–c). This observation may appear at odds with the fact that an increased amount of charged carboxylate groups in the monolayer necessarily involves a concomitant decrease in the number of neutral carboxylic acid moieties. Thus, the lack of changes in the SF intensity indicates that the orientation of the remaining CO groups must vary as the monolayer deprotonates. Had the orientation remained constant, the decrease in the vCO band should have been particularly noticeable. Due to the expected squared dependence of the SF signal with the number of oscillators, a reduction of 16% in the number of carboxylic groups should have represented a 30% decrease in the SF signal.
C–OH) is observed as a broadband centered at ∼1310 cm−1 in the SSP spectrum,17,69 while the carbonyl stretch at ∼1720 cm−1 is resolved in all three polarization combinations. In the PPP spectrum the center position of vCO appears blue-shifted due to interference with the non-resonant background and the water bending mode at ∼1650 cm−1 (Fig. 3a), the latter also observed as a shoulder in the SSP spectrum.17 Upon exchanging the subphase to 100 mM NaCl (Fig. 3b), the hydrated carboxylate at ∼1408 cm−1 (vs![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) COO−) band becomes evident not only in the SSP, but also in the PPP polarization combination, as the monolayer partly deprotonates. Importantly, the features linked to the remaining uncharged moieties, which constitute still ∼85% of the surface molecules, overall decrease but at contrasting rates: the vsC–OH sharply drops to less than half of the intensity in water, while the carbonyl band vCO decreases by ∼25% in SSP, ∼50% SPS, and almost completely vanishes in the PPP spectrum (Fig. 3b). At higher salt concentrations (Fig. 3c) as the monolayer further deprotonates, the broad vsC–OH at ∼1310 cm−1 decreases to just ∼5–10% of the original intensity in water, while the vCO at ∼1720 cm−1 is no longer observable in PPP. It, however, remains mostly constant in the SSP polarization combination, and only slightly decreases in the SPS spectrum.
COO−) band becomes evident not only in the SSP, but also in the PPP polarization combination, as the monolayer partly deprotonates. Importantly, the features linked to the remaining uncharged moieties, which constitute still ∼85% of the surface molecules, overall decrease but at contrasting rates: the vsC–OH sharply drops to less than half of the intensity in water, while the carbonyl band vCO decreases by ∼25% in SSP, ∼50% SPS, and almost completely vanishes in the PPP spectrum (Fig. 3b). At higher salt concentrations (Fig. 3c) as the monolayer further deprotonates, the broad vsC–OH at ∼1310 cm−1 decreases to just ∼5–10% of the original intensity in water, while the vCO at ∼1720 cm−1 is no longer observable in PPP. It, however, remains mostly constant in the SSP polarization combination, and only slightly decreases in the SPS spectrum.
 | ||
| Fig. 3 VSF spectra collected in the polarization combinations SSP, SPS, and PPP of a dAA Langmuir monolayer with (a) pure water, (b) 100 mM, and (c) 500 mM NaCl in the subphase at a pH of 6 (EDTA 4 μM). (d) Theoretical curves showing the expected variations of the SF intensity of the vsCO mode as a function of carbonyl tilt angle (θCO) from the surface normal for the three polarization combinations, assuming a delta distribution (solid lines) and Gaussian distribution with standard variation of 15° (dashed lines). The ratio SSP/SPS is also shown in (d) (right axis). Shaded areas centered at θCO = 75° and 60°, correspond to ∼0% and ∼20% deprotonation, respectively. See text and Section S7 of ESI† for details. | ||
As mentioned above, the magnitude of the SF signal typically depends on both the average orientation and the number density of interfacial molecules.70 For the arachidic acid Langmuir monolayer, the total number of molecules, as well as the relative proportion of charged species, are known with high accuracy. Thus, the observed behavior necessarily results from a change in the orientation of the neutral carboxylic acid group, as confirmed by the contrasting intensity variations observed at different polarization combinations (Fig. 3). A deeper insight into these changes can be obtained by performing an orientational analysis.71–75 The procedure consists in comparing the experimentally determined amplitudes under different polarization combinations (i.e., SSP, PPP, and SPS), with theoretical curves. These curves model changes of spectral amplitudes and ratios as a function of the orientation of the functional group under consideration; i.e., the tilt of the carbonyl group (CO).
The orientational analysis of the carbonyl group has already been the subject of a detailed study,76 and here we follow a similar approach with one distinction: the ratio between the two independent non-zero hyperpolarizability tensor elements, β(2)aac/β(2)ccc = 0.27 ± 0.02, was estimated from the experimentally determined Raman depolarization ratio (see Sections S2 and S7 in ESI† for details). In short, the transformation of the hyperpolarizability elements to laboratory coordinates (x, y, z) using an Euler rotation matrix, allows us to theoretically determine the variation of the SF intensity as a function of the center tilt angle and assumed angular distribution (i.e., delta or Gaussian). The theoretical curves depicting the expected variations of the SF intensity for the polarization combinations SSP, SPS, and PPP are shown in Fig. 3d (left axis) assuming both delta (solid curves) and a Gaussian distribution (dashed curves correspond to a standard deviation of 15°). In the same figure, the SSP/SPS intensity ratio is also shown for reference (Fig. 3d – right axis).
Comparing the theoretically calculated curves and the experimentally determined fitted amplitudes (see Section S8 in ESI† for details), indicates that in pure water the carbonyl lies, on average, relatively close to the surface plane with a tilt angle θCO of ∼75° ± 5. The SSP intensity is 1.6± 0.2 higher than in SPS, and the PPP contribution is small yet non-zero (Fig. 3a). With the addition of salt and the partial deprotonation of the monolayer, the orientation of the remaining carbonyl groups shifts to angles closer to the surface normal. The estimated θCO center values for 100 mM and 500 mM are ∼65° ± 5, ∼60° ± 5, respectively. The angular shift is consistent with an increase of the SSP/SPS intensity ratio (from ∼1.6 toward ∼2.5) and importantly, explains the apparent constancy of the vCO in the SSP spectra with increasing salt (Fig. 2). The expected increase in absolute intensity in the SSP and SPS polarization combination upon a change of orientation of the carbonyl tilt from ∼75° to ∼60° (Fig. 3d), compensates for the decrease in the number of neutral carboxylic acid moieties with deprotonation (∼85% at 100 mM and ∼80% at 500 mM NaCl). The shift is also consistent with the disappearance of the vCO in the PPP spectra. From the values of θCO, the tilt angle of the accompanying single C–O bond of the carboxylic acid headgroup can be estimated to be ∼45° in pure water, shifting closer to the surface plane (i.e., θC–OH ∼60°) upon charging of the monolayer. This change in orientation towards higher angles reinforces the effect of decreasing numbers, explaining the fast decrease of the vsC–OH band observed with the addition of NaCl (Fig. 3a–c). It is worth noting that the headgroup orientation in these Langmuir monolayers is significantly different from that observed in shorter chain carboxylic acids at the liquid/vapor interface,76,77 an effect most likely associated to the packing constraints imposed by the long alkyl chains.
To obtain atomistic details on the measured orientational angles, all-atom MD simulations were performed using two different force fields: all-atom OPLS and charmm36. The OPLS force field, which yields relatively flexible acid molecules, was used to determine the CO orientation of the uncharged acid moieties in monolayers with three different degrees of deprotonation: 0% (fully uncharged), 30% and 45%. In qualitative agreement with the experimental results, the tilt angle for the fully uncharged case was found to be close to the surface plane (θCO centered at 83°), shifting to lower tilt angles upon deprotonation (θCO = 78° and θCO = 74° for 30% and 45% charge, respectively). Consistent with the SF spectra in the CD stretching region, the change in orientation of the headgroup has minimal influence on the tilt angle of the monolayer alkyl chain. This was determined by measuring the angle between the surface normal and the vector between the last and the first carbon of the arachidic acid molecules in the monolayer (see inset in Fig. 4). The θC20–C1 varied from 21.8° when fully uncharged, to 20.6° at 45% deprotonation. Upon deprotonation, a smaller peak around 30° arises in the distribution of tilt angles for the CO bond, indicating a higher flexibility of the head group, due to rotation around the C1–C2 axis. However, neither an orientation of θCO ∼ 30° nor the large distribution of orientational angles suggested by the molecular simulations in Fig. 4, are compatible with the SF experimental data. The former would have implied a significant increase of the vCO at ∼1720 cm−1 in the PPP spectra (Fig. 3d), and the latter an overall decrease of the signal in all polarization combinations. Simulations using the alternative charmm36 force field that yields stiffer carboxylic acid molecules, resulted in distributions with the absence of the peak at 30°. Further, charmm36 also predicts an orientation shift of the carbonyl group towards lower tilt angles upon deprotonation; however, the predicted shift is limited to <2° (see Section S4 in ESI†). These findings indicate that none of the force fields fully match the experimental conditions to generate quantitative predictions (the OPLS force field generates too flexible monolayers while in charmm36 the headgroups are too stiff).
 | ||
| Fig. 4 Orientation of the carbonyl group of the arachidic acid as a function of deprotonation of the Langmuir monolayer determined from all-atom MD simulations using the OPLS force fields. Average angles between the carbonyl group and the surface normal are depicted in black (fully uncharged surface), red (30% deprotonated surface), and blue (45% deprotonated surface). Orientation of the fatty acid alkyl chain is shown as an inset using the same color notation; i.e., average angles between the surface normal and the vector between the first and the last carbon of the arachidic acid (θC20–C1). | ||
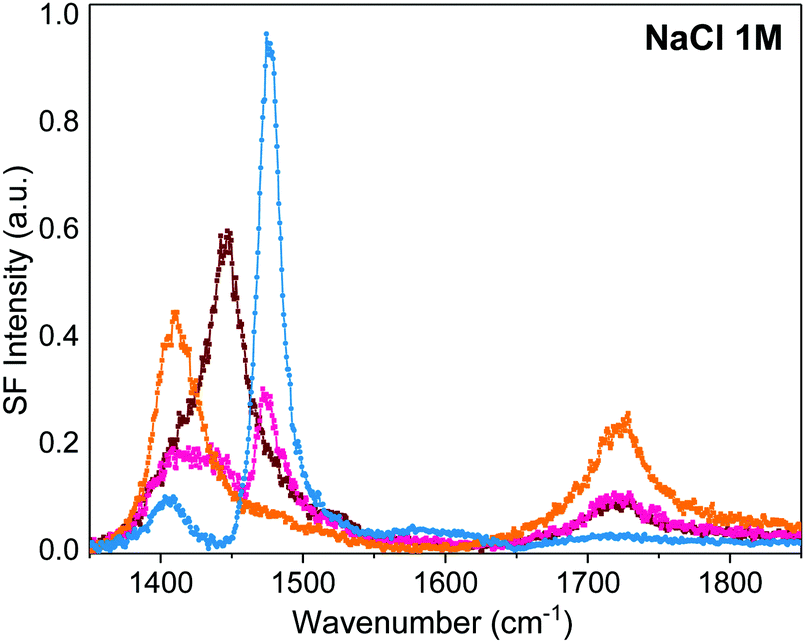 | ||
| Fig. 5 VSF spectra collected under the SSP polarization combination of dAA Langmuir monolayer on a 1 M NaCl (99.999%, trace metal basis) subphase in the absence of EDTA. The irreproducibility of the spectral features is ascribed to trace amounts of polyvalent cations in the high purity salts used. The spectra result from solutions prepared with salt crystals from either different suppliers, different batches or even the same batch (see Section S1 in ESI† for details). | ||
The data shows that if no active measures are taken to remove traces of polyvalent metal ion impurities, they can dominate the interactions at charged interfaces and have adverse implications in the interpretation of the results. This is particularly relevant for flat interfaces (i.e., Langmuir monolayers), where the surface to volume ratios are low, and surface-active contaminants can easily compete for adsorption sites. For example, previous SF studies16 of similar fatty acid monolayers interacting with the monovalent alkali metals Na+ and K+ revealed multiple peaks in the carboxylate region (we note that in the absence of multivalent ions, K+ and Na+ salts exhibit a single carboxylate band at natural pH. See Fig. 2 and Section S1 in ESI† for details). In the light of these conclusions, the interpretation of many such studies16,28,29 may need to be reconsidered.
The VSF spectra of a per-protonated arachidic acid monolayer on a subphase with increasing concentration of NaCl are shown in Fig. 6a. The sharp peaks centred at 2875 cm−1 and 2940 cm−1, together with the shoulder at ∼2960 cm−1, are all assigned to methyl vibrations: the symmetric stretch (r+), corresponding Fermi resonance (rFR+), and asymmetric methyl stretch (r−), respectively. The weak methylene stretch at 2845 cm−1 (d+) indicates that the alkyl chains in the monolayer are well ordered, with only few, if any, gauche defects (see Fig. 6a). This is in accordance to the interpretation made above for the per-deuterated acid. Besides the apparent small increase observed in the rFR+ mode, which results from interference with neighbouring water bands, the CH stretching modes are unaffected by the charging of the monolayer.
 | ||
| Fig. 6 (a) VSF spectra of a AA Langmuir monolayer collected in the CH/OH stretching region for different NaCl salt concentrations under the SSP polarization combination. The water OH stretches are magnified in the inset (EDTA concentration: 0.5 μM. pH ∼ 5.8). (b) Integrated OH band intensity (left axis) and surface potential (right axis), as a function of salt concentration. OH intensity data for NaCl concentrations <10 mM is taken from ref. 17. Red line is a theoretical model prediction of the OH intensity as a function salt concentration that accounts for contributions of water molecules in the immediate surface and diffuse double layer (χ(2)s/χ(3) = −0.12 (1 − 2α) see Section S9 in ESI† for details). The surface potential was estimated from the experimental determined surface charge using the Grahame equation (PB) and a MPB model that accounts for steric effects (a = 7.5 Å). | ||
In contrast, the OH bands show clear differences (inset Fig. 6a). The overall intensity decreases with the addition of NaCl in the subphase. Most of this signal in the OH stretching region, particularly for the lowest NaCl concentrations shown in Fig. 6a, originates from water molecules within the electrical double layer perturbed by the field (χ(3) response), which can extend several nanometers away from the surface (up to 4κ−1, i.e., ∼12 nm for 10 mM NaCl).82 Water molecules in the diffuse layer display only a minute polar orientational preference (i.e., rotating dipoles with non-zero angle average potentials), however, their summed contribution to the SF signal becomes significant due to their large numbers, which is typically orders of magnitude higher than those in the immediate surface. This χ(3) response is expected to be proportional to the surface potential,28,82,84 which in turn can be independently estimated from the experimentally determined surface charge using either the Grahame equation39 or a more elaborate MPB expression that accounts for steric effects that are non-negligible at high salt concentrations (see Experimental section).40 Although the surface charge increases with the addition of salt (Fig. 2d), the surface potential decreases due to the screening effect of the growing number of ions in solution (Fig. 6b). The decrease in potential is less pronounced for the MPB model since when taking into account the effective ion size, the ion concentration at the surface reaches a saturation level for the highest ionic strengths. The drop in intensity of the OH bands observed in Fig. 6a is then qualitatively explained by a lowering of the surface potential.
A more quantitative insight has been obtained by comparing the experimental results with a model that predicts the contributions from water molecules next to the charge interface as a function of ionic strength and surface potential.28,82,83Fig. 6b shows the data points of integrated OH band intensities measured for various NaCl concentration in the subphase (data for <10 mM was taken from ref. 17 and included for completion), and in red, the theoretical prediction based on the surface potentials derived from the MPB model (see Section S9 in ESI† for details). At concentrations below ∼1 mM contributions from molecules in the diffuse double layer partially, or totally cancel due to interference (i.e., Δkz−1 is comparable or smaller than κ−1).17,82 For the pure water subphase the spectral features observed are essentially only linked to water molecules directly interacting with the carboxylic acid headgroup. Recent MD simulations indicate that the broadband centred at ∼3275 cm−1 originates from water molecules accepting a hydrogen bond from the uncharged fatty acid OH group, while the band centred at ∼3510 cm−1 is associated to those donating a hydrogen bond to the CO group.36 The maximum in the integrated OH intensities is reached at ∼1 mM NaCl (Fig. 6b), when contributions from water molecules in the diffuse double layer are the largest. This transition is also observed in the spectral features since the lower frequency band centred at ∼3200 cm−1 becomes relatively more intense at 10 mM NaCl when compared to pure water (Fig. 6a). As expected, the implication is that the hydrogen bond environment in the diffuse layer is different from that in direct proximity to the carboxylic acid groups. At higher ionic strengths the model deviates significantly from the experimental data (Fig. 6b), with water bands decreasing at a faster rate with concentration. The spectral features for 1 M NaCl, although close to the detection limit, are remarkably similar those of pure water with the highest intensity observed at ∼3550 cm−1 (Fig. 6a). This suggest that the signal in the 1 M spectrum originates from molecules in direct contact to the monolayer (χ(2)s response) with little or no contribution from water in the diffuse layer (χ(3) response). Thus, the discrepancy with the model prediction could be explained by the fact that at high salt concentrations, when the Debye length is limited to <1 nm, the decaying surface electric field is no longer probing water molecules in the diffuse layer, but instead mainly those hydrating the cations in the more centrosymmetric environment of the condensed surface layer (see Section S9 in ESI† for details). We note that although the degree of monolayer charging at high salt concentrations depends on the nature of the cation, no significant differences were observed when exchanging the subphase for CsCl or LiCl solutions at the same pH (data not shown).
3.3. Specific monovalent cation interactions: pH dependence
The interactions of the monovalent cations with the carboxylic acid group were also investigated at different pHs for a constant salt concentration. The SF spectra of dAA monolayers at three selected pHs with subphases of 1 M CsCl, NaCl, and LiCl, are shown in Fig. 7. As expected, the overall degree of deprotonation, judged by the intensity of the hydrated vsCOO− band, increases with pH. However, a closer inspection at any given pH shows differences depending on the nature of the cations. At pH 4.5 (Fig. 7a), the deprotonation of the monolayer is highest for solutions containing Cs+, followed by Na+, then Li+. At pH 6 (Fig. 7b) this trend remains, yet the differences diminish. Upon further pH increase, the trend reverses (Fig. 7c) and the cations order instead as Li+ > Na+ > Cs+. The change in the degree of deprotonation with pH is summarized in Fig. 7d (for all cases the area per molecule remained approximately constant at ∼20 ± 1 Å2). The experimental results imply that Cs+ ions have a larger preference, relative to Na+ and Li+, to interact with the fatty acid monolayer when it is less charged (i.e., more neutral carboxylic acids present). In contrast, as the number of charged carboxylate groups increases with pH, the series reverses, with Li+ ions displaying the strongest interactions. This latter ordering is in agreement with the Collins empirical law of matching water affinities.85
 | ||
| Fig. 7 VSF spectra in the carboxylate/carbonyl stretching region of a dAA Langmuir monolayer on a subphase consisting of 1 M CsCl, NaCl, and LiCl solutions at (a) pH 4.5 (b) pH 6.0 and (c) pH 9.0. The inset highlights the blue shift of ∼7 cm−1 of the vsCOO− peak for LiCl at pH 9.0. All spectra collected in the polarization combination SSP. EDTA concentration: 20 μM. The pH was adjusted with a solution of NaOH. (d) The degree of deprotonation as a function of pH estimated from the intensity of the carboxylate band. | ||
The reversal of the cationic Hofmeister series with pH for a carboxylic acid monolayer was predicted in a recent publication20 based on a modified Poisson–Boltzmann modeling approach. Therein, salts are ordered according to their efficiency in stabilizing solutes against precipitation. The reversal of the Hofmeister series is explained by a turnaround in the cationic affinities to the protonated/deprotonated forms of carboxylic acid.
We now present a coarse grained simulation approach which captures the dynamic acid–base equilibrium of the Langmuir monolayer (see Fig. 1). Using grand canonical Monte Carlo (GCMC) simulations, we calculate the average deprotonation of the arachidic acid monolayer as a function of pH, type, and concentration of salt. The advantage of this model is that the protonation states of carboxylic acid moieties fluctuate while the pH and the salt chemical potential are kept constant, allowing us to directly simulate the experimental conditions. The simulated apparent pKa value of the monolayer in pure water reproduces the experimental value of 10.8.17 The simulation results, summarized in Fig. 8, show that in accord with experiment, the deprotonation increases with the salt concentration (Fig. 2) as well as with pH (Fig. 7).
 | ||
| Fig. 8 Degree of deprotonation of the Langmuir monolayer as a function of salt concentration and pH obtained by coarse grained, grand canonical Monte Carlo simulations at constant pH and salt chemical potential. Highlighted data points at 1 M concentration are directly comparable with the experimental results in Fig. 7d. | ||
The GCMC simulations accurately predict the ability of cations to deprotonate the surface: at pH 4.5 the classical Hofmeister order is obtained, i.e., Cs+ > Na+ > Li+, with a swap between pH 6 and 7, and a total reversal at pH 9: Li+ > Na+ > Cs+. The change in cation preference results from the fact that activity coefficients (which were used to parametrize the model. See Section 2.7) follow the reversed order depending on whether the fatty acid is neutral or charged: aLi–COOH > aNa–COOH > aCs–COOH while aLi–COO < aNa–COO < aCs–COO; the lower the activity coefficient, the stronger the interaction between the species. GCMC further reproduce the relative differences between the cations, being larger at high pH than at low pH values. Relative to experiments, however, the GCMC simulations overestimate the absolute degree of deprotonation at high pH values (Fig. 7). This can be attributed to the simplistic continuum electrostatic model used and the fact that no attempts were made to optimize the model parameters to experiment, relying instead on bulk activity coefficient data and MD results for ion–ion and ion–headgroup interactions. Nevertheless, the coarse grained approach yields good qualitative agreement for ion-specificity, salt concentration, and pH variations (Fig. 8).
To validate an earlier assumption that at constant pH, a higher percentage of the deprotonation in the monolayer is linked to a stronger affinity of a given cation to the carboxylic acid moiety, all-atom MD simulations were also performed. The Langmuir monolayer here has a fixed charge and the amount of cations interacting with the monolayer at any specified state can therefore be established. Three cases with fixed degrees of deprotonation were considered: a fully protonated monolayer (0% charged), 30% deprotonated, and 45% deprotonated. Fig. 9 shows how the different cations interact in terms of the calculated excess at the three selected charged scenarios. The analysis provides the number of cations in each layer parallel to the monolayer according to the z-axis of the simulation box, i.e., starting at the water/acid interface and ending in bulk. Bulk values were set to unity to facilitate the determination of ion excess or depletion at the water/carboxylic acid interface.
 | ||
| Fig. 9 MD simulations showing the interactions of cations with fully protonated (a), 30% (b), and 45% (c) deprotonated surfaces of arachidic acid. The “excess” or bulk normalized number densities are calculated along the z-axis, where 0 is the position of the water/carboxylic acid interface and 3 nm is the bulk solution. The OPLS force field for the monolayer and van der Vegt force field45 for ions were used. | ||
In Fig. 9a for the fully protonated monolayer, the cationic excess curves show that both lithium and sodium ions are depleted from the surface, while cesium is attracted. This situation changes when the arachidic acid monolayer is partly deprotonated. Due to the introduced negative surface charge, all cations are now attracted to the surface. Comparing the cations, we observe that in the range extending from 10 to 30% deprotonation, the preferential cation interaction follows: Cs+ > Na+ > Li+. As the number of deprotonated moieties in the monolayer increases, the differences between the cations decrease (Fig. 9b for 30% deprotonation). This contrasting behaviour results from an interplay between an ion specific electrostatic attraction to the charged carboxylates (Li+ > Na+ > Cs+), and an attraction (Cs+) or repulsion (Li+ > Na+) towards the uncharged carboxylic acid groups. By further increasing the amount of arachidates in the monolayer, we reach a point when all cations are similarly attracted to the acid/water interface, followed by a swap in the order of attractive interactions at higher deprotonation levels, with Li+ > Na+ > Cs+ (see Fig. 9c for 45% deprotonation). Due to electrostatic arguments, the overall amount of any cations at the surface increases when compared to the previous cases. However, the absolute increase in the populations of cations at the surface is lowest for cesium due to its reduced affinity towards the carboxylates. Lithium interacts stronger with arachidates and is more depleted from arachidic acids, leading to different binding patterns when compared to sodium, as discussed in the following section. Repeating all MD simulations with another ionic force-field based on charge scaling46,47 gave identical trends (see Section S4 in ESI†).
The all-atom MD simulations qualitatively agree with coarse grained GCMC. At low pH (Fig. 8 top left), corresponding to low degree of deprotonation in all-atom simulations (Fig. 9a), we observe the classical Hofmeister order, Cs+ > Na+ > Li+. Above 30% of surface deprotonation the order swaps, corresponding to pH 6 to 7 in the coarse grained simulations. Finally, at 45% deprotonation (Fig. 9c) the reversed series, Li+ > Na+ > Cs+, is obtained as with pH 9 in GCMC, and in fair agreement with experimental observations (Fig. 7). The combination of coarse grained and all-atom simulations, embraces both the dynamic aspect of the experiment and the atomistic detail, thus providing a strong platform for microscopically rationalizing the observed behaviour. The main conclusion from the simulations is the direct correlation between the amount of cations attracted to the surface and the degree of deprotonation of the monolayer. This helps to confirm one of the key assumptions we made when interpreting the experimental results.
COO− assignment. Interestingly, however, for the specific case of 1 M LiCl at pH 9.0, the center position of this band was reproducibly blue-shifted to ∼1415 cm−1 with no major changes in its bandwidth (see inset in Fig. 7c). The shift towards higher frequencies indicates a strengthening of the bonds involved in the vibration,86 which would take place, for example, upon formation of a contact ion pair.12 We thus assign the ∼1415 cm−1 band to the bound or contact ion-pair between the carboxylate and the Li+ ion (vsCOO−–Li+). We note that the same peak position has been observed for lithium acetate in the bulk and assigned to an ionic bond type.79
One of the direct implications of this finding is that the preference of a monovalent cation to interact with the carboxylate moiety through either solvent-separated (or solvent-shared) pairs, contact pairs, or bound complexes would depend on the ratio of charged species found at the interface. The specific ratio would depend on the identity of the cation and could be modified, for example, by an increase in either pH and/or salt concentration. Indeed, as will be discussed elsewhere,87 a similar shift is observed for Na+ ions at even higher pHs. This fine dependence on solution conditions could explain the contrasting claims found in the literature on the type of pair formation formed between carboxylic acids and monovalent cations.11,12,14,20,45
 | ||
| Fig. 10 Cationic density maps (Li+ in black, Na+ in green, and Cs+ in violet), around arachidic acid (top) and arachidates (bottom) with the isovalues of 4 and 1.5 show binding patterns of cations at the interface. | ||
Cation binding around the charged arachidates shows significant differences (Fig. 10, bottom section). Cesium is only present as a solvent-shared cation and does not perturb the first solvation shell of the carboxylate. In contrast, sodium and lithium can either be found in a solvent-shared configuration (most external umbrella volumes) or directly bound to the carboxylate. When bound, the configuration differs between the two cations. As sodium is slightly less repelled from the arachidic acid moiety, and in general has a weaker solvation shell than lithium, it binds at all possible places in the neighbourhood of the carboxylate: that is in a monodentate or bidentate chelating pattern. Lithium on the other hand, being much more directional, binds mainly as a bidentate chelating cation. These results are in partial agreement with previous X-ray and simulations studies11 where it was nonetheless suggested that in bulk, sodium binds more strongly with the acetate than lithium and potassium.
When compared to the arachidic acid experimental results, the MD simulations with van der Vegt force field45 provide a fairly good agreement, i.e., a solvent-shared ion pair for cesium and mostly a solvent-shared ion pair for lithium and sodium. However, the direct sodium-carboxylate contact ion pair formation is overestimated. It is worth noting that all other ionic force fields tested (OPLS,42 charmm36, scaled charges46,47), resulted in poorer agreement with experiments by strongly overestimating the formation of contact ion pairs for all cations. With the exception of LiCl at pH 9, the SF spectra indicate that for all conditions studied, the cations are not found in direct contact to the carboxylate moiety, but instead in a solvent shared or hydrated configuration. It is only for LiCl at pH 9, when the vsCOO− band blue-shifts to ∼1415 cm−1, that a first sign is seen of a more direct interaction between the cation and the carboxylate moiety. This suggests that the binding patterns inferred from simulations are probably expected at more basic conditions than the ones considered here.
4. Summary and conclusions
The distinctive SF spectral features of carboxylate vibrations together with molecular area information extracted from a Langmuir trough technique were used to measure the charge density of a fatty acid monolayer. At a constant pH of 6, the degree of deprotonation as a function of salt concentration was found, starting from 100 mM, to depart from the prediction of the Gouy–Chapman theory, and depend on the identity of the alkali metal cation, highlighting the appearance of ion specific effects. Better agreement was found using a modified Poisson–Boltzmann model which accounts for steric effects through an effective ion size parameter that restricts the artificial ion accumulation at the surface.41 At pH 6, this parameter ranges between 7.5 Å to 9 Å and follows the same trend as the hydrated diameters, with Li+ being the largest and leading to the lowest monolayer charge. We note that the fitted sizes are largely empirical and likely enclose multiple physical phenomena not explicitly covered by the MPB model used, such as accounting for ion-dependent dielectric response,66 dispersion interactions,67 or water molecules as explicit point-like dipoles,68 just to mention a few. Additionally, the MPB model used does not predict the change in cations affinities observed at higher pH. This could be circumvented by combining atomistically resolved ion–surface interactions with PB theory as shown by Netz and coworkers for a surface composed of carboxylic acids,20 or in principle, also by combining non-electrostatic potentials with PB theory and considering ion size, and hydration.88 The latter theory, however, has not yet been fully developed for a surface exposing only carboxylic acid groups to solution.89At a constant salt concentration of 1 M the degree of deprotonation dependence with pH was found to be cation specific with an order that inverts around neutral pH. At pH 4.5 the monolayer is more deprotonated in the presence of Cs+, followed by Na+, and finally Li+. At pH 9.0 the series is inverted with Li+ > Na+ > Cs+, and a cross-over occurring at pH ∼ 7. Experiments together with simulations confirm that the degree of deprotonation is linked to the amount of cations attracted to the surface. In qualitative agreement with the experimental data, the coarse grained and all-atom simulations detail the origin of the contrasting affinities observed at different pH. Cesium cations interact with the uncharged carboxylic acid moiety, in particular with the CO group, in contrast to Li+ and Na+ that are repelled. All cations are attracted to the charged carboxylate group, following an inverted order, Li+ > Na+ > Cs+. This dependence on solution conditions could be one of the causes of the contrasting claims found in the literature on the ordering of monovalent alkali metal cation interactions with the carboxylic acid moiety.11,12,14,20,45 Note that the pH range where the cation affinity cross-over occurs will be lower for soluble carboxylic acids in the bulk. The surface potential associated with the charged monolayers raises the surface concentration of hydronium ions, resulting in an apparent pKa that can be several orders of magnitude higher that thermodynamic pKa of ∼5.17
The SF spectral features from the carboxylate headgroup also provided information on the type of ion pair formed. With the exception of LiCl at pH 9, the cations do not directly interact with the carboxylate headgroup. They remain hydrated, in either a solvent-separated (2SIP) or solvent-shared (SIP) ion-pair configuration. For the specific case of Li+ at pH 9, a blue shift of ∼7 cm−1 was interpreted as the formation of a contact ion pair (CIP). In contrast, the all-atom molecular simulations performed overestimated the presence of CIPs for all cations, and reflected a very high sensitivity to the choice of the ionic force field used. Experimentally, CIPs are indeed favored at very high monolayer charge densities (i.e., by further increasing the pH and/or the salt concentration), indicating that the type of ion pairs formed depend on subtle changes in solution conditions.
In contrast to the charged carboxylate moiety, the orientation of the uncharged carboxylic acid headgroups was sensitive to the monolayer degree of deprotonation, with the carbonyl group tilting closer to the surface normal (from ∼75° to ∼60°) when increasing the charge of the monolayer. In qualitative agreement with experimental SF data, all-atom simulations using two alternative force fields (OPLS42 and charmm3643), also predicted a CO orientation close to the surface plane when fully uncharged and a tilt towards the surface normal upon charging of the monolayer. Nonetheless, quantitative agreement is not reached in either case. The OPLS force field results in too flexible headgroups, while it is significantly underestimated by the stiffer charmm36 force field. This suggests that new force fields need to be developed for fatty acid monolayers.
Finally, sub-ppm impurities of divalent and trivalent cations were found to readily displace monovalent ions from carboxylic acid moieties, even when using high purity salts (>99.99% trace metals). Unless active measures are taken (here we use the chelating agent EDTA), they could lead to artifacts, easy to misinterpret. The implications of these results cast doubt on previous studies of carboxylic acid–monovalent ion interactions at medium to high salt concentrations carried out on single interfaces where no measures were taken to remove ion impurities (i.e., in a Langmuir trough or planar solid/liquid interfaces).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Swedish Foundation for Strategic Research (SSF-FFL-5 program) and the Swedish Research Council (VR). The authors thank Robert Corkery for insightful discussions. We thank the Crafoord Foundation for support and LUNARC in Lund for providing computational resources.References
- P. Lo Nostro and B. W. Ninham, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed.
- B. W. Ninham and P. Lo Nostro, Molecular Forces and Self Assembly, Cambridge University Press, US, 2010 Search PubMed.
- W. Kunz, Specific Ion Effects, World Scientific, 2010 Search PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247–260 Search PubMed.
- P. Jungwirth and P. S. Cremer, Nat. Chem., 2014, 6, 261 CrossRef CAS PubMed.
- Y. Marcus, Chem. Rev., 2009, 109, 1346–1370 CrossRef CAS PubMed.
- A. W. Omta, M. F. Kropman, S. Woutersen and H. J. Bakker, Science, 2003, 301, 347 CrossRef CAS PubMed.
- Y. Zhang and P. S. Cremer, Annu. Rev. Phys. Chem., 2010, 61, 63–83 CrossRef CAS PubMed.
- K. Werner, Specific Ion Effects, World Scientific Publishing, Singapore, 2010 Search PubMed.
- K. D. Collins, G. W. Neilson and J. E. Enderby, Biophys. Chem., 2007, 128, 95–104 CrossRef CAS PubMed.
- E. F. Aziz, N. Ottosson, S. Eisebitt, W. Eberhardt, B. Jagoda-Cwiklik, R. Vácha, P. Jungwirth and B. Winter, J. Phys. Chem. B, 2008, 112, 12567–12570 CrossRef CAS PubMed.
- J. S. Uejio, C. P. Schwartz, A. M. Duffin, W. S. Drisdell, R. C. Cohen and R. J. Saykally, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6809–6812 CrossRef CAS PubMed.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- M. Pastorczak, S. T. van der Post and H. J. Bakker, Phys. Chem. Chem. Phys., 2013, 15, 17767–17770 RSC.
- N. F. A. van der Vegt, K. Haldrup, S. Roke, J. Zheng, M. Lund and H. J. Bakker, Chem. Rev., 2016, 116, 7626–7641 CrossRef CAS PubMed.
- C. Y. Tang and H. C. Allen, J. Phys. Chem. A, 2009, 113, 7383–7393 CrossRef CAS PubMed.
- E. Tyrode and R. Corkery, J. Phys. Chem. C, 2018, 122, 28775–28786 CrossRef CAS.
- Y. Marcus and G. Hefter, Chem. Rev., 2006, 106, 4585–4621 CrossRef CAS PubMed.
- P. Ganguly, P. Schravendijk, B. Hess and N. F. A. van der Vegt, J. Phys. Chem. B, 2011, 115, 3734–3739 CrossRef CAS PubMed.
- N. Schwierz, D. Horinek and R. R. Netz, Langmuir, 2015, 31, 215–225 CrossRef CAS PubMed.
- N. Schwierz, D. Horinek, U. Sivan and R. R. Netz, Curr. Opin. Colloid Interface Sci., 2016, 23, 10–18 CrossRef CAS.
- G. L. Gaines, Insoluble Monolayers at Liquid-Gas Interfaces, Wiley (Interscience), New York, 1966 Search PubMed.
- E. Pezron, P. M. Claesson, J. M. Berg and D. Vollhardt, J. Colloid Interface Sci., 1990, 138, 245–254 CrossRef CAS.
- V. B. Fainerman, D. Vollhardt and R. Johann, Langmuir, 2000, 16, 7731–7736 CrossRef CAS.
- F. Leveiller, D. Jacquemain, L. Leiserowitz, K. Kjaer and J. Als-Nielsen, J. Phys. Chem., 1992, 96, 10380–10389 CrossRef CAS.
- E. Le Calvez, D. Blaudez, T. Buffeteau and B. Desbat, Langmuir, 2001, 17, 670–674 CrossRef CAS.
- E. Tyrode, P. Niga, M. Johnson and M. W. Rutland, Langmuir, 2010, 26, 14024–14031 CrossRef CAS PubMed.
- Y.-C. Wen, S. Zha, X. Liu, S. Yang, P. Guo, G. Shi, H. Fang, Y. R. Shen and C. Tian, Phys. Rev. Lett., 2016, 116, 016101 CrossRef PubMed.
- C. Y. Tang, Z. Huang and H. C. Allen, J. Phys. Chem. B, 2010, 114, 17068–17076 CrossRef CAS PubMed.
- W. Sung, S. Krem and D. Kim, J. Chem. Phys., 2018, 149, 163304 CrossRef.
- S. Ye, H. Noda, T. Nishida, S. Morita and M. Osawa, Langmuir, 2004, 20, 357–365 CrossRef CAS.
- P. B. Miranda, Q. Du and Y. R. Shen, Chem. Phys. Lett., 1998, 286, 1–8 CrossRef CAS.
- C. Y. Tang, Z. Huang and H. C. Allen, J. Phys. Chem. B, 2011, 115, 34–40 CrossRef CAS PubMed.
- J. Lovrić, D. Duflot, M. Monnerville, C. Toubin and S. Briquez, J. Phys. Chem. A, 2016, 120, 10141–10149 CrossRef PubMed.
- W. Lin, A. J. Clark and F. Paesani, Langmuir, 2015, 31, 2147–2156 CrossRef CAS PubMed.
- S. K. Reddy, R. Thiraux, B. A. Wellen Rudd, L. Lin, T. Adel, T. Joutsuka, F. M. Geiger, H. C. Allen, A. Morita and F. Paesani, Chem, 2018, 4, 1629–1644 CAS.
- J. F. D. Liljeblad and E. Tyrode, J. Phys. Chem. C, 2012, 116, 22893–22903 CrossRef CAS.
- C. D. Bain, P. B. Davies, T. H. Ong, R. N. Ward and M. A. Brown, Langmuir, 1991, 7, 1563–1566 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Elsevier, Oxford, 2011 Search PubMed.
- M. S. Kilic, M. Z. Bazant and A. Ajdari, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 021502 CrossRef PubMed.
- I. Borukhov, D. Andelman and H. Orland, Phys. Rev. Lett., 1997, 79, 435–438 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- S. Lee, A. Tran, M. Allsopp, J. B. Lim, J. Hénin and J. B. Klauda, J. Phys. Chem. B, 2014, 118, 547–556 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- B. Hess and N. F. A. van der Vegt, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13296–13300 CrossRef CAS PubMed.
- E. Pluhařová, P. E. Mason and P. Jungwirth, J. Phys. Chem. A, 2013, 117, 11766–11773 CrossRef PubMed.
- M. Kohagen, P. E. Mason and P. Jungwirth, J. Phys. Chem. B, 2016, 120, 1454–1460 CrossRef CAS PubMed.
- A. Habartová, M. Roeselová and L. Cwiklik, Langmuir, 2015, 31, 11508–11515 CrossRef PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- M. Lund, M. Trulsson and B. Persson, Source Code Biol. Med., 2008, 3, 1 CrossRef PubMed.
- R. A. Robinson and R. H. Stokes, Electrolyte Solutions: Second Revised Edition, Dover Publications, 2002 Search PubMed.
- M. Lund and B. Jönsson, Biochemistry, 2005, 44, 5722–5727 CrossRef CAS PubMed.
- V. M. Kaganer, H. Möhwald and P. Dutta, Rev. Mod. Phys., 1999, 71, 779–819 CrossRef CAS.
- R. Johann and D. Vollhardt, Mater. Sci. Eng., 1999, 8, 35–42 CrossRef.
- R. Johann, D. Vollhardt and H. Möhwald, Langmuir, 2001, 17, 4569–4580 CrossRef CAS.
- S. Kundu and D. Langevin, Colloids Surf., A, 2008, 325, 81–85 CrossRef CAS.
- A. Angelova, D. Vollhardt and R. Ionov, J. Phys. Chem., 1996, 100, 10710–10720 CrossRef CAS.
- E. Tyrode and J. Hedberg, J. Phys. Chem. C, 2012, 116, 1080–1091 CrossRef CAS.
- K. Dill and S. Bromberg, Molecular Driving Forces. Statistical Thermodynamics in Biology, Chemistry, Physics, and Nanoscience, Garland Science, New York, 2010 Search PubMed.
- A. G. Lambert, P. B. Davies and D. J. Neivandt, Appl. Spectrosc. Rev., 2005, 40, 103–145 CrossRef CAS.
- D. Ben-Yaakov, D. Andelman, R. Podgornik and D. Harries, Curr. Opin. Colloid Interface Sci., 2011, 16, 542–550 CrossRef CAS.
- D. Ben-Yaakov, D. Andelman and R. Podgornik, J. Chem. Phys., 2011, 134, 074705 CrossRef PubMed.
- B. W. Ninham and V. Yaminsky, Langmuir, 1997, 13, 2097–2108 CrossRef CAS.
- A. Abrashkin, D. Andelman and H. Orland, Phys. Rev. Lett., 2007, 99, 077801 CrossRef PubMed.
- C. M. Johnson, E. Tyrode, S. Baldelli, M. W. Rutland and C. Leygraf, J. Phys. Chem. B, 2005, 109, 321–328 CrossRef CAS PubMed.
- C. D. Bain, in Surfactant Science Series, Dekker, New York, 1999, vol. 83, pp. 335–373 Search PubMed.
- C. Hirose, N. Akamatsu and K. Domen, Appl. Spectrosc., 1992, 46, 1051–1072 CrossRef.
- X. Zhuang, P. B. Miranda, D. Kim and Y. R. Shen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12632–12640 CrossRef CAS.
- H.-F. Wang, W. Gan, R. Lu, Y. Rao and B.-H. Wu, Int. Rev. Phys. Chem., 2005, 24, 191–256 Search PubMed.
- G. R. Bell, Z. X. Li, C. D. Bain, P. Fischer and D. C. Duffy, J. Phys. Chem. B, 1998, 102, 9461–9472 CrossRef CAS.
- E. Tyrode, C. M. Johnson, M. W. Rutland, J. P. R. Day and C. D. Bain, J. Phys. Chem. C, 2007, 111, 316–329 CrossRef CAS.
- E. Tyrode, C. M. Johnson, S. Baldelli, C. Leygraf and M. W. Rutland, J. Phys. Chem. B, 2005, 109, 329–341 CrossRef CAS PubMed.
- C. M. Johnson, E. Tyrode, A. Kumpulainen and C. Leygraf, J. Phys. Chem. C, 2009, 113, 13209–13218 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- J. E. Tackett, Appl. Spectrosc., 1989, 43, 483–489 CrossRef CAS.
- R. Johann, D. Vollhardt and H. Möhwald, Colloids Surf., A, 2001, 182, 311–320 CrossRef CAS.
- A. Sthoer, E. M. Adams, S. Sengupta, H. C. Allen and E. Tyrode, 2019, in preparation.
- G. Gonella, C. Lütgebaucks, A. G. F. de Beer and S. Roke, J. Phys. Chem. C, 2016, 120, 9165–9173 CrossRef CAS.
- P. E. Ohno, H.-f. Wang and F. M. Geiger, Nat. Commun., 2017, 8, 1032 CrossRef PubMed.
- S. Ong, X. Zhao and K. B. Eisenthal, Chem. Phys. Lett., 1992, 191, 327–335 CrossRef CAS.
- K. D. Collins, Biophys. J., 1997, 72, 65–76 CrossRef CAS PubMed.
- N. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Academic Press, San Diego, 1990 Search PubMed.
- A. Sthoer and E. Tyrode, 2018, in preparation.
- D. F. Parsons, M. Bostrom, N. P. Lo and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- D. F. Parsons, T. Duignan and A. Salis, Interface Focus, 2017, 7, 20160137 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Langmuir isotherms for various salt concentrations and pH, detection of polyvalent impurities in the SF spectra, VSFS fitting parameters, details of the orientational analysis of the carbonyl vibration, details of the modified Poisson Boltzmann model, parametrization of the coarse grain model, and MD results using other tested force fields. See DOI: 10.1039/c9cp00398c |
| ‡ Authors contributed equally. |
| This journal is © the Owner Societies 2019 |