
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The fate of O2 in photocatalytic CO2 reduction on TiO2 under conditions of highest purity†
Martin
Dilla
a,
Alina
Jakubowski
a,
Simon
Ristig
 *a,
Jennifer
Strunk
*b and
Robert
Schlögl
ac
*a,
Jennifer
Strunk
*b and
Robert
Schlögl
ac
aMax Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany. E-mail: simon.ristig@cec.mpg.de
bLeibniz Institute for Catalysis, 18059 Rostock, Germany. E-mail: jennifer.strunk@catalysis.de
cFritz Haber Institute of the Max Planck Society, 14195 Berlin, Germany
First published on 2nd May 2019
Abstract
Although the photocatalytic reduction of CO2 to CH4 by using H2O as the oxidant presupposes the formation of O2, it is often not included in the product analysis of most of the studies dealing with photocatalytic CO2 reduction or it is reported to be not formed at all. The present study aims to clarify the absence of O2 in the photocatalytic gas phase CO2 reduction on TiO2. By modifying P25-TiO2 with IrOx co-catalysts it was possible to observe photocatalytic water splitting, i.e. the formation of gaseous O2 and H2 in almost stoichiometric amounts, without the use of sacrificial agents, while bare P25-TiO2 showed no activity in H2 and O2 formation under similar reaction conditions. Investigating the effect of improved H2O oxidation properties on the photocatalytic CO2 reduction revealed that the CH4 formation on P25 from CO2 was completely inhibited as long as the H2O splitting reaction proceeded. Furthermore, we found that a certain amount of O2 is consumed under conditions of photocatalytic water oxidation. A quantification showed it to be in the same order of magnitude as the oxygen which is missing as a byproduct from photocatalytic CO2 conversion. A detailed interpretation of the results in the context of the general understanding of the photocatalytic CO2 reduction with H2O on TiO2 allows the hypothesis that P25-TiO2 undergoes a stoichiometric reaction, meaning that the CH4 formation is not based on a true catalytic cycle and runs only as long as TiO2 can consume oxygen.
Introduction
The photocatalytic reduction of CO2 attracted lots of attention within the last thirty years. Forming mobile energy sources by chemical reactions with CO2 and the help of (sun-)light would be an important achievement for a more sustainable energy system that helps to protect the environment.1 Due to the massive use of fossil fuels for energy supply and the depletion of natural reservoirs in the near future,2 it is desirable to commercialize alternative ways for providing humanity with renewable energy sources.3 Another important issue is the noticeable accumulation of CO2 in the atmosphere as a result of the total combustion of fossil fuels.3,4 An increasing CO2 level accelerates the greenhouse effect and is associated with global warming and changes of the climatic conditions. Making use of CO2 as a raw material could contribute to decreasing the production of anthropogenic CO2 and help to use carbon-containing energy carriers in a closed cycle. An often used term for such a reaction is “artificial photosynthesis”.5An efficient photocatalyst needs to perform at least two functions. The first function is that of a photoabsorber, which means that the material generates charge carriers upon absorption of photons. These charge carriers must have adequate oxidation and reduction potential for the desired redox reactions.6,7 It is also necessary that the charge carriers become spatially separated from each other to avoid their immediate recombination, and that they can migrate to the surface of the material. The second function is that of a heterogeneous catalyst. Therefore, it is necessary that the material exposes active sites in order to activate the reactants sufficiently to facilitate charge carrier transfer reactions. Such an activation can be initiated by adsorption of the reactants on the catalyst surface.8 After decades of research TiO2 is still one of the most promising and most studied materials as photocatalyst for reduction of CO2.1 Indeed a number of alternative materials have been tried, but the activity of TiO2,9 the non-toxicity, stability against photocorrosion10,11 and the commercial availability maintain it as a meaningful candidate for photocatalytic approaches. However, performing photocatalytic CO2 reduction experiments in order to gain knowledge about the mechanistic details is a challenge.12,13 The examination of elementary steps, for instance, the identification of kinetic barriers or the finding of details about the structure–function relationships of photocatalysts is often limited by their low activity in product formation and the detection limit of many standard analytical methods and tools.14 For this reason the mechanism of this highly complex reaction is not well understood.15–18
In many reports the main products of photocatalytic CO2 reduction are CH4, CO and methanol.14,19–21 Given that CH4 is formed from CO2 in a photocatalytic reduction, it is necessary that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds are cleaved and four C–H bonds are formed. This process requires the transfer of eight electrons from TiO2 to the carbon atom. For this reason a second reactant, which can provide these charge carriers and the source of hydrogen is needed. H2O is the most desirable oxidant for this purpose. It is commonly believed that the process of photocatalytic H2O oxidation on TiO2 is associated with the transfer of four holes (h+) to form an O2 molecule (1). At the same time the bound hydrogen is liberated for a subsequent hydrogenation reaction (1).
O double bonds are cleaved and four C–H bonds are formed. This process requires the transfer of eight electrons from TiO2 to the carbon atom. For this reason a second reactant, which can provide these charge carriers and the source of hydrogen is needed. H2O is the most desirable oxidant for this purpose. It is commonly believed that the process of photocatalytic H2O oxidation on TiO2 is associated with the transfer of four holes (h+) to form an O2 molecule (1). At the same time the bound hydrogen is liberated for a subsequent hydrogenation reaction (1).
 | (1) |
 | (2) |
In our previous studies32,33 we performed continuous flow CO2 reduction experiments where we demonstrated that CH4 formation is dependent on the availability of CO2 and H2O. Without dosing of H2O, the product formation diminished after a couple of hours and initiated again as a pulse of H2O was given into the reactor. It was concluded that the reaction proceeds initially by the contribution of physisorbed H2O residues, which are usually available on the surface of TiO2.34,35 The product formation ceases once they are consumed for hydrogenation or light-induced thermal desorption. Then the pulse-dosed H2O molecules serve as the source of hydrogenation. Although H2O appears to participate in the photoinduced CH4 formation, it was not possible to detect any O2 or oxygen-derived species as products of H2O oxidation. On this account the present study focuses on the clarification of the fate of O2 as the by-product of the overall photocatalytic CO2 reduction.
The most prominent materials for H2O oxidation in electrochemical approaches are IrOx and CoOx. It has been found that these transition metal oxides exhibit an outstanding activity in this reaction because of their relatively low overpotential.36–39 Investigations on IrOx and CoOx materials have shown that the activity in the OER is dependent on the concentration of lattice defects.36,40 A study on hydrated amorphous Ir oxyhydrates40 revealed that IrOx species having defects in the cationic and anionic lattice structure are more active in the OER. The authors concluded that weakly bound electrophilic oxygen species play a key role in the superior activity. In order to form such electrophilic oxygen species the surrounding Ir lattice requires flexible oxidation states.40 The electrophilic oxygen species are prone to nucleophilic attacks of H2O. It is assumed that these species participate in the potential limiting step, namely the O–O bond formation of the OER.40,41 Furthermore the photocatalytic activity of IrOx as a co-catalyst for the O2 evolution reaction has also been tested. It is proposed that photogenerated holes in TiO2 are transferred to the IrOx particles,42,43 where the H2O oxidation reaction takes place. In this way TiO2 represents the photoabsorber and IrOx the heterogeneous catalyst. Such IrOx/TiO2 materials showed an improved O2 evolution activity compared to bare TiO2.42 On this account it appears to be a suitable candidate for modification of P25 TiO2 with an O2 evolution co-catalyst, in order to evaluate if improved H2O oxidation conditions have an effect towards the overall product formation of photocatalytic CO2 reduction to CH4. At the same time the impact of O2 evolution on CH4 formation can be investigated. This elucidation helps to verify if product formation is a true catalytic cycle. Furthermore, it contributes to a better understanding of this complicated process and assists the development of reaction-based modification strategies for photocatalyst preparation.
Results
Photocatalytic cleaning and H2O splitting with IrOx/P25
The results of photocatalytic H2O splitting experiments with the 0.05 wt% IrOx/P25 samples calcined at 400 and 200 °C, respectively are shown in Fig. 1 and 2. A significant H2 formation can be observed for both samples once the illumination is started at 0 h (Fig. 1 and 2). It can also be seen that the H2 formation rates scale with the flow rates of H2O and stabilize over the course of the experiment. In contrast O2 formation shows a different development. O2 is only detected after 6 h of illumination in the first experiment of the 400 °C sample (Fig. 1A). For this reason a second H2O splitting experiment with this sample is conducted (Fig. 1B). In this experiment the apparent O2 formation rate increases strongly from 0 to 3.75 h. From 3.75 h on, the formation rate of O2 appears to be unsteady and the ratio between the evolution of O2 and H2 is approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.
:3.
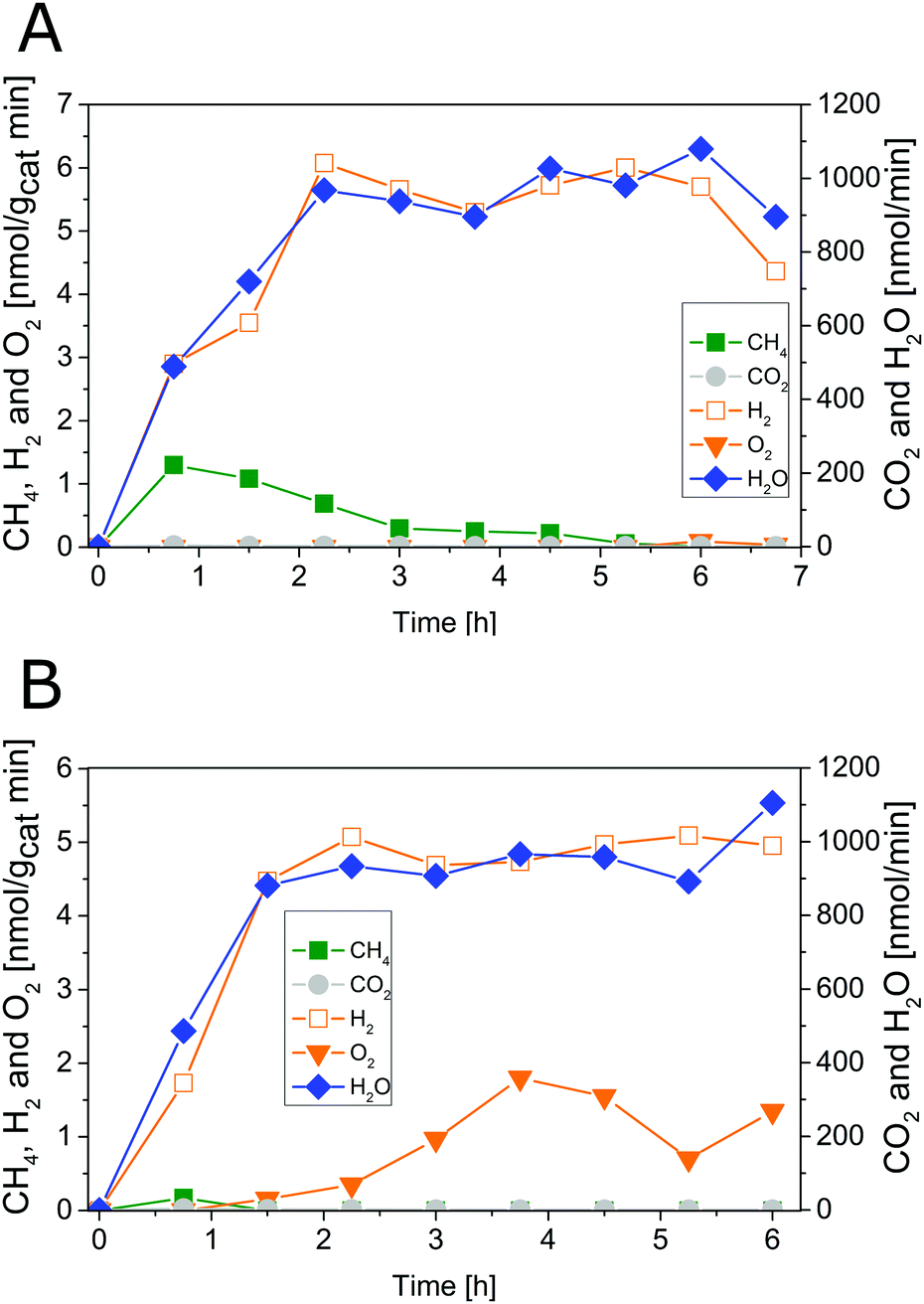 | ||
| Fig. 1 Photocatalytic H2O splitting with 0.05 wt% IrOx/P25 calcined at 400 °C, irradiation time: 6.75 h; (A) first experiment, (B) second experiment. Lines are included in order to guide the eye. | ||
 | ||
| Fig. 2 Photocatalytic H2O splitting with 0.05 wt% IrOx/P25 calcined at 200 °C. Irradiation time: 6.75 h. Lines are included in order to guide the eye. | ||
The 200 °C calcined sample shows also an increase of the apparent O2 formation rate from 0 to 3.75 h (Fig. 2). Moreover, the product formation rates after 3.75 h reveal a O2:H2 ratio of approximately 1:2.
From 5.25 to 6.75 h (Fig. 2) the product formation becomes relatively stable so that steady-state conditions are almost reached. However, it is questionable why the apparent O2 formation starts delayed to the H2 formation. Another important observation is the difference in the overall activity of the two samples. It can be noticed that the 200 °C sample is about 30 times more active in H2 formation than the 400 °C sample. Thus, the calcination temperature seems to have a distinct influence on the catalytic properties of the IrOx/P25 samples.
The reference P25 samples were also tested with respect to their performance in photocatalytic H2O splitting. From Fig. 3A and B it can be observed that the samples show no measureable activity in the formation of O2 and H2. The results of the modified samples indicate that IrOx positively influences the formation of H2 and O2.
 | ||
| Fig. 3 Photocatalytic H2O splitting with (A) P25 reference sample calcined at 200 °C. (B) P25 reference sample calcined at 400 °C. Irradiation time: 6.75 h. Lines are included in order to guide the eye. | ||
Photocatalytic cleaning and H2O splitting with CoOx/P25
In contrast to the IrOx-modified P25 samples, the CoOx modified samples show a significantly different activity in the photocatalytic H2O splitting reaction. From Fig. S1A and B (ESI†) it can be observed that only the sample calcined at 200 °C showed activity in H2 formation. Furthermore it can be seen that the activity in H2 formation ceases over the time of illumination (Fig. S1A, ESI†). Both samples, the 200 °C and 400 °C sample, did not exhibit any activity in O2 formation. The 400 °C samples displayed no activity in the H2 and O2 formation. Due to the absence of activity in O2 formation, the samples were not further tested in the photocatalytic CO2 reduction.Photocatalytic CO2 reduction with IrOx/P25
Fig. 4 shows results of combined flow cleaning and photocatalytic CO2 reduction experiments with the IrOx/P25 samples. Both samples show significant activity in the formation of H2 and O2 before the CO2 dosing was initiated. The formation rate of these products decreases once CO2 is flushed through the reactor, although the water concentration remained fairly constant. In case of the sample calcined at 400 °C the rate of H2 was more than halved within 3.25 h of CO2 dosing, while O2 was completely absent (Fig. 4A) in the products. These observations are less pronounced for the 200 °C sample (Fig. 4B) so that only a slight decrease of both rates was observed. However, under the applied reaction conditions it was not possible to detect CH4 as a product of CO2 reduction. It rather seems that CO2 has primarily a negative impact on the H2 and O2 formation in a way that generation of products is markedly disturbed. The dosing of H2O was stopped after 3.25 h of CO2 reduction, to probe if the absence of this reactant makes a difference in the overall selectivity. From Fig. 4 it can be seen that the absence of H2O in the gas flow strongly affects the H2 and O2 formation. A clear decrease of both formation rates can be seen. More specific, it can be noticed that no H2 and O2 evolution takes place for the 400 °C sample. In case of the 200 °C sample, the O2 formation declines completely while that of H2 drops down to a constant rate. However, after the H2O dosing was stopped it was possible to observe formation of CH4, at least for the 400 °C sample. Thereby the rate increased from 5.25 to 6.75 h (Fig. 4A). In contrast the 200 °C sample showed no CH4 formation under the applied reaction conditions.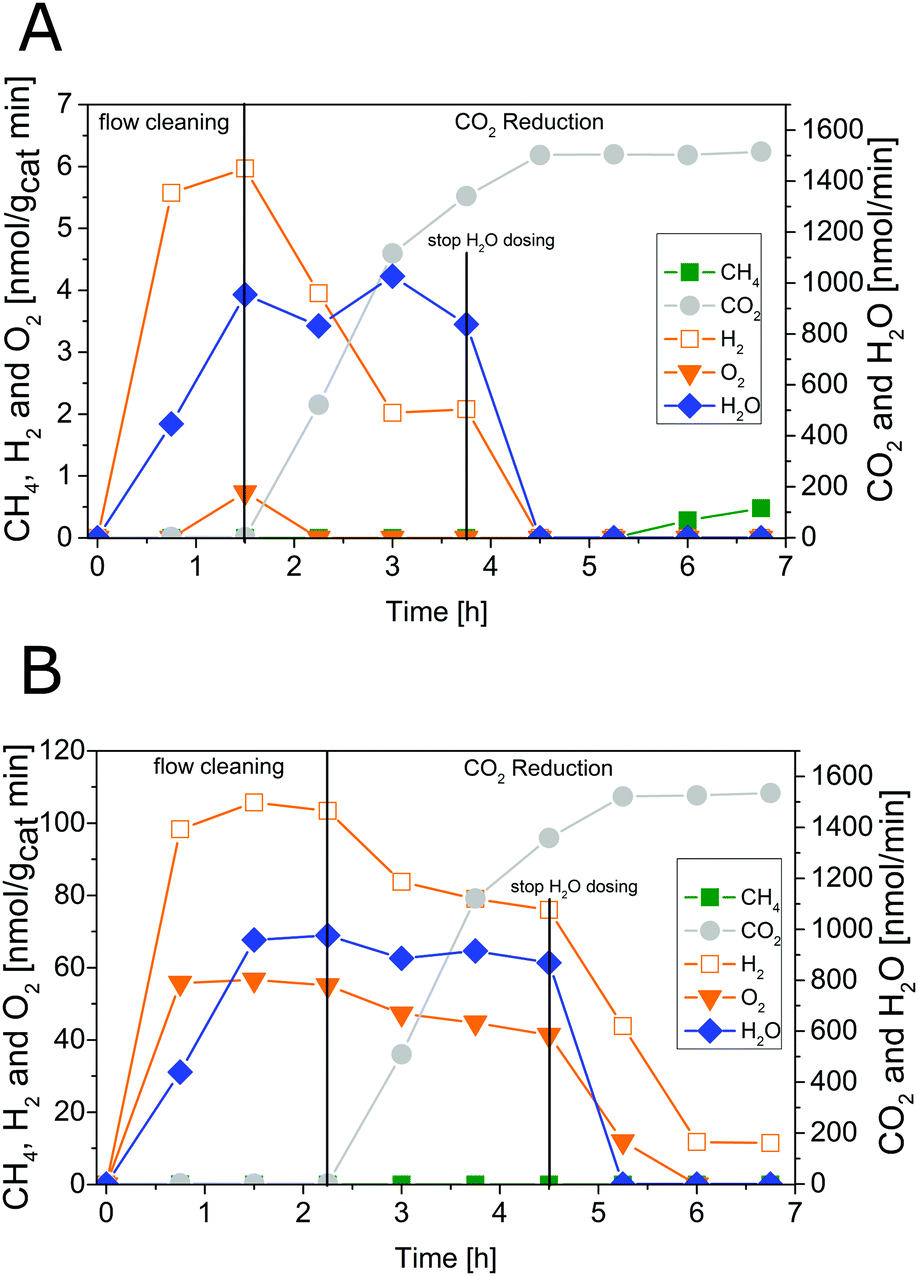 | ||
| Fig. 4 Photocatalytic CO2 reduction with 0.05 wt% IrOx/P25 calcined at (A) 400 °C (flow cleaning from 0 to 1.5 h, CO2 reduction from 1.5 h to 6.75 h, H2O dosing from 0 to 3.75 h) and (B) 200 °C (flow cleaning from 0 to 2.25 h, CO2 reduction from 2.25 h to 6.75 h, H2O dosing from 0 to 4.5 h). Lines are included in order to guide the eye. | ||
The intensive dosing of H2O during activity studies could result in an accumulation of this reactant on the surface of TiO2 based photocatalysts, which can limit its activity in CO2 reduction.32 In order to investigate this potential limitation, a similar experiment with the 200 °C sample was carried out with a reduced H2O flow rate of ∼25 nmol min−1. With these conditions bare P25 showed the highest CO2 reduction activity in our previous studies.32 The results of the flow cleaning and CO2 reduction experiments are displayed in Fig. S2 (ESI†) and Fig. 5, respectively, and reveal a significant activity in the H2 and O2 formation with the reduced amounts of H2O. After adding CO2 to the gas flow the apparent O2 formation ceases completely, while H2 formation decreases to a stable rate. It was again not possible to detect any CH4 formation (Fig. 5). The pure P25 references, without IrOx or CoOx, showed significant activity in CH4 formation when CO2 was given to the reactor (Fig. S3A and B, ESI†). The activity of the sample calcined at 200 °C (Fig. S3A, ESI†) was higher than that of the 400 °C sample (Fig. S3B, ESI†). This observation is similar to the activity of the IrOx/P25 samples in H2O splitting (Fig. 1 and 2). The P25 reference samples however exhibited no activity in H2 and O2 formation at all (Fig. 3). These findings are in accordance with our previous studies on photocatalytic CO2 reduction with P25.32,33 In a consecutive experiment with the P25 references it was investigated if a similarly delayed initiation of O2 evolution can be observed during CH4 formation, as it was observed for the IrOx/P25 samples. Therefore a combined flow cleaning and CO2 reduction experiment was carried out with the sample calcined at 400 °C (Fig. 6) for an extended period of 18 h. To prevent an inhibition of CH4 formation by extensive accumulation or a complete lack of H2O, repetitive pulse dosing of H2O was performed in order to provide suitable conditions to run CH4 formation over an appropriate period of time. It can be obviously seen in Fig. 6 that after about 6 h the pulses of H2O initially decrease the rate of CH4 formation. However, a significant increase of the CH4 formation can be observed in each of the subsequent GC analysis cycles. In total it was possible to observe CH4 formation over a period of 14.25 h (Fig. 6). Under the applied reaction conditions the CH4 formation rate decreases over the course of the CO2 reduction experiment. There was no activity left after 17.75 h (Fig. 6). As no delayed O2 formation could be detected during the experiment it seems that, in contrast to the H2O splitting experiments on IrOx/P25, bare P25 does not release any molecular oxygen in a similar range of time.
 | ||
| Fig. 5 Photocatalytic CO2 reduction with 0.05 wt% IrOx/P25 calcined at 200 °C (flow cleaning from 0 to 1.5 h, CO2 reduction from 1.5 h to 6.75 h, ∼25 nmol min−1 H2O dosing from 0 to 6.75 h). Lines are included in order to guide the eye. | ||
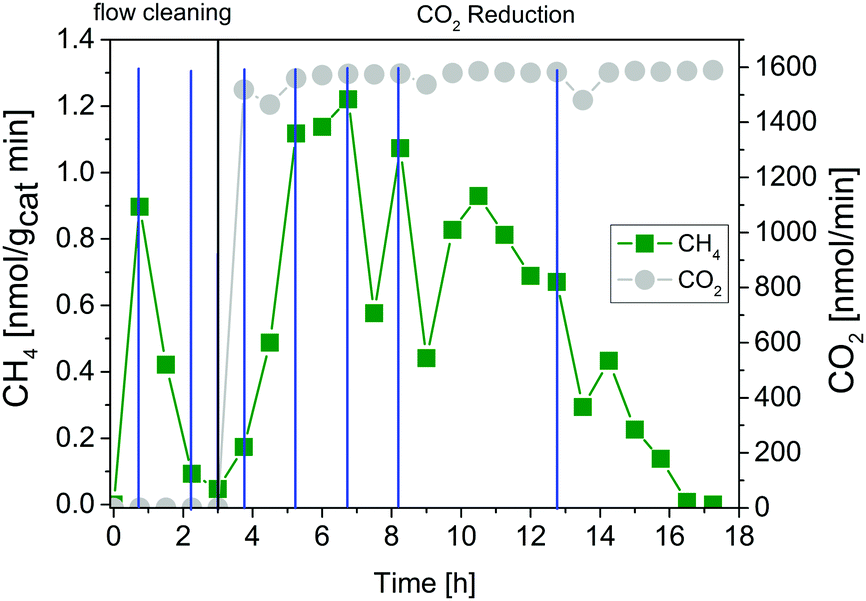 | ||
| Fig. 6 Photocatalytic CO2 reduction with P25 calcined at 400 °C (flow cleaning from 0 to 3 h, CO2 reduction from 3 h to 17.75 h, pulse dosing of H2O indicated by blue dashes). Lines are included in order to guide the eye. | ||
Discussion
The results can be discussed in the context of the general understanding of photocatalytic energy conversion reactions on TiO2-based materials. Then, a detailed insight into the mode of action can be obtained for IrOx/TiO2 and some hypotheses on the reaction progress on TiO2 can be formulated.Photocatalytic cleaning and H2O splitting on IrOx/P25
The IrOx-modified P25 samples showed a noticeable activity in the formation of H2 and O2. Due to the strong dependence of the activity from the dosing of H2O and the O2:H2 ratio of approximately 1:2, it is assumed that the product formation originates from overall photocatalytic H2O splitting (4). This is a meaningful result showing that the stoichiometric H2O splitting in a gas–solid process has been realized, since no external bias or any sacrificial agents were used in order to improve the formation rates of H2 and O2. Furthermore, it is possible to observe the simultaneous formation of both products in one high-purity reaction chamber. However, it is questionable how the product formation is realized on IrOx/P25 and which charge carrier dynamic processes are involved. A hypothetical approach to explain the activity of IrOx/P25 can be based on the two main functions of a photocatalyst. In this sense TiO2 represents the photoabsorber, where mobile charge carriers are generated under absorption of UV photons. Some of the photogenerated h+ escape from recombination and migrate to the IrOx particles, where they catalyze the oxidation of H2O to O2(1). In this way IrOx represents the function of the heterogeneous catalyst. This has been well documented in liquid-phase H2O splitting. The separation of charge carriers over the interface of TiO2 and IrOx enhances their lifetime, thus it is beneficial for the light efficiency of the overall H2O splitting reaction (4). The H+ formed from H2O oxidation (1) are then reduced to H2 gas (3). Since the electrons are left behind in TiO2, this reaction is likely to proceed on the TiO2 surface. For this purpose TiO2 must expose sites which can transfer electrons. It appears that such sites are available and their accessibility is maintained even under continuous flow conditions and accumulation of H2O on the surface of TiO2, since the activity of H2 formation is almost stable during the H2O splitting experiments.| 2H+ + 2e− → H2 E0 = 0 V | (3) |
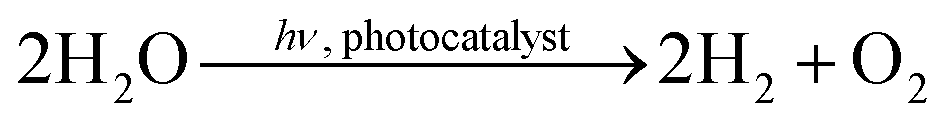 | (4) |
According to the hypothetical approach in the previous chapter to explain the activity of IrOx/P25 it is expected that the H2O oxidation reaction similarly proceeds on CoOx and the formation of H2 on TiO2. In Fig. S1A and B (ESI†) it was shown that only the sample calcined at 200 °C showed activity in H2 formation. However, the fact that H2 was formed indicates that a hydrogen source has been provided by photocatalytic H2O oxidation. The interface between TiO2 and CoOx can potentially cause an insufficient hole transfer between the photoabsorber and the co-catalyst which lowers the activity of H2O oxidation. Furthermore the reaction pathway of photocatalytic H2O oxidation on CoOx will be different compared to IrOx. Therefore it is possible that specific steps in the overall reaction mechanism cannot be activated sufficiently to allow an efficient O2 formation. As a consequence H2O is only oxidized partly, so that the resulting oxygen-derived intermediates remain on CoOx, without being further oxidized to O2 gas. In this way the oxygen-derived intermediates might prevent further oxidation of H2O, which lowers the activity over time as it was observed in Fig. S1A (ESI†).
Potential reasons for the lack of CH4 formation on IrOx/TiO2
The CO2 reduction experiments with the IrOx/P25 samples displayed no measurable activity in CH4 formation. Instead, O2 and H2 formation are the dominant processes and only small amounts of CH4 can be formed if H2O dosing is stopped (Fig. 4A). This observation is in stark contrast to the reference samples. It is highly probable that the improved H2O splitting conditions by the IrOx modification are responsible for the absence of CH4 formation. In the following it will be elucidated in which ways the H2O oxidation reaction might affect the CO2 reduction reaction.The oxidation of H2O to O2 is often represented by four single step reactions (5)–(8).31,46–48
| H2O + h+ → OHads + H+ | (5) |
| OHads + h+ → Oads + H+ | (6) |
| H2O + Oads + h+ → OOHads + H+ | (7) |
| OOHads + h+ → O2 + H+ | (8) |
Aside from hydrocarbon oxidation, another explanation for the absence of CH4 formation could be based on a competition for charge carriers. For instance the reduction of O2(9) and the generation of H2(3) are also electron consuming reactions. It is questionable which of these processes is associated with the lowest barrier and is thus more likely to proceed. Due to a lack of knowledge on activation barriers, the redox potentials of the particular reactions are regarded as a first approximation.
| O2 + e− → O2˙− E0 = −0.137 V | (9) |
| CO2 + e− → CO2˙− E0 = −1.9 V | (10) |
The absence of CH4 formation in the presence of a highly active co-catalyst for O2 evolution due to reactive oxygen species and competitive charge transfer reaction appears to be plausible.
As a third possible explanation it should be considered that at least one potential mechanism suggested for CO2 reduction to CH4 on TiO2, namely the glyoxal pathway,18 contains not only reductive but also oxidative elementary steps. If such a mechanism is in operation, it will no longer be possible on TiO2 if all holes are transferred to IrOx.
H2O oxidation on IrOx/TiO2 and fate of oxygen
What makes the results of the IrOx/P25 sample significantly more interesting is the activity in the H2 formation as a consequence of the improved H2O oxidation properties, together with the delayed O2 formation rate in the initial phase of the flow cleaning experiments. The obtained results provide specific information about the photocatalytic properties of TiO2. Those will later be used to explain product formation of photocatalytic CO2 reduction on TiO2.The results with IrOx/P25 (Fig. 1, 2 and 4) indicated that not all O2 produced from H2O oxidation was detected over the course of the experiment. In case of CO2 reduction on P25 it was even not possible to detect any O2. For this reason the deficient amount of O2 was determined for the IrOx/P25 and P25 samples calcined at 400 °C. At first the absent amount of O2 is calculated for IrOx/P25. For this purpose the overall H2 formation and the O2:H2 ratio of 1:2 as proposed in reaction (4) are used. The difference between the theoretically expected (O2 theoretical) and the detected amount (O2 detected) corresponds to the amount of O2 which was not found (O2 absent) in the gas-phase of the photoreactor (Table 1).
| Molar amount [nmol] | |
|---|---|
| H2 | 247 |
| O2 theoretical | 123.5 |
| O2 detected | 20 |
| O2 absent | 103.5 |
In order to realize a reliable comparison to the absent amount of O2 in CO2 reduction, it would be necessary to run the reaction until stoichiometric formation of both products can be observed, too. However, the CH4 formation ceased after 14.25 h of CO2 reduction (Fig. 6). Consequently it is solely possible to determine the theoretically expected amount of O2 to this point of time. The calculation was performed under consideration of the stoichiometry in reaction (2). Thereby the molar amount of O2 corresponds to twice the molar amount of CH4 (Table 2).
| Molar amount [nmol] | |
|---|---|
| CH4 | 39 |
| O2 absent | 78 |
The comparison of the balances (Tables 1 and 2) reveals that the absent amounts of O2 in both the H2O splitting reaction with IrOx/P25 (Fig. 1) and the CO2 reduction with bare P25 (Fig. 6) are at least in the same order of magnitude. On this account it appears realistic that the observed phenomena are based on the same effect. The present study uses a GC as the analytical device so that only the gas phase of the photoreactor can be analyzed. Hence, the absence of O2 in the analysis is either indicative for the absence of its formation or the consumption by the sample. The obtained results are the basis to hypothesize the fate of oxygen in photocatalytic reaction on TiO2.
Plausible conclusions on the mode of action of TiO2 in photocatalytic CO2 reduction
In contrast to the IrOx-modified samples, the reference P25 samples exhibited a significant activity in the CH4 formation. At the same time there was no observable activity in the H2O splitting reaction. In general, the photocatalytic H2O oxidation is thermodynamically possible on bare TiO2. Kinetic barriers in the overall reaction cycle might result in a very small apparent rate when no co-catalyst is used. Although no indication for water splitting is observed for bare TiO2 our previous work has shown that the presence of water is required for CH4 formation from photocatalytic CO2 reduction. Therefore, water is still expected to deliver hydrogen for CH4 formation. Considering that the proposed elementary steps of H2O oxidation (5)–(8) all liberate H+, a hydrogen source is generated even if the final O2 evolution step is kinetically hindered. From the results with the IrOx/P25 samples it can be concluded that such a source of hydrogen should be rapidly reduced to H2 on TiO2. However, on bare TiO2 we did not observe H2 formation, so it is questionable if any of the generally assumed steps of H2O oxidation occurs under the applied reaction conditions. If this is not the case, a different source of protons than photocatalytic H2O oxidation needs to participate in C–H bond formation. Moreover, the results of the present study revealed that CH4 formation only proceeds when no H2O is oxidized and the by-product O2 is absent in the gas-phase. This correlation is in conflict with the proposed reaction eqn (2).Based on our results, product formation in photocatalytic CO2 reduction coupled to oxygen consumption by TiO2 is suggested as likely explanation. One feasible way for such consumption is the replenishment of oxygen vacancies (OV). From experimental studies53–55 it is known that O2 dissociates and can undergo reactions with OV on the surface of TiO2 under room temperature conditions. Since all experiments of this study were performed under such conditions and the amount of absent O2 is very small, it is proposed that the replenishment of OV may explain the fate of this by-product. The obtained results of the IrOx/P25 samples showed that O2 formation started delayed to the H2 formation. The replenishment of OV may be the reason why fractions of O2 were temporarily not detectable in the gas phase. Once all defects are healed the release of the entire amount of O2 from the surface of the photocatalyst initiates, so the formation rates converged to the stoichiometric ratio of H2O splitting. Another explanation for the initially missing gas-phase oxygen not further discussed here could be a formation of peroxide species adsorbed on the catalyst surface. The adsorption56,57 and reactions57 of peroxide species, possibly also formed in a gas-phase H2O splitting reaction on TiO2, has been examined in detail by comprehensive theoretical studies. Hydrogen peroxide has also been identified as origin for an oxygen deficit in liquid-phase photocatalytic water splitting over Pt/TiO2.58
The overall CO2 reduction is proposed to include two processes forming O2. On the one hand side the formation of CH4 by reduction of CO2, on the other hand side the oxidation of H2O to generate the source of hydrogen for C–H bond formation. Assuming that oxygen vacancies are responsible for the absence of O2, these point defects are strongly associated with the photocatalytic activity of TiO2. In one of our studies33 it was verified that the presence of O2 in the gas-phase of the reaction gas atmosphere induces an inhibition of CH4 formation. Since oxygen-derived species from CO2 reduction are also suspected to affect the CH4 formation negatively, a replenishment of OV might avoid that gaseous O2 and potentially reactive oxygen-derived intermediates inhibit the CH4 formation.
Even though H2O oxidation according to the generally accepted mechanism is not likely to occur on bare P25-TiO2, the CH4 formation has been proven to require the presence of H2O.32 For this reason an alternative pathway for liberation of the hydrogen in H2O must be considered. In general, surface hydroxyl groups are naturally present on the surface of TiO2 and can be formed by dissociative adsorption of H2O in a OV(11).
| H2O + OV + Obr → 2OHbr | (11) |
Anyway, the irreversible replenishment of vacant positions by excess oxygen from CO2 or H2O is an example for a stoichiometric reaction of the photocatalyst. Most likely the photocatalytic activity in CO2 reduction will be diminished as soon as all vacancies are healed. The observed termination of the CH4 formation in the CO2 reduction after 14.25 h (Fig. 6) underlines this hypothesis. Overall, the results indicate that the CH4 formation is presumably not based on a complete catalytic cycle including both the photocatalytic CO2 reduction and H2O oxidation.
Experimental
High-purity gas-phase photoreactor set-up
The photocatalytic CO2 reduction experiments were carried out with a reactor set-up which works under conditions of highest purity. A detailed description of the set-up was given by Mei et al.12 In brief, the reactor is made from stainless steel components which are usually used for ultra-high vacuum application. All tube connections are realized by VCR and CF connections, so that sealing of the overall system works grease-free. Purging of the reactor is performed by 99.9999% He gas (He 6.0). A diluted CO2 in He mixture (7000 ppm CO2 in He 6.0) is used as the reactant gas. Dosage of H2O into the gas-phase is realized by a stainless steel saturator, which is temperature controlled. The H2O concentration in the gas flow is adjusted by the temperature of the saturator. Mass flow controllers are used for purging and dosing of the reactants. The powdered sample is finely distributed in a quartz vessel, which is placed in the center of the reactor. Illumination is conducted by a 200 W Hg/Xe lamp from Oriel instruments. A water-based filter is utilized to remove the IR radiation from the lamp spectrum. Product analysis is executed by a Shimadzu Tracera GC 2010 Plus. This GC is equipped with a barrier discharge ionization detector (BID), which allows quantifying CO2, CO, CH4, C2H6, H2O, O2 and H2 in the 0.1 ppm range. On the basis of this high sensitivity, the BID is a suitable analytical device for an application in continuous-flow photocatalytic CO2 reduction.Sample preparation
Sample modification of P25 with IrOx and CoOx was performed via photodeposition in a semi-batch reactor made from quartz glass. On-top illumination was realized by a 1000 W Hg/Xe lamp. 400 mg P25 was dispersed in 200 mL H2O and ultra-sonicated for one minute. Then the dispersion was given into the reactor and 20 mL of methanol were added. The precursors iridium acetate or cobalt acetate were added to the dispersion amounting to a nominal loading of 0.05 wt% Ir and Co, respectively. The reactor was sealed and deaerated with pure He gas for 1.5 h. After purging the reactor, the He flow rate was set to 20 mL min−1 and the illumination was initiated. The overall deposition time was 3 h. Subsequently the dispersion was filtered and dried overnight. The obtained powder was then divided into two equal fractions. One fraction was calcined in synthetic air for 3 h at 200 °C, and the other at 400 °C, respectively, with a heating rate of 5 K min−1. Reference samples without Ir and Co have been prepared by the same procedure. Calcination of the samples is the first step to remove adsorbed carbon-containing species from the surface to prevent their potential contribution to the product formation leading to an overestimated activity.Sample pretreatment and CO2 reduction
70 mg of each sample has been tested in a CO2 reduction experiment. As the initial calcination steps are not sufficient to remove all of the carbon-containing species from the sample, further purification has been carried out before the activity test. This pretreatment step was performed in the photoreactor set-up. A He gas flow only including H2O was given through the reactor under illumination. The flow rate of H2O is adjusted to ∼1000 nmol min−1. This pretreatment under photocatalytic reaction conditions is further termed as flow cleaning. Although such an experiment is mainly performed to purify the sample from carbonaceous impurities, it simultaneously provides conditions for photocatalytic H2O splitting. Thus the activity in the H2O splitting reaction is determined before CO2 reduction is studied. GC measurements are conducted all 45 minutes to monitor the cleaning progress and the formation of H2 and O2. After the concentration of products from the flow cleaning procedure (CH4 and CO2) are sufficiently low the CO2 reduction experiment is initiated. Therefore, the gas flow is changed from pure He to 1700 nmol min−1 CO2 in He. The H2O flow rate in the CO2 reduction experiments is either held at ∼1000 nmol min−1 or changed to ∼25 nmol min−1. From our previous study32 we know that high H2O flow rates can inhibit product formation. A flow of ∼25 nmol min−1 H2O is sufficient to observe significant product formation.Conclusions
In this study the effect of improved H2O oxidation properties on the gas-phase photocatalytic CO2 reduction was studied by modifying P25 with co-catalysts for H2O oxidation. Simultaneously, potential reasons for the absence of gaseous O2 as the by-product of photocatalytic CO2 reduction on pure TiO2 were investigated The IrOx/P25 samples exhibited a significant activity in the overall photocatalytic H2O splitting, which was realized here in a pure gas–solid process under high-purity reaction conditions. It was possible to detect both products H2 and O2 in nearly stoichiometric amounts without the use of sacrificial agents. Photocatalytic CO2 reduction experiments with IrOx/P25 samples revealed that CH4 formation is only possible when no activity in H2 and O2 formation is observed. Competition for charge carriers by H2 formation and the backward reaction of CO2 reduction due to the presence of highly reactive oxygen species formed on IrOx appear to be responsible for the absence of CH4 formation. If photogenerated electrons are favorably consumed by H2 formation, but this product is not formed on bare TiO2, this might indicate that H2O oxidation is not the source of hydrogen for C–H bond formation in CO2 reduction. Furthermore, the results of the H2O splitting experiments on IrOx/P25 revealed that O2 and O-derived species are initially likely consumed by TiO2 under photocatalytic reaction conditions. A quantification of the missing amounts of O2 in the gas phase showed that a consumption of O2 by TiO2 could be responsible for the absence of this by-product in the CO2 reduction reaction. Under this circumstance TiO2 undergoes a stoichiometric reaction and the CH4 formation is not a true catalytic cycle. Instead, it runs only as long as TiO2 can consume oxygen. In addition the inhibiting effect of O2 on the product formation of CO2 reduction reveals that the consumption of this by-product is essential for the activity of TiO2 in this reaction. Only then the inhibiting effect of O2 and O-derived species in the photocatalytic CO2 reduction is prevented.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Open Access funding provided by the Max Planck Society.Notes and references
- E. V. Kondratenko, et al. , Energy Environ. Sci., 2013, 6, 3112 RSC.
- N. Armaroli and V. Balzani, Energy for a sustainable world: From the oil age to a sun-powered future, Wiley-VCH, Weinheim, 2011 Search PubMed.
- S. C. Roy, et al. , ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- R. Lal, Energy Environ. Sci., 2008, 1, 86–100 RSC.
- S. Sato, T. Arai and T. Morikawa, Inorg. Chem., 2015, 54, 5105–5113 CrossRef CAS PubMed.
- B. Ohtani, J. Photochem. Photobiol., C, 2010, 11, 157–178 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- J. Li and N. Wu, Catal. Sci. Technol., 2015, 5, 1360–1384 RSC.
- A. Di Paola, et al. , J. Phys. Chem. C, 2009, 113, 15166–15174 CrossRef CAS.
- M. Anpo and J. M. Thomas, Chem. Commun., 2006, 3273–3278 RSC.
- J. Nowotny, Oxide Semiconductors for Solar Energy Conversion: Titanium Dioxide, CRC Press, 2016 Search PubMed.
- B. Mei, A. Pougin and J. Strunk, J. Catal., 2013, 306, 184–189 CrossRef CAS.
- M. Anpo, et al. , J. Electroanal. Chem., 1995, 396, 21–26 CrossRef.
- A. Pougin, M. Dilla and J. Strunk, Phys. Chem. Chem. Phys., 2016, 10809 RSC.
- D. Lee and Y. Kanai, J. Am. Chem. Soc., 2012, 134, 20266–20269 CrossRef CAS PubMed.
- I. A. Shkrob, et al. , J. Phys. Chem. C, 2012, 116, 9461–9471 CrossRef CAS.
- C. Amatore and J. M. Saveant, J. Am. Chem. Soc., 1981, 103, 5021–5023 CrossRef CAS.
- I. A. Shkrob, et al. , J. Phys. Chem. C, 2012, 116, 9450–9460 CrossRef CAS.
- M. Anpo, et al. , J. Phys. Chem. B, 1997, 101, 2632–2636 CrossRef CAS.
- N. G. Moustakas and J. Strunk, Chem. – Eur. J., 2018, 24, 12739–12746 CrossRef CAS PubMed.
- J. Schneider, et al. , Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- E. Korovin, D. Selishchev and D. Kozlov, Top. Catal., 2016, 59, 1292–1296 CrossRef CAS.
- K. Teramura, et al. , Angew. Chem., Int. Ed., 2012, 51, 8008–8011 CrossRef CAS PubMed.
- K. Iizuka, et al. , J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- J.-C. Wang, et al. , ACS Appl. Mater. Interfaces, 2015, 7, 8631–8639 CrossRef CAS PubMed.
- L. Liu, et al. , ACS Catal., 2012, 2, 1817–1828 CrossRef CAS.
- F. Saladin and I. Alxneit, J. Chem. Soc., Faraday Trans., 1997, 93, 4159–4163 RSC.
- J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 130, 13885–13891 CrossRef CAS PubMed.
- H. Belhadj, et al. , Phys. Chem. Chem. Phys., 2015, 17, 22940–22946 RSC.
- Z. Dohnalek, et al. , J. Phys. Chem. B, 2006, 110, 6229–6235 CrossRef CAS PubMed.
- J. Rossmeisl, et al. , J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- M. Dilla, et al. , ChemCatChem, 2017, 9, 4345–4352 CrossRef CAS.
- M. Dilla, R. Schlögl and J. Strunk, ChemCatChem, 2017, 9, 696–704 CrossRef CAS.
- D. A. Panayotov and J. T. Yates, Chem. Phys. Lett., 2005, 410, 11–17 CrossRef CAS.
- R. L. Kurtz, et al. , Surf. Sci., 1989, 218, 178–200 CrossRef CAS.
- Z. Cai, et al. , Adv. Energy Mater., 2018, 8, 1701694 CrossRef.
- H. Tueysuez, et al. , Nano Res., 2013, 6, 47–54 CrossRef CAS.
- C. C. L. McCrory, et al. , J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- F. A. Frame, et al. , J. Am. Chem. Soc., 2011, 133, 7264–7267 CrossRef CAS PubMed.
- V. Pfeifer, et al. , Chem. Sci., 2016, 7, 6791–6795 RSC.
- V. Pfeifer, et al. , Chem. Sci., 2017, 8, 2143–2149 RSC.
- W.-H. Ryu, et al. , J. Mater. Chem. A, 2014, 2, 5610–5615 RSC.
- B. H. Meekins and P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 2304–2310 CrossRef CAS.
- Y. K. Kho, et al. , J. Phys. Chem. C, 2010, 114, 2821–2829 CrossRef CAS.
- D. Y. C. Leung, et al. , ChemSusChem, 2010, 3, 681–694 CrossRef CAS PubMed.
- Y.-F. Li, et al. , J. Am. Chem. Soc., 2010, 132, 13008–13015 CrossRef CAS PubMed.
- Y.-F. Li and A. Selloni, ACS Catal., 2016, 6, 4769–4774 CrossRef CAS.
- A. Valdes, et al. , J. Phys. Chem. C, 2008, 112, 9872–9879 CrossRef CAS.
- R. van de Krol and M. Grätzel, Photoelectrochemical Hydrogen Production, Springer US, 2011 Search PubMed.
- J. Petlicki and T. G. M. van de Ven, J. Chem. Soc., Faraday Trans., 1998, 94, 2763–2767 RSC.
- W. H. Koppenohl and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef.
- N. M. Dimitrijevic, et al. , J. Am. Chem. Soc., 2011, 133, 3964–3971 CrossRef CAS PubMed.
- M. A. Henderson, et al. , J. Phys. Chem. B, 2003, 107, 534–545 CrossRef CAS.
- Y. Du, Z. Dohnalek and I. Lyubinetsky, J. Phys. Chem. C, 2008, 112, 2649–2653 CrossRef CAS.
- S. Tan, et al. , J. Am. Chem. Soc., 2011, 133, 2002–2009 CrossRef CAS PubMed.
- H. Alghamdi, et al. , Surf. Sci., 2018, 669, 103–113 CrossRef CAS.
- W.-F. Huang, et al. , J. Comput. Chem., 2011, 32, 1065–1081 CrossRef CAS PubMed.
- V. Daskalaki, et al. , Chem. Eng. J., 2011, 170, 433–439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp07765g |
| This journal is © the Owner Societies 2019 |