
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structure and reactivity of Fe(IV)oxo species in metal–organic frameworks
Fernan
Saiz
 *a and
Leonardo
Bernasconi
*b
*a and
Leonardo
Bernasconi
*b
aInstitut de Ciència de Materials de Barcelona, Bellaterra, Barcelona, 08193, Spain. E-mail: fsaiz@icmab.es
bCenter for Research Computing, University of Pittsburgh, 312 Schenley Place, 4420 Bayard Street, Pittsburgh, PA 15260, USA. E-mail: leonardo.bernasconi@pitt.edu
First published on 4th February 2019
Abstract
We investigate the potential use of Fe(IV)oxo species supported on a metal–organic framework in the catalytic hydroxylation of methane to produce methanol. We use periodic density-functional theory calculations at the 6-31G**/B3LYP level of theory to study the electronic structure and chemical reactivity in the hydrogen abstraction reaction from methane in the presence of Fe(IV)O(oxo) supported on MOF-74. Our results indicate that the Fe(IV)O moiety in MOF-74 is characterised by a highly reactive (quintet) ground-state, with a distance between Fe(IV) and O(oxo) of 1.601 Å, consistent with other high-spin Fe(IV)O inorganic complexes in the gas phase and in aqueous solution. Similar to the latter systems, the highly electrophilic character (and thus the reactivity) of Fe(IV)O in MOF-74 is determined by the presence of a low-lying anti-bonding virtual orbital (3σ*), which acts as an electron acceptor in the early stages of the hydrogen atom abstraction from methane. We estimate an energy barrier for hydrogen abstraction of 50.77 kJ mol−1, which is comparable to the values estimated in other gas-phase and hydrated Fe(IV)O-based complexes with the ability to oxidise methane. Our findings therefore suggest that metal–organic frameworks can provide suitable supports to develop new solid-state catalysts for organic oxidation reactions.
I. Introduction
Natural gas has attracted attention in the last few decades for its potential use in the production of energy, as a clean and sustainable alternative to oil and coal and as a means to provide important feedstock for the chemical industry on a planetary scale. Although global reserves of natural gas are estimated to be of the order of 1011 m3,1 the direct use of methane gas, its main component, is severely limited by transport costs from production sites to consumption areas. Currently, natural gas is transported through high-pressure pipelines or in liquefied natural gas (LNG) carriers, which suffer from high compression and refrigeration costs. The conversion of methane to liquid species, e.g. dimethyl ether, formaldehyde, acetic acid or liquid fuels, via Fischer–Tropsch catalysis offers an appealing alternative to the direct transportation of methane gas.2Methane can be oxidised to methanol using several multi-step industrial processes.3 Some of these reactions require catalysts, while others occur in the absence of a catalyst. For instance, in a two-step process methane is first decomposed into synthesis gas (CO + H2) via e.g. dry reforming
| CH4 + CO2 ⇔ 2CO + 2H2 (ΔH° = 247.0 kJ mol−1) | (1) |
| CH4 + 0.5O2 ⇔ CO + 2H2 (ΔH° = −36.0 kJ mol−1), | (2) |
| CH4 + H2O ⇔ CO + 3H2 (ΔH° = 206.2 kJ mol−1) | (3) |
| CO + 2H2 ⇔ CH3OH (ΔH° = −90.9 kJ mol−1) | (4) |
The production of synthesis gas via steam reforming is an expensive method, as it requires high temperature and pressure. Considerable effort has therefore been devoted to developing more cost-effective methods for the methane-to-methanol conversion at mild temperature and pressure. Compounds based on transition metals offer a potential route towards this goal, and Fe-based catalysts are particularly appealing in view of the abundance and minimal environmental impact of Fe-species. In particular, Fenton-like reactions8–10 have been investigated in view of their applicability to oxidise several organic species, including methane. Although the origin and nature of the active catalytic species in Fenton oxidation are still being discussed, it is now well established that, in a variety of experimental conditions, an oxidoiron(IV) (ferryl) intermediate, [(H2O)5FeO]2+,11–19 is the main species responsible for hydrogen abstraction from substrate molecules in aqueous solution, as part of a two-step rebound oxidation mechanism.9,10,20
These findings have motivated the modelling of Fenton-like gas-phase complexes of various compositions.21–26 Density-functional theory (DFT) calculations have revealed that the highly reactive high-spin (quintet) state is favoured by weak (e.g. oxygen-based) coordination environments.27–30 In optimal conditions, these species can affect the abstraction of a hydrogen atom from a methane molecule with enthalpy barriers of the order of only 50 kJ mol−1. The ability of Fe(IV)O complexes to oxidise methane in aqueous solution has also been examined, and the presence of the solvent has been shown to play a crucial role in favouring the reaction.31–33 In the presence of suitable coordination environments for hydrogen abstraction reactions carried out in water solution at room temperature, free-energy barriers as low as ca. 30 kJ mol−1 have been predicted on the basis of DFT calculations, to be compared to an estimate of ca. 90 kJ mol−1 for the “Fenton catalyst” ([(H2O)5FeO]2+) in water solution.34 These results provide evidence that Fenton-like species with suitably engineered Fe-ion coordination environments can indeed be the basis to create new classes of homogeneous catalysts for hydrocarbon oxidation under mild working conditions.
The aim of this study is to explore the suitability of metal–organic frameworks (MOFs) as supports for Fe(IV)oxo species of catalytic relevance. MOFs are important in catalytic processes because of their large internal surface areas, tuneable topologies and potential applications as chemical reagents.35 Thanks to these properties, MOFs have been recently employed e.g. for the catalytic capture and degradation of alkanes, alkenes, aromatic hydrocarbons and oxygenated volatile organic compounds36 to reduce atmospheric emissions as well as the hydrogenation of CO2 to methanol.37–39 They have also been shown to adsorb and photocatalytically degrade organic compounds in water.40 In addition, the structure of MOFs can be tuned to oxidise light alkanes. Quantum-mechanical calculations have predicted that magnesium-diluted MOF-74 catalyses the oxidation of ethane in the presence of N2O.41 Similar calculations have also proposed that MOF-74 containing an Fe(IV)O species (CPO-27-Fe) can hydroxylate ethane to ethanol,42 as these framework materials have been shown to separate mixtures of light hydrocarbons at their Fe(II) coordination sites43 and to produce phenol with efficiencies as high as 60% using H2O2 as an oxidant.44 Recent studies have reported results concerning the oxidation of methane to methanol catalysed by Cu–Oxo clusters stabilized in NU-1000 MOFs,45 and of MOFs with design inspired to particulate methane monooxygenase (pMMO).46
In this work we study the ability of an Fe(IV)O species in MOF-74 to promote the direct oxidation of methane. As mentioned above, it has been shown that the catalytic activity of Fe(IV)O species is strongly influenced by the coordination environment of the Fe ion.19,21,31 In particular, it has been demonstrated that an oxygen-rich Fe coordination environment stabilises the most reactive (quintet) spin state of the Fe(IV)O moiety and decreases the energy of its lowest virtual orbital (3σ*), which is responsible for the electrophilic character, and therefore for the catalytic activity, of Fe(IV)O in hydrocarbon oxidation.21 The structure of MOF-74 offers, in principle, an ideal coordination framework capable of stabilising highly reactive Fe(IV)O species. In ref. 29 it has for instance been shown that Fe(II) ions can be included as O-coordinated stable dopants in MOF-74, and the resulting system exhibits excellent hydrocarbon separation properties. On the basis of these results, we will consider here the existence, structure, and reactivity of analogous solid-state systems containing Fe(IV)O ions. We will model Fe(IV)O/MOF-74 using the neutron diffraction data presented in ref. 29 and use DFT calculations at the B3LYP level on the crystalline system to examine the properties of the hypothetical Fe(IV)O/MOF-74 and its reactivity in the oxidation of methane.
The manuscript is organised as follows. In Section II, we describe the methods employed to model the Fe(IV)O/MOF-74 structure and to calculate various electronic properties of interest. Results concerning these properties and the reactivity of Fe(IV)O/MOF-74 in methane oxidation are described in Section III. We pay particular attention to identifying the most stable ground spin state among the several competing ones, as this is the crucial factor driving the reactivity of Fe(IV)O. We also present an analysis of the orbital structure of this system. Our results are summarised in Section IV.
II. Simulation methods
We create an initial structure for Fe(IV)O/MOF-74 using information obtained for acetylene/MOF-74 available from the Cambridge Crystallographic Database47 using the Materials Studio suite package.48 The space group is R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (148) representing a unit cell of dimensions of 25.92 × 25.92 × 6.95 Å3 with angles α = β = 90° and γ = 120°, containing 180 atoms and corresponding to 18 irreducible cells. The irreducible unit cell of Fe(IV)O/MOF-74 contains 10 atoms: one Fe, three framework O's, four C's, one H and one O(oxo). We replace the acetylene molecule present in the original structure with an oxygen atom O(oxo), at a distance of 1.68 Å from the Fe atom, which is slightly larger than the typical Fe(IV)–O(oxo) distances determined for gas-phase complexes (1.60–1.62 Å).21 We then optimise the atomic positions with the COMPASS2 force field. After optimisation, the Fe(IV)–O(oxo) bond length decreases to 1.65 Å.
(148) representing a unit cell of dimensions of 25.92 × 25.92 × 6.95 Å3 with angles α = β = 90° and γ = 120°, containing 180 atoms and corresponding to 18 irreducible cells. The irreducible unit cell of Fe(IV)O/MOF-74 contains 10 atoms: one Fe, three framework O's, four C's, one H and one O(oxo). We replace the acetylene molecule present in the original structure with an oxygen atom O(oxo), at a distance of 1.68 Å from the Fe atom, which is slightly larger than the typical Fe(IV)–O(oxo) distances determined for gas-phase complexes (1.60–1.62 Å).21 We then optimise the atomic positions with the COMPASS2 force field. After optimisation, the Fe(IV)–O(oxo) bond length decreases to 1.65 Å.
The atomic positions from the resulting configurations are then optimised with the DFT code CRYSTAL17.49 The DFT calculations are carried out using periodic boundary conditions with the hybrid B3LYP exchange–correction functional. We use this functional because it provides a reasonably accurate description of high-spin Fe(IV) states,50,51 which is also more adequate than most generalised-gradient approximations (e.g. PBE52 and BLYP53) in the solid state. We do however notice that, for gas-phase or solvated systems, the OPBE functional54 may also provide an accurate alternative to B3LYP (ref. 51). A standard all-electron 6-31G**55,56 basis set is used to represent the local atomic orbitals in terms of primitive Cartesian Gaussian functions. Polarization functions (p-functions for hydrogens and d-functions for carbons, oxygens, and silicons) are used to ensure that the orbitals can distort from their original atomic symmetry, and to better adapt to the molecular surroundings leading to a more accurate estimate of the total energy of the system with a high hydrogen content. Accurate truncation thresholds for the tolerances of the Coulomb and exchange bielectronic series are used in all calculations57 to improve the convergence rate during the self-consistent solution of the Kohn–Sham equations. Brillouin zone integrations are carried out using a Monkhorst–Pack net of 2 × 2 × 2 k-points, and a ground-state energy convergence is enforced of 1 × 10−5 Hartree. Long-range forces are included by adding the van der Waals dispersions given by Grimme's DF2 scheme.58
III. Results and discussion
III.1. Electronic structure of Fe(IV)O/MOF-74
Our analysis of the electronic structure of Fe(IV)O/MOF-74 begins with a structural optimisation of its primitive unit cell (shown in Fig. 1). This optimisation employs CRYSTAL17's symmetry operators and is carried out for the singlet, triplet, and quintet spin configurations of all six Fe(IV) atoms. Therefore, the difference between the number of α and β electrons is 12 for the triplet and 24 for the quintet. In Table 1 we list the absolute energies of the optimised structure of Fe(IV)O/MOF-74 obtained using three approximations for the exchange–correlation functional (LDA, BLYP and B3LYP). In all cases we find that the quintet is the most stable state, followed by the triplet and the singlet. A quintet ground state has also been reported for Fe(IV)O metal-proteins with oxidative activity, including taurine/α-ketoglutarate dioxygenase (TauD)59 and methane monooxygenase (MMO).60 Furthermore, our results indicate that for each level of theory the distance between Fe(IV) and O(oxo) in the quintet and triplet atoms changes by less than 1% with respect to the singlet. The calculated optimised distance is 1.601 Å, which is only 1.25% shorter than the values found in other gas-phase Fe(IV)O complexes.21 Considering that ref. 51 reported that functionals like LDA and BLYP disfavour high-spin states, while hybrid functionals provide the correct spin ground state of iron complexes, we will only use B3LYP to investigate further the electronic structure of Fe(IV)O/MOF-74. A more thorough study of the influence of the exchange–correlation model on the electronic properties of Fe(IV)O systems will be presented elsewhere.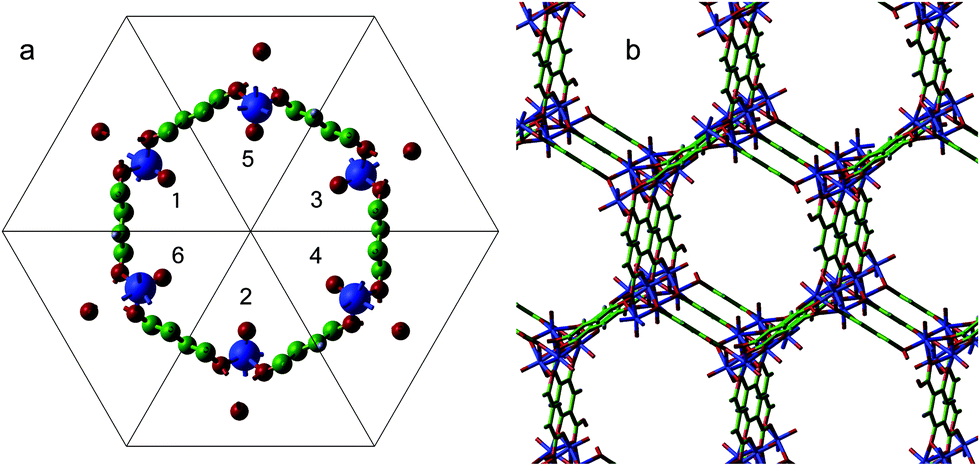 | ||
| Fig. 1 Representation of the primitive unit cell of Fe(IV)O/MOF-74 (a). Each triangle forming the hexagon is labelled 1–6 for reference. Each triangle contains 10 atoms. The Fe(IV) atoms are blue, the C atoms green, the O atoms red, and the H atoms grey. Panel b illustrates the interatomic bonds of the replicated unit cell in perspective view. | ||
| Functional | Spin | Total energy (Ha) | Difference in total energy w.r.t. singlet per Fe–O site (eV) | Bond Fe–O(oxo) (Å) |
|---|---|---|---|---|
| LDA | Singlet | −10270.070 | 0.000 | 1.588 |
| LDA | Triplet | −10270.196 | −0.571 | 1.594 |
| LDA | Quintet | −10270.205 | −0.615 | 1.597 |
| BLYP | Singlet | −10305.355 | 0.000 | 1.632 |
| BLYP | Triplet | −10305.528 | −0.782 | 1.628 |
| BLYP | Quintet | −10305.558 | −0.919 | 1.632 |
| B3LYP | Singlet | −10303.370 | 0.000 | 1.588 |
| B3LYP | Triplet | −10303.669 | −1.356 | 1.597 |
| B3LYP | Quintet | −10303.776 | −1.843 | 1.601 |
In Table 2 we list the energies of the lowest unoccupied orbitals localised on Fe atoms for the triplet state. The lowest unoccupied orbital has an energy minimum (−3.7788 eV) at k = (0.6265, 0.5326, 0.2760) Å−1 for the first iron atom. We also note that the orbital energies have the same values in more Fe atoms and k-points. For example, the LUMO energy at the gamma point is −3.6549 eV for the third and fourth Fe atoms and −3.3966 eV for the fifth and sixth Fe atoms. In the quintet state (Table 3) the spin α-LUMO (the 3σ* acceptor orbital, with an energy of −4.1615 eV at the Gamma point) is substantially more stable (0.7649 eV) than in the singlet, consistent with the results of ref. 21. This lower energy indicates that Fe(IV)O/MOF-74 can indeed exhibit strong oxidation ability.
| Spin | k-Point coordinates (Å−1) | Fe1 | Fe2 | Fe3 | Fe4 | Fe5 | Fe6 |
|---|---|---|---|---|---|---|---|
| ↑ | 0.0000, 0.0000, 0.0000 | −3.7179 | −3.6923 | −3.6549 | −3.6549 | −3.3966 | −3.3966 |
| ↑ | 0.1481, −0.0741, −0.0741 | −3.7528 | −3.7351 | −3.7263 | −3.624 | −3.4774 | −3.3801 |
| ↑ | 0.1481, 0.0542, −0.2023 | −3.7108 | −3.7024 | −3.6591 | −3.5794 | −3.4836 | −3.3878 |
| ↑ | 0.6265, 0.5326, 0.2760 | −3.7788 | −3.7788 | −3.6334 | −3.5912 | −3.5912 | −3.3068 |
| ↓ | 0.0000, 0.0000, 0.0000 | −3.2987 | −3.2987 | −3.1408 | −3.1408 | −3.0715 | −3.0715 |
| ↓ | 0.1481, −0.0741, −0.0741 | −3.4498 | −3.3347 | −3.1796 | −3.1757 | −3.1105 | −3.0676 |
| ↓ | 0.1481, 0.0542, −0.2023 | −3.2950 | −3.2436 | −3.1997 | −3.1489 | −3.0695 | −3.0679 |
| ↓ | 0.6265, 0.5326, 0.2760 | −3.5441 | −3.1947 | −3.1818 | −3.1155 | −3.1155 | −3.0864 |
| Spin | k-Point coordinates (Å−1) | Fe1 | Fe2 | Fe3 | Fe4 | Fe5 | Fe6 |
|---|---|---|---|---|---|---|---|
| ↑ | 0.0000, 0.0000, 0.0000 | −4.1615 | −4.1402 | −3.8518 | −3.8518 | −3.5548 | −3.5548 |
| ↑ | 0.1481, −0.0741, −0.0741 | −4.0824 | −4.0595 | −4.0219 | −3.9639 | −3.6361 | −3.5178 |
| ↑ | 0.1481, 0.0542, −0.2023 | −4.1558 | −4.1485 | −3.8384 | −3.7345 | −3.6619 | −3.5674 |
| ↑ | 0.6265, 0.5326, 0.2760 | −4.0782 | −4.0782 | −3.9676 | −3.9676 | −3.7850 | −3.4074 |
| ↓ | 0.0000, 0.0000, 0.0000 | −3.7128 | −3.7104 | −3.432 | −3.432 | −3.3555 | −3.3555 |
| ↓ | 0.1481, −0.0741, −0.0741 | −3.8155 | −3.7939 | −3.7691 | −3.6535 | −3.5057 | −3.4092 |
| ↓ | 0.1481, 0.0542, −0.2023 | −3.7123 | −3.7116 | −3.4296 | −3.3981 | −3.3796 | −3.3597 |
| ↓ | 0.6265, 0.5326, 0.2760 | −3.8182 | −3.8182 | −3.6882 | −3.6507 | −3.6507 | −3.3301 |
Fig. 2(a) shows that 90% of the LUMO's charge is distributed symmetrically in the primitive cell of Fe(IV)O/MOF-74 around each iron site. A closer look to one of the Fe atoms (Fig. 2(b)) indicates that the LUMO indeed exhibits the typical nodal structure of a 3σ* orbital, consistent with what has been found in a number of reactive gas-phase complexes containing Fe(IV)O groups (see e.g. Fig. 3 in ref. 21). This orbital has a lobe extending on the oxygen side of the Fe(IV)O group, which makes it an ideal acceptor of one electron from the incoming hydrogen atom of an alkane molecule located in the MOF pore.
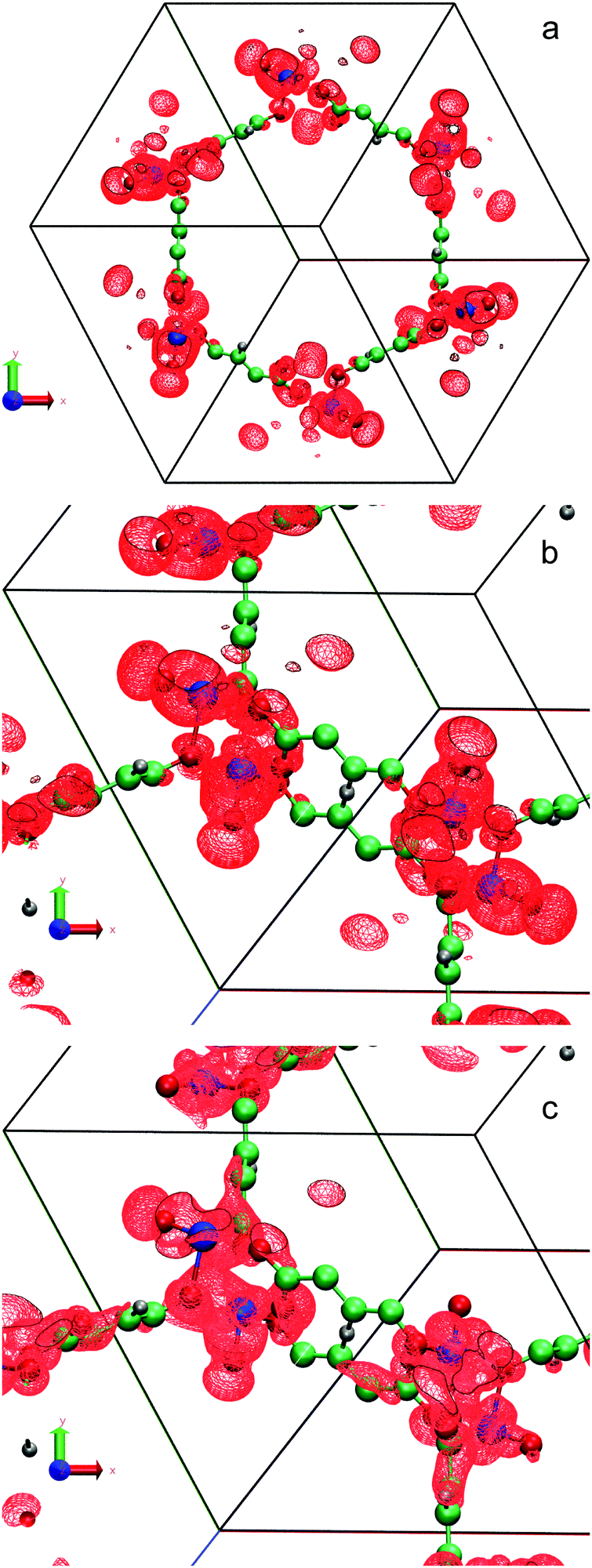 | ||
| Fig. 2 Symmetric representation (a) of the LUMO's charge density obtained with B3LYP using an isosurface value of 2.90 × 10−4 electrons per Bohr3. Zoomed-in view of the LUMO charge density (b) and wavefunction (c) with a value of 2.90 × 10−2 (electrons per Bohr3)1/2 showing the typical 3σ* anti-bonding nodal structure. Fe atoms are pink and O atoms blue. C and H atoms are green and grey respectively. | ||
The magnetic couplings between unpaired electrons on individual Fe atoms in the quintet state are also evaluated using broken-symmetry DFT calculations.61,62 We consider 64 (=26) possible spin arrangements, in which the spins of all six unpaired electrons in the 3d orbital of each of the 6 Fe atoms are simultaneously inverted. Table 4 lists the spins uninverted ↑ and inverted ↓ in each case and the total energy of the system with respect to case 1, which corresponds to the pure ferromagnetic coupling of all Fe centres with all uninverted 24 spins ↑. The second pure ferromagnetic coupling corresponds to inverting all 24 spins ↓ (case 64), and its energy is only 0.090 × 10−3 eV lower than that of case 1. The energy decreases more significantly (ca. two orders of magnitude) for the other 62 configurations. The lowest energy is found in case 29 (6.176 × 10−3 eV lower than in case 1). This decrease is achieved by flipping the spins in the first, fifth, and sixth Fe atoms. A similar reduction of (−6.155 × 10−3 eV) is obtained in case 22, where we reverse the spins of the first, third, and fifth Fe atoms. Therefore, although the energy difference is very small in all cases, we verify that the antiferromagnetic coupling is the most stable configuration for Fe(IV)O/MOF-74, as shown by case 29. The small energy differences between different spin configurations do however indicate that the magnetic Fe centres are virtually uncoupled, likely as a consequence of the large distance between them (7.94 Å) and/or of the inability of the MOF framework to act as a suitable channel for superexchange.
| Case | SFe1 | SFe2 | SFe3 | SFe4 | SFe5 | SFe6 | ΔE (×103 eV) | Case | SFe1 | SFe2 | SFe3 | SFe4 | SFe5 | SFe6 | ΔE (×103 eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | 0.00000 | 33 | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | −3.30782 |
| 2 | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | −3.28842 | 34 | ↓ | ↑ | ↑ | ↑ | ↑ | ↓ | −6.11714 |
| 3 | ↑ | ↑ | ↑ | ↑ | ↓ | ↑ | −3.28410 | 35 | ↓ | ↑ | ↑ | ↑ | ↓ | ↑ | −6.11698 |
| 4 | ↑ | ↑ | ↑ | ↑ | ↓ | ↓ | −3.28205 | 36 | ↓ | ↑ | ↑ | ↑ | ↓ | ↓ | −6.17347 |
| 5 | ↑ | ↑ | ↑ | ↓ | ↑ | ↑ | −3.28586 | 37 | ↓ | ↑ | ↑ | ↓ | ↑ | ↑ | −5.73200 |
| 6 | ↑ | ↑ | ↑ | ↓ | ↑ | ↓ | −3.30589 | 38 | ↓ | ↑ | ↑ | ↓ | ↑ | ↓ | −5.82871 |
| 7 | ↑ | ↑ | ↑ | ↓ | ↓ | ↑ | −3.30589 | 39 | ↓ | ↑ | ↑ | ↓ | ↓ | ↑ | −5.82604 |
| 8 | ↑ | ↑ | ↑ | ↓ | ↓ | ↓ | −0.51796 | 40 | ↓ | ↑ | ↑ | ↓ | ↓ | ↓ | −3.06970 |
| 9 | ↑ | ↑ | ↓ | ↑ | ↑ | ↑ | −3.30761 | 41 | ↓ | ↑ | ↓ | ↑ | ↑ | ↑ | −3.30336 |
| 10 | ↑ | ↑ | ↓ | ↑ | ↑ | ↓ | −5.73355 | 42 | ↓ | ↑ | ↓ | ↑ | ↑ | ↓ | −5.82596 |
| 11 | ↑ | ↑ | ↓ | ↑ | ↓ | ↑ | −6.12008 | 43 | ↓ | ↑ | ↓ | ↑ | ↓ | ↑ | −6.15260 |
| 12 | ↑ | ↑ | ↓ | ↑ | ↓ | ↓ | −5.82890 | 44 | ↓ | ↑ | ↓ | ↑ | ↓ | ↓ | −5.93012 |
| 13 | ↑ | ↑ | ↓ | ↓ | ↑ | ↑ | −6.11472 | 45 | ↓ | ↑ | ↓ | ↓ | ↑ | ↑ | −5.82971 |
| 14 | ↑ | ↑ | ↓ | ↓ | ↑ | ↓ | −5.82680 | 46 | ↓ | ↑ | ↓ | ↓ | ↑ | ↓ | −5.54089 |
| 15 | ↑ | ↑ | ↓ | ↓ | ↓ | ↑ | −6.17475 | 47 | ↓ | ↑ | ↓ | ↓ | ↓ | ↑ | −5.93328 |
| 16 | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | −3.07356 | 48 | ↓ | ↑ | ↓ | ↓ | ↓ | ↓ | −2.96105 |
| 17 | ↑ | ↓ | ↑ | ↑ | ↑ | ↑ | −3.31465 | 49 | ↓ | ↓ | ↑ | ↑ | ↑ | ↑ | −3.30184 |
| 18 | ↑ | ↓ | ↑ | ↑ | ↑ | ↓ | −6.11679 | 50 | ↓ | ↓ | ↑ | ↑ | ↑ | ↓ | −6.17570 |
| 19 | ↑ | ↓ | ↑ | ↑ | ↓ | ↑ | −5.73567 | 51 | ↓ | ↓ | ↑ | ↑ | ↓ | ↑ | −5.82805 |
| 20 | ↑ | ↓ | ↑ | ↑ | ↓ | ↓ | −5.82846 | 52 | ↓ | ↓ | ↑ | ↑ | ↓ | ↓ | −5.92680 |
| 21 | ↑ | ↓ | ↑ | ↓ | ↑ | ↑ | −6.11796 | 53 | ↓ | ↓ | ↑ | ↓ | ↑ | ↑ | −5.80977 |
| 22 | ↑ | ↓ | ↑ | ↓ | ↑ | ↓ | −6.15461 | 54 | ↓ | ↓ | ↑ | ↓ | ↑ | ↓ | −5.93034 |
| 23 | ↑ | ↓ | ↑ | ↓ | ↓ | ↑ | −5.82789 | 55 | ↓ | ↓ | ↑ | ↓ | ↓ | ↑ | −5.53602 |
| 24 | ↑ | ↓ | ↑ | ↓ | ↓ | ↓ | −3.07326 | 56 | ↓ | ↓ | ↑ | ↓ | ↓ | ↓ | −2.96205 |
| 25 | ↑ | ↓ | ↓ | ↑ | ↑ | ↑ | −3.30494 | 57 | ↓ | ↓ | ↓ | ↑ | ↑ | ↑ | −0.51987 |
| 26 | ↑ | ↓ | ↓ | ↑ | ↑ | ↓ | −5.82922 | 58 | ↓ | ↓ | ↓ | ↑ | ↑ | ↓ | −3.07234 |
| 27 | ↑ | ↓ | ↓ | ↑ | ↓ | ↑ | −5.82895 | 59 | ↓ | ↓ | ↓ | ↑ | ↓ | ↑ | −3.07318 |
| 28 | ↑ | ↓ | ↓ | ↑ | ↓ | ↓ | −5.53913 | 60 | ↓ | ↓ | ↓ | ↑ | ↓ | ↓ | −2.96284 |
| 29 | ↑ | ↓ | ↓ | ↓ | ↑ | ↑ | −6.17614 | 61 | ↓ | ↓ | ↓ | ↓ | ↑ | ↑ | −3.07158 |
| 30 | ↑ | ↓ | ↓ | ↓ | ↑ | ↓ | −5.92900 | 62 | ↓ | ↓ | ↓ | ↓ | ↑ | ↓ | −2.96292 |
| 31 | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ | −5.92876 | 63 | ↓ | ↓ | ↓ | ↓ | ↓ | ↑ | −2.96154 |
| 32 | ↑ | ↓ | ↓ | ↓ | ↓ | ↓ | −2.96562 | 64 | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | −0.09086 |
We now consider case 29 and its fully-symmetric counterpart to examine their spin populations. Our intention is to measure the fraction of spin polarisation that is initially transferred from the iron atoms to other atoms in the ground state. For the DFT simulations with full symmetry we find that 82.28% of the initial spin is retained by the Fe atoms, whereas 12.69% is transferred to the O(oxo) atom (see Table 5). For case 29 with broken-symmetry DFT we obtain that the initial spin retained by Fe atoms is 83.88% for spin polarisation ↑ and 84.09% for polarisation ↓, whereas the spin transferred to O(oxo) atoms is 21.41% with spin ↑ and 21.46% with spin ↓. These four values indicate that the initial polarisation is not inverted at self-consistency. The added oxygen is covalently bound to Fe with a double bond, and therefore some spin polarisation is transferred to the O atom because of the Fe–O orbital overlap. Additionally, no major differences in spin polarisation are observed for the oxygens, carbons, and hydrogens forming the MOF structure. The absence of spin polarisation transfer to the framework indicates that the framework-Fe bond is largely ionic, which suggests that the bonding in the MOF is similar to other FeO complexes in the gas phase (or in solution), and that the reactivity should therefore follow similar rules. Moreover, we observe only negligible (0.02 eV) energy differences in the energies of the HOMOs and LUMOs during the broken-symmetry DFT calculations.
| Symmetry | Broken symmetry (case 29) | ||||||
|---|---|---|---|---|---|---|---|
| Atom | 1–6 | 1 | 2 | 3 | 4 | 5 | 6 |
| Fe | 3.291 | 3.286 | −3.313 | −3.316 | −3.314 | 3.285 | 3.29 |
| O | 0.034 | 0.032 | −0.028 | −0.040 | −0.033 | 0.026 | 0.039 |
| O | 0.069 | −0.049 | 0.046 | −0.067 | 0.049 | −0.046 | 0.066 |
| O | 0.074 | 0.017 | −0.017 | −0.075 | −0.017 | 0.017 | 0.075 |
| C | 0.004 | 0.003 | −0.005 | −0.002 | −0.003 | 0.005 | 0.002 |
| C | 0.012 | −0.005 | 0.006 | −0.014 | 0.005 | −0.006 | 0.014 |
| C | 0.002 | −0.003 | 0.002 | 0.005 | 0.003 | −0.002 | −0.004 |
| C | 0.006 | −0.002 | 0.003 | −0.005 | 0.002 | −0.003 | 0.005 |
| H × 103 | 0.089 | −0.145 | 0.018 | −0.100 | 0.134 | −0.032 | 0.089 |
| O(oxo) | 0.508 | 0.507 | −0.475 | −0.483 | −0.478 | 0.504 | 0.511 |
| Fe/All (%) | 82.28 | 84.77 | 84.39 | 83.33 | 85.24 | 83.90 | 82.96 |
| O(oxo)/All (%) | 12.69 | 13.09 | 12.09 | 12.15 | 12.29 | 12.88 | 12.89 |
The Fe, O(oxo), and MOF atom projected densities of states are shown in Fig. 3. We observe notable differences in the spin channel distribution in the purely ferromagnetically coupled case. The spin polarisation of the Fe(IV)O group affects the Fe and O atoms in the 7–15 eV range and C–H sp3 bands in the 7–18 eV range, whereas the C–C sp3 bonds (>20 eV) are less affected. In contrast, in the antiferromagnetic case, the two spin components of the density of states are virtually identical.
 | ||
| Fig. 3 Electronic density of states of the purely ferromagnetic and antiferromagnetic states projected on Fe (a and d), O(oxo) (b and e), and the atoms forming the MOF structure (c and f). Curves are adjusted such that the Fermi level is set to 0.0 eV. | ||
III.2. Reactivity of Fe(IV)O/MOF-74 with methane
We examine the reactivity of Fe(IV)O/MOF-74 with methane by computing the reaction energy barrier for the interaction of a methane molecule in the MOF pore with one Fe(IV)O unit. A methane molecule is initially placed at the centre of the MOF pore with one of the C–H bonds pointing toward the O(oxo) atom, and the total energy of the system after geometry optimisation with the O(oxo)–H constrained to its initial value is calculated. We then reduce the constrained distance in steps of 0.089 Å, to obtain the reaction energy profile for the abstraction of an H atom from methane. This procedure is repeated until the O(oxo)–H distance reaches ca. 1 Å. At each distance, we calculate the DFT (B3LYP) optimised energy and the long-range dispersion energy, as well as the distance between the reactive hydrogen and the carbon atom of methane.Fig. 4 illustrates the reaction profile between 3.56 and 0.98 Å. All energies are plotted relative to their values at the initial O(oxo)–H distance of 3.56 Å. The behaviour of the total energy indicates that, as the hydrogen approaches the O(oxo) site, the former will initially displace the rest of the methane molecule with it. As observed in simulations of this reaction in the gas phase and in solution, an initial minimum in the energy is observed at an O(oxo)–H distance of ca. 2.4 Å. This minimum corresponds to the formation of a reactant complex in which the Fe(IV)O units and the substrate molecule are weakly bound (ca. 5 kJ mol−1). When the hydrogen atom is sufficiently close to the O(oxo) atom (ca. 1.25 Å), the H–CH3 bond starts to break and, simultaneously, an O(oxo)–H bond is established. The latter process is exothermic. We observe that the O(oxo)–H distance of 1.25 Å is very close to the separation of the reactive hydrogen and the central carbon (1.26 Å) and is ca. 0.19 Å larger than the equilibrium H–CH3 bond length. The dispersion forces increase up to an O(oxo)–H distance of 1.09 Å, after which they decrease by ca. 1 kJ mol−1. It is however clear from Fig. 4 that van der Waals interactions represent an important component of the overall energy profile, to which they contribute by as much as ca. 10 kJ mol−1, especially near the barrier maximum, where the methane molecule reaches the most unfavourable location before the H–CH3 bond is cleaved. As the H–CH3 bond breaks, the CH3 radical group is effectively free to move away from the reaction centre (Fig. 5). At the end of the reaction, the CH3 group appears to be located at a distance of 1.94 Å from the hydrogen atom now bound to the Fe(IV)O group. The angles of the CH3 moiety are between 119.2 and 119.6 degrees, which are close to the value of an sp2 hybridized structure, consistent with the radical nature of this group. The bond lengths between the central carbon and the three hydrogens (1.09 Å) are also consistent with a methyl radical structure. We observe that the global HOMO has a substantial contribution from MOF orbitals and its spatial distribution remains virtually unchanged as the methane molecule reacts with the Fe(IV)O moiety (Fig. 6). The overall energy barrier for the reaction (taken as the difference between the total energy at O(oxo)–H distances of 1.25 and 2.40 Å) is 50.77 kJ mol−1. This value indicates that an Fe(IV)O unit supported by an MOF-74 framework exhibits a reactivity in methane hydroxylation comparable to high-spin Fe(IV)O moieties in oxygen-rich coordination environments in the gas phase and in water solution. We also note that the methyl radical exhibits a high degree of mobility, at variance with what is observed in hydroxylation reactions carried out in water solution, in which, following the H abstraction step, this group remains pinned in the vicinity of the Fe(IV) centre and favourably oriented for the rebound step. Whether this larger mobility has a noticeable influence on the overall reaction mechanism and what is the fate of the methyl moiety produced after the C–H bond activation will be subjects of future work based on ab initio molecular dynamics simulations and free-energy calculations. Our results do however indicate that the rebound step of the hydroxylation reaction in a MOF environment can occur with more complex modalities than in water solution, because of the absence of the solvent cage effect. We also note that, according to our results, the C–H activation is an endergonic process, and it is therefore not thermodynamically favourable. It is however conceivable that the formation of a C–OH bond in the second step of the rebound mechanism can make the overall reaction thermodynamically favourable. We will examine this possibility in future work based on ab initio molecular dynamics free-energy calculations.
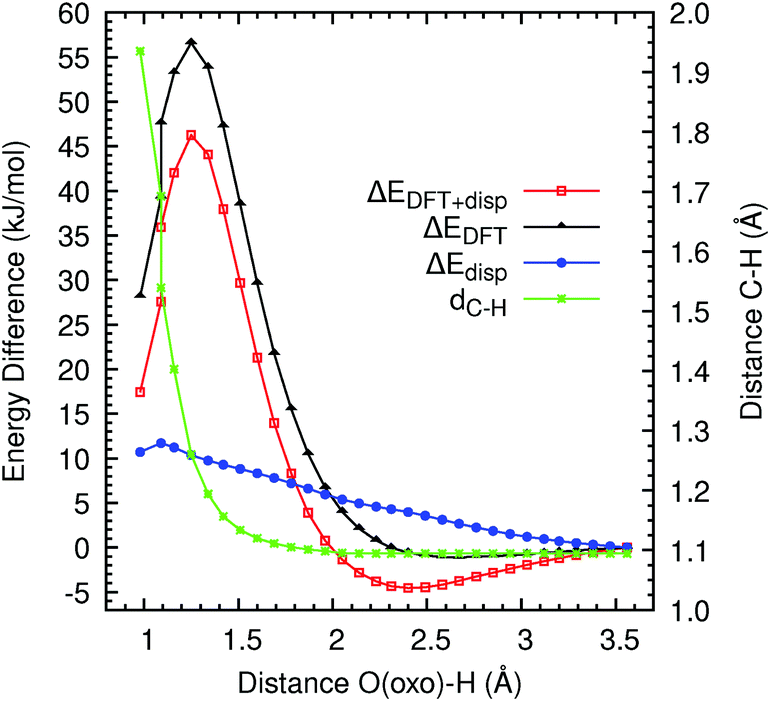 | ||
| Fig. 4 Difference of the total (ΔEDFT+disp), DFT (ΔEDFT), and long-range dispersion (ΔEdisp) energies from their values at 3.56 Å, and distance between the central C atom of the methane molecule and the reactive hydrogen (dC–H) vs. the distance between O(oxo) and the methane's H. All calculations are performed at the B3LYP level. | ||
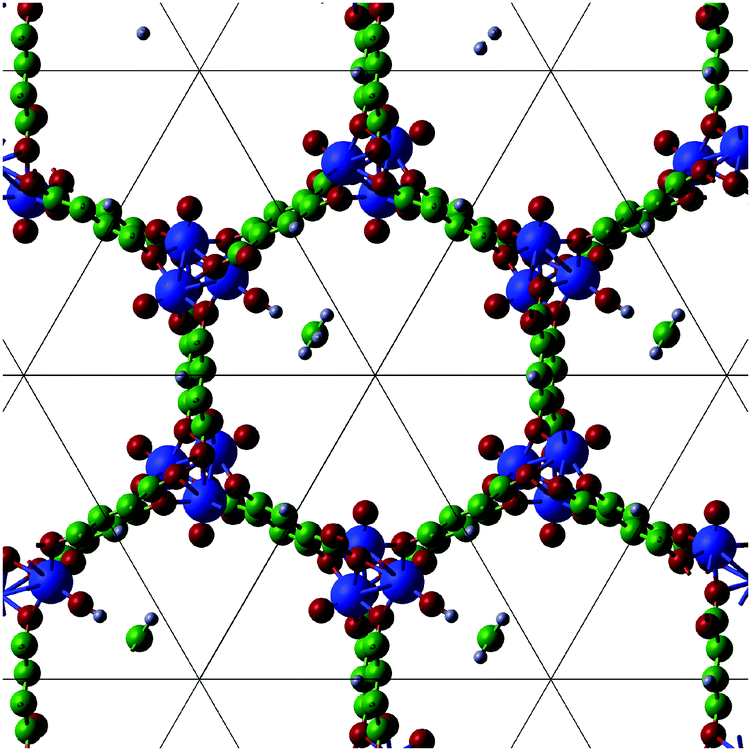 | ||
| Fig. 5 Representation of a replicated unit cell of Fe(IV)O/MOF-74 with an H atom bound to the O-end of the Fe(IV)O group. The H–O(oxo) distance is 0.98 Å and the H–CH3 distance is 1.94 Å. | ||
 | ||
| Fig. 6 Representation of the highest-occupied molecular orbital (HOMO) obtained using isosurface values of 1.49 × 10−4 electron per Bohr3 for an O(oxo)–H separation of 3.56 Å (a) and 1.53 × 10−4 electron per Bohr3 for 0.98 Å (b). | ||
Finally, we analyse the spin populations of the Fe(IV), O(oxo), the methane's carbon and the abstracted hydrogen. Fig. 7 shows the behaviour of these populations for O(oxo)–H distances between 0.98 and 3.56 Å. The spin population of H is almost negligible far away from the O(oxo) atom (7.14 × 10−6 at 3.56 Å) and shows an increase at around 2.00 Å reaching a maximum value of 0.019 at 1.16 Å, which corresponds to the maximum of the total energy. After this peak, the spin of H decreases to −0.017 when this atom forms a bond with O(oxo). The O(oxo)'s spin is also negative and drops by −0.150 upon H abstraction. The largest transfers of spin polarisation are observed on the Fe atom (+0.952) and on the methyl's carbon (−1.072). The former value represents the gain of one shared electron with the Fe atom from its bond with the O(oxo), while the latter corresponds to the loss of one common electron by the methyl carbon from its bond with the abstracted hydrogen. Fig. 8 illustrates the distribution of the density of the ↑ and ↓ spins within the methyl group upon abstraction of the hydrogen atom by O(oxo). This distribution shows that ↓ spin concentrates on the central C atom and its three surrounding hydrogens carry a ↑ spin.
 | ||
| Fig. 7 Spin populations of Fe and O(oxo) in the MOF and of the methane's H and C atoms as a function of the O(oxo)–H distance. | ||
 | ||
| Fig. 8 Representation of the spin density data on the methyl group obtained using isosurface values of 5.0 × 10−4 Bohr−3 (a) and −5.0 × 10−4 Bohr−3 for an O(oxo)–H separation of 3.56 Å (b). | ||
IV. Conclusions
We have used DFT calculations at the 6-31G**/B3LYP level to investigate the potential of Fe(IV)oxo species in metal–organic frameworks to act as methane-to-methanol conversion catalysts. Our DFT calculations indicate that the ground state of Fe(IV)O(oxo)/MOF-74 is in a high-spin (quintet) configuration, with an Fe(IV)–O distance of 1.601 Å. In the quintet state, the α LUMO exhibits the typical nodal structure of a 3σ* anti-bonding orbital, with an energy substantially lower than in the triplet and singlet states, indicating, by analogy with previous work on Fe(IV)O complexes, that Fe(IV)O/MOF-74 can have strong oxidation properties. Our calculations show that the antiferromagnetic and ferromagnetic configurations of the quintet have very similar total energies, with differences of less than 0.01 eV. In contrast, significant differences are observed in the electronic density of states projected on the Fe(IV), O(oxo), and the rest of the atoms forming the MOF skeleton and in the spin polarisation distribution for the antiferromagnetic relative to the ferromagnetic case. The calculated H-abstraction barrier from methane for the quintet ground state amounts to 50.77 kJ mol−1, indicating a reactivity comparable to high-spin Fe(IV)O moieties in oxygen-rich coordination environments and superior to that of the Fenton catalyst in water solution. Dispersion interactions are found to contribute sizably (up to ca. 10 kJ mol−1) to the overall reaction barrier.Our B3LYP calculations indicate that the triplet state of Fe(IV)O/MOF-74 is substantially higher in energy (ca. 281 kJ mol−1, Table 1) than the quintet ground state. Assuming that the Fe(IV)O centres contribute equally to the overall triplet–quintet energy difference, we can estimate that, at each reactive site, the triplet state is only ca. 46 kJ mol−1 higher in energy than the quintet. This value is comparable to our calculated H-abstraction barrier. It is therefore possible that the triplet state contributes to the reactivity of Fe(IV)O/MOF-74 in methane oxidation, at least at some stages of the reaction. By contrast, single-state (quintet) reactivity has been observed in Fe(IV)O complexes in the gas phase and in water solution.31 A more detailed analysis of two- (or multi-) state reactivity for Fe(IV)O/MOF-74 (which can affect, inter alia, the mechanism and rate constant of the hydroxylation reaction58) will be presented elsewhere. Future work will also be devoted to studying the effect of the exchange–correlation approximation on the electronic structure and on the reactivity of Fe(IV)O/MOF-74 as well as to compute free-energies of reaction at room temperature.
Finally, we observe that, under typical working conditions, the Fe(IV)O active group in MOF-74 has to be regenerated every time a methane molecule is converted to methanol (a process which, furthermore, can occur sub-stoichiometrically). Typically, the generation of Fe(IV)O from O2 requires high temperatures, which can make temperature-resilient MOFs (e.g. NU-1000) more suitable than MOF-74 under actual conditions, even though the formation of Fe(IV)O from O2 at room temperature and pressure (in water solution) has also been reported (see e.g. ref. 31). Our work nonetheless provides strong evidence for the existence of highly reactive Fe(IV)O centers stabilized by a MOF structure, which may pave the way for further theoretical and experimental work on the mechanistic details of hydrocarbon hydroxylation in a solid state environment.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by STFC through a Service Level Agreement with EPSRC and by the HPC Materials Chemistry Consortium (grant EP/L000202). This research was supported in part by the University of Pittsburgh Center for Research Computing through the resources provided.References
- R. H. Crabtree, Chem. Rev., 1995, 95, 987 CrossRef CAS.
- T. Mokrani and M. Scurrell, Catal. Rev., 2009, 51, 1–145 CrossRef CAS.
- M. J. da Silva, Fuel Process. Technol., 2016, 145, 42–61 CrossRef.
- See S. Ojala, N. Koivikko, T. Laitinen, A. Mouammine, P. K. Seelam, S. Laassiri, K. Ainassaari, R. Brahmi and R. L. Keiski, Catalysts, 2015, 5, 1092–1151 CrossRef CAS and references therein.
- M. C. Alvarez-Galvan, N. Mota, M. Ojeda, S. Rojas, R. M. Navarro and J. L. G. Fierro, Catal. Today, 2011, 171, 15–23 CrossRef CAS.
- S. S. Bharadwaj and L. D. Schmidt, Fuel Process. Technol., 1995, 42, 109–127 CrossRef CAS.
- J. M. Bermúdez, B. Fidalgo, A. Arenillas and J. A. Menéndez, Fuel, 2012, 94, 197–203 CrossRef.
- H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- J. T. Groves and M. Van Der Puy, J. Am. Chem. Soc., 1974, 96, 5274–5275 CrossRef CAS.
- J. T. Groves and G. A. McClusky, J. Am. Chem. Soc., 1976, 98, 859–861 CrossRef CAS.
- T. Løgager, J. Holcman, K. Sehested and T. Pedersen, Inorg. Chem., 1992, 31, 3523 CrossRef.
- O. Pestovsky and A. Bakac, J. Am. Chem. Soc., 2004, 126, 13757–13764 CrossRef CAS PubMed.
- O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. Münck, L. Que Jr. and A. Bakac, Angew. Chem., Int. Ed., 2005, 44, 6871–6874 CrossRef CAS PubMed.
- F. Buda, B. Ensing, M. C. M. Gribnau and E. J. Baerends, Chem. – Eur. J., 2001, 7, 2775–2783 CrossRef CAS.
- F. Buda, B. Ensing, M. C. M. Gribnau and E. J. Baerends, Chem. – Eur. J., 2003, 9, 3436–3444 CrossRef CAS PubMed.
- B. Ensing, F. Buda, P. Blöchl and E. J. Baerends, Angew. Chem., Int. Ed., 2001, 40, 2893–2895 CrossRef CAS PubMed.
- B. Ensing, F. Buda, P. Blöchl and E. J. Baerends, Phys. Chem. Chem. Phys., 2002, 4, 3619–3627 RSC.
- B. Ensing, F. Buda, M. C. M. Gribnau and E. J. Baerends, J. Am. Chem. Soc., 2004, 126, 4355–4365 CrossRef CAS PubMed.
- M. J. Louwerse and E. J. Baerends, Phys. Chem. Chem. Phys., 2007, 9, 156–166 RSC.
- J. T. Groves, J. Inorg. Biochem., 2006, 100, 434–447 CrossRef CAS PubMed.
- L. Bernasconi, M. J. Louwerse and E. J. Baerends, Eur. J. Inorg. Chem., 2007, 3023–3033 CrossRef CAS.
- L. Que and Y. Dong, Acc. Chem. Res., 1996, 29, 190–196 CrossRef CAS.
- P. Belanzoni, L. Bernasconi and E. J. Baerends, J. Phys. Chem. A, 2009, 113, 11926–11937 CrossRef CAS PubMed.
- S. Ye, G. Xue, I. Krivokapic, T. Petrenko, E. Bill, L. Que Jr and F. Neese, Chem. Sci., 2015, 6, 2909–2921 RSC.
- N. Lehnert, F. Neese, R. Y. N. Ho, L. Que Jr. and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 10810–10822 CrossRef CAS PubMed.
- N. Lehnert, R. Y. N. Ho, L. Que Jr. and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 12802–12816 CrossRef CAS PubMed.
- F. Neese, J. Inorg. Biochem., 2006, 100, 716–726 CrossRef CAS PubMed.
- A. Decker, J.-U. Rohde, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2004, 126, 5378–5379 CrossRef CAS PubMed.
- N. Lehnert, R. Y. N. Ho, L. Que Jr and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 8271–8290 CrossRef CAS PubMed.
- G. Ricciardi, E. J. Baerends and A. Rosa, ACS Catal., 2016, 6, 568–579 CrossRef CAS.
- L. Bernasconi, P. Belanzoni and E. J. Baerends, Phys. Chem. Chem. Phys., 2011, 13, 15272–15282 RSC.
- L. Bernasconi and E. J. Baerends, J. Am. Chem. Soc., 2013, 135, 8857–8867 CrossRef CAS PubMed.
- A. Kazaryan and E. J. Baerends, ACS Catal., 2015, 5, 1475–1488 CrossRef CAS.
- M. J. Louwerse and E. J. Baerends, Phys. Chem. Chem. Phys., 2007, 9, 156–166 RSC.
- L. Peng, M. Asgari, P. Mieville, P. Schouwink, S. Bulut, D. T. Sun, Z. Zhou, P. Pattison, W. van Beek and W. L. Queen, ACS Appl. Mater. Interf., 2017, 9, 23957–23966 CrossRef CAS PubMed.
- K. Vellingiri, P. Kumar and K. H. Kim, Nano Res., 2016, 9, 3181–3208 CrossRef CAS.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- F. N. Al-Rowaili, A. Jamal, M. S. Ba Shammakh and A. Rana, ACS Sustain. Chem. Eng., 2018, 6, 15895–15914 CrossRef CAS.
- M. Müller, S. Hermes, K. Kähler, M. W. E. van den Berg, M. Muhler and R. A. Fischer, Chem. Mater., 2008, 20, 4576–4587 CrossRef.
- E. M. Dias and C. Petit, J. Mater. Chem. A, 2015, 3, 22484–22506 RSC.
- P. Verma, K. D. Vogiatzis, N. Planas, J. Borycz, D. J. Xiao, J. R. Long, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2015, 137, 5770–5781 CrossRef CAS PubMed.
- H. Hirao, W. K. H. Ng, A. M. P. Moeljadi and S. Bureekaew, ACS Catal., 2015, 5, 3287–3291 CrossRef CAS.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS.
- S. Bhattacharjee, J.-S. Choi, S.-T. Yang, S. B. Choi, J. Kim and W.-S. Ahn, J. Nanosci. Nanotechnol., 2010, 10, 135–141 CrossRef CAS.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- https://www.ccdc.cam.ac.uk/structures/Search?Doi=10.5517%2Fccy2j0j, accessed 22 November 2017. SARGID: catena-(bis(η2-dideuteroacetylene)-(μ8-2,5-dioxido-1,4-benzenedicarboxylate)-di-iron(II)).
- Dassault Systèmes BIOVIA, Materials Studio Suite, v.5.5.2, Dassault Systèmes, San Diego, 2017.
- R. Dovesi, R. Orlando, A. Erba, C. M. Zicovich-Wilson, B. Civalleri, S. Casassa, L. Maschio, M. Ferrabone, M. De La Pierre, P. D’Arco, Y. Noel, M. Causa, M. Rerat and B. Kirtman, Int. J. Quantum Chem., 2014, 114, 1287 CrossRef CAS.
- S. Choomwattana, T. Maihom, P. Khongpracha, M. Probst and J. Limtrakul, J. Phys. Chem. C, 2008, 112, 10855–10861 CrossRef CAS.
- M. Swart, A. R. Groenhof, A. W. Ehlers and K. Lammertsma, J. Phys. Chem. A, 2004, 108, 5479–5483 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef.
- N. C. Handy and A. J. Cohen, Mol. Phys., 2001, 99, 403 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081 CrossRef CAS.
- R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, P. D’Arco, M. Llunell, M. Causà, Y. Noël, L. Maschio, A. Erba, M. Rerat and S. Casassa, CRYSTAL17 User's Manual, University of Torino, Torino, 2017 Search PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger Jr and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS PubMed.
- Y. Dong, L. Que Jr., K. Kauffmann and E. Muenck, J. Am. Chem. Soc., 1995, 117, 11377–11378 CrossRef CAS.
- L. Noodleman and E. R. Davidson, Chem. Phys., 1986, 109, 131–143 CrossRef.
- L. Noodleman and D. A. Case, Adv. Inorg. Chem., 1992, 38, 423–470 CrossRef CAS.
| This journal is © the Owner Societies 2019 |