
Possible B–C bonding in the hydroboration of benzonitrile by an external electric field†
Ming-Xia
Zhang
and
Hong-Liang
Xu
 *
*
Institute of Functional Material Chemistry, Department of Chemistry, National & Local United Engineering Laboratory for Power Battery, Northeast Normal University, Changchun 130024, Jilin, People's Republic of China. E-mail: hlxu@nenu.edu.cn
First published on 23rd November 2018
Abstract
Generally, the hydroboration of benzonitrile produces B–N containing compounds. However, an unprecedented B–C bond may be formed in the presence of a suitable external electric field (EEF). In reactions of benzonitrile with borane, when the EEF is oriented parallel to the positive direction (N → C) of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond, the barriers to Markovnikov hydroboration are decreased gradually, meaning the pathway for generating B–C bonds becomes more favorable. Accordingly, hydroboration could be influenced and its selectivity could be controlled by changing the magnitude and direction of an applied EEF.
C bond, the barriers to Markovnikov hydroboration are decreased gradually, meaning the pathway for generating B–C bonds becomes more favorable. Accordingly, hydroboration could be influenced and its selectivity could be controlled by changing the magnitude and direction of an applied EEF.
Boron-containing compounds, as a series of important intermediates, are widely utilized in many fields,1 such as organic synthesis, materials chemistry, optoelectronic devices and hydrogen storage. Catalytic hydroboration has played a powerful role in the preparation of organoboron compounds since it was used in the reduction of alkenes and alkynes, originally pioneered by Herbert C. Brown.2 Now, hydroboration mainly focuses on the reactions of unsaturated carbon (C)–carbon bonds (alkene, and alkyne),3 and carbon–oxygen (O) bonds (aldehyde, ketone, and ester).3c,4 Moreover, hydroboration addition to carbon—nitrogen (N) bonds is an essential process in the preparation of dyes, agrochemicals, and pharmaceutical compounds,5 and so on. Notably, only a handful of reduction reactions of organic nitriles5d,6 have been reported, even though synthesized unsaturated amine borane complexes have been known as isolable molecules for many years.7
On the other hand, hydroboration reactions of multiple C–N materials have been reported using different catalysts8 (transition metal complexes, the main-group catalysts, and the Lewis acid/Lewis acid–base pairs, etc.), which may have associated problems of cost, environmental pollution, by-products, and so on. Thus, seeking a new economical, environmentally friendly catalytic strategy is a requirement for the future. An external electric field (EEF), as an “invisible catalyst” used to accelerate/inhibit reaction rates and control selectivity, has been demonstrated theoretically9 and experimentally.10 Corresponding examples from theoretical and experimental investigations have sufficiently demonstrated the above conclusion. In 2010, Shaik et al.9c calculated that oriented electric fields could accelerate Diels–Alder reactions when the external field was oriented along the “reaction axis”, and control the endo/exo selectivity when it was oriented perpendicular to the “reaction axis”. Several years later, Aragonès et al.10b successfully designed a surface model system for the purpose of probing the Diels–Alder reaction, and coupled it with a scanning tunnelling microscopy break-junction approach.11
Inspired by the above key points, we selected the hydroboration of benzonitrile (PhCN) for the present study (see Scheme 1), to explore the influences of an EEF on the reaction rate and selectivity by using the QCISD(T)/6-311++G**//B3LYP/6-31G* method12 coupled with the Gaussian 09 program.13 Benzonitrile was selected as the reactant because it was used in the first experimental examples of the hydroboration of nitriles.14 Since then, it has also been selected as the reactant for hydroboration in other experiments.15 In order to verify that our method was reliable, three other methods (for example, the CCSD(T) method) were also utilized for some calculations. These results are shown in the ESI† (Tables S1–S3 and Fig. S1–S3). The directions of the EEF were parallel (FZ, Scheme 1b) and perpendicular (FX, Scheme 1c) to the N–C bond. An EEF along the FX direction could just change the reaction rate but not the selectivity (more details are shown in the ESI†) of hydroboration across the nitrogen–carbon triple bond. In order to examine the solvent effects on activation barriers, C6H6 (dielectric constant, ε = 2.27), THF (ε = 7.4), and DMSO (ε = 46.8) were employed. Furthermore, the synergistic effects of the EEF and solvent were also considered. These investigations indicated that solvents are unfavourable for this kind of hydroboration, and the results are presented in the ESI† (Fig. S6). The exploration of the hydroboration of PhCN in the gas phase and the changes caused by the EEF are discussed below.
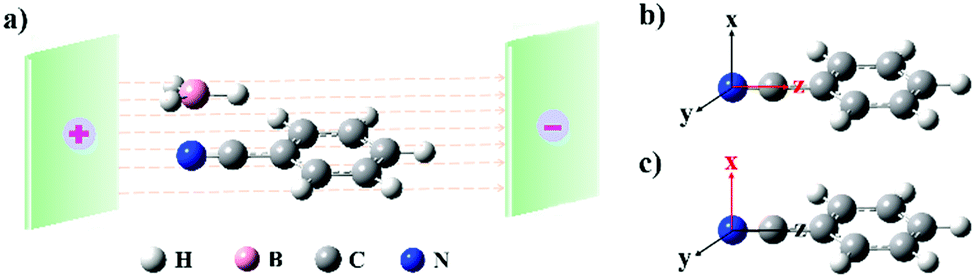 | ||
| Scheme 1 (a) Reaction model for the hydroboration of benzonitrile under an EEF (white balls denote hydrogen atoms, pink denotes boron, grey denotes carbon, and blue denotes nitrogen); (b) the positive direction (red arrow) of the EEF when it is oriented parallel to the nitrogen–carbon triple bond (the direction from N towards C atoms is denoted as FZ); (c) the positive direction (red arrow) of the EEF when it is oriented perpendicular to the nitrogen–carbon triple bond, denoted as FX. | ||
As shown in Fig. 1, two possible pathways exist for the hydroboration reactions of borane (BH3) with PhCN in the gas phase. The barrier height of path AM (a traditional anti-Markovnikov hydroboration reaction in which the B atom is added to the N atom) is 62.13 kJ mol−1 lower than that of path M (a Markovnikov hydroboration in which the B atom is added to the C atom). Thus, path AM is the more favorable reaction channel. When the transition states (TSs) of the two pathways are optimized, one can see that the –BH2 group is added to the N atom in TSAM, while it is bound to the C atom in TSM. The number and values of the vibration frequencies of the TSs are listed in the ESI† (Table S5). Compared with the structural parameters (Fig. 1) of TSAM, the bond lengths of N–C and B–H in TSM are respectively longer by 0.033 and 0.010 Å, indicating that these two bonds are much impaired in TSM. Accompanied by the newly formed bonds (N–H and C–B) in TSM have stronger interactions than the N–B and C–H bonds in TSAM. On the other hand, both for TSAM and TSM, the approach of BH3 to PhCN is asymmetric from the point view of bond distance. Intrinsic reaction coordinate (IRC)16 analyses for the two pathways were calculated to verify the rationality of the TSs, with the total energies and key bond lengths given in the ESI† (Fig. S4a and b). Fig. 2 shows the changes in the N–C and N–B bonds of TSAM, and the N–C and C–B bonds of TSM. Both the N–C bonds are elongated slowly as the reactions proceed, while the N–B and C–B bonds are respectively shortened for path AM and path M. On the other hand, one can clearly see that the slope of the C–B bond curve is larger than the slope of the N–B bond curve. That is to say, path M needs to overcome a higher reaction barrier in order to proceed. In addition, the variations of Mayer bond orders17 for these key bonds, plotted in the ESI† (Fig. S1c and d), further confirm the above conclusion. Electron localization function (ELF) isosurfaces18 (see ESI,† Fig. S4e and f) for IRC were employed to clearly visualize the process of the formation and rupture of key bonds, using the Multiwfn program.19 For TSAM, the isosurface of the NC bond shrinks gradually, and the B–H bond vanishes at the end, while the isosurfaces of the N–B and C–H bonds take shape as the hydroboration proceeds. The same explanation holds for TSM (Fig. S4f, ESI†).
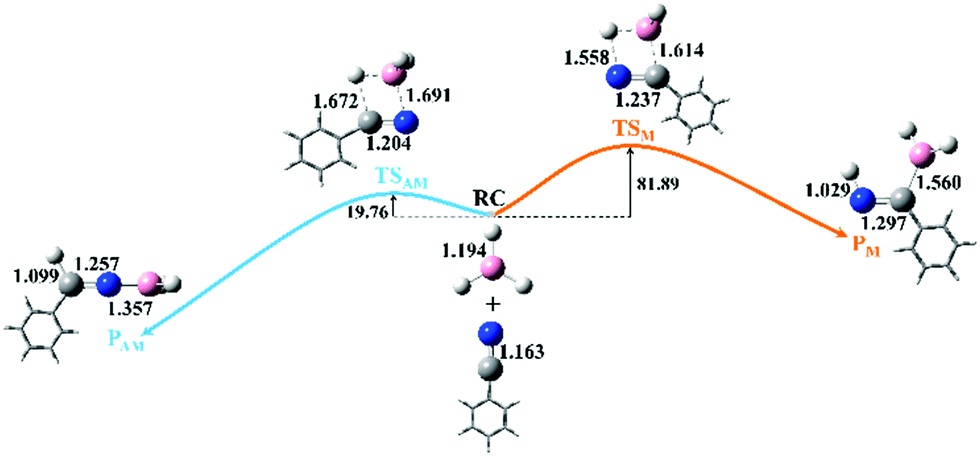 | ||
| Fig. 1 Two possible pathways of hydroboration of benzonitrile with borane in the gas phase (the orange line indicates path M, whereas the light blue line is for path AM; the energies are given in kJ mol−1). The geometries with key bond lengths (in Å) of the reactants, transition states (TSAM and TSM), and products (PAM and PM) of the two pathways are also shown. (The color coding of the balls is the same as that used in Scheme 1. For the sake of clarity, the phenyl group is represented in all figures by its diagrammatic drawing.) | ||
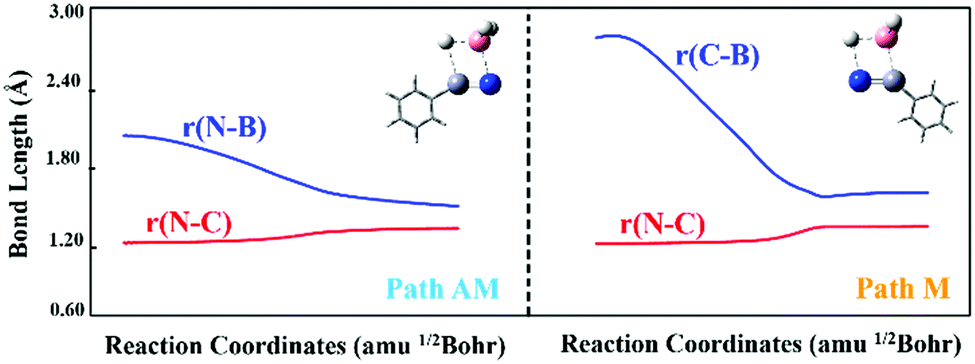 | ||
| Fig. 2 Variations in bond lengths of N–B and N–C for path AM, and of C–B and N–C for path M along the reaction coordinates. (The left panel represents the path AM, while the right panel represents the path M; the color coding of the balls is the same as that used in Scheme 1.) | ||
Interestingly, similar structures but very different conclusions were reached when the EEF was applied for the hydroboration of PhCN. Firstly, all of the geometries of the stationary points along the potential energy surfaces were optimized when a series of EEF were oriented along the Z axis. The selected TS structures with key bond lengths are shown in Fig. 3 and Table 1. Compared with those in a vacuum, these TSs maintained similar geometrical structures. Therein the largest change is found for the C–H bond in TSAM at FZ = −50 (× 10−4) a.u.; the bond is shortened by 0.022 Å. Then, the harmonic vibration frequencies were calculated for every TS with the EEF to verify that they each had only one imaginary frequency. These frequencies results are also presented in the ESI† (Table S5). Subsequently, we listed the values of the dipole moments as the EEF increased from FZ = 0 to 300 (× 10−4) a.u. so as to ascertain the root cause of the EEF effect. As seen in Table 2, the dipole moment decreases gradually with increasing EEF along the +FZ direction. Thus, we can conclude that the charges in the N atom are partially transferred to the −CPh group. The atomic dipole moment corrected Hirshfeld population (ADCH)20 charges (see Table 2) prove this above conclusion. It can be inferred that as the EEF is oriented to +FZ, the hydroboration of PhCN via path M should occur more easily than happens in the gas phase. From the point view of kinetics, the barrier height for path M in the EEF is indeed decreased, which is clearly shown in Fig. 4, and the opposite effect is obtained for path AM. Interestingly, in this +FZ region, the barrier height of path M is decreasing, while it is increasing for path AM, thus, a possible competition between the pathways could emerge as the EEF becomes larger. In other words, the path M would compete with path AM at some +FZ, to give a possible B–C bond formation in this hydroboration reaction. Because there is not an explicit definition of the normal catalytic range of an EEF, we increased the FZ to 300 (× 10−4) a.u. Although the barrier height of path M is still higher than that of path AM, the difference is reducing more and more. Thus, the EEF not only accelerates/inhibits the hydroboration of PhCN, but might possibly also control its selectivity.
 | ||
| Fig. 3 Geometries of the transition states (TSAM and TSM) with key bond lengths (in Å) at FZ = ± 50 (× 10−4) a.u.; the Z-directions of the EEF are also given. (The pink arrow represents +FZ, while the green arrow represents −FZ; the color coding of the balls is the same as that used in Scheme 1. Both N atoms are located at the coordinate origin.) | ||
| Structure | 0 | 50 | −50 | |
|---|---|---|---|---|
| TSAM | N–C | 1.204 | 1.202 | 1.208 |
| N–B | 1.691 | 1.702 | 1.678 | |
| C–H | 1.672 | 1.692 | 1.650 | |
| B–H | 1.269 | 1.269 | 1.270 | |
| C–C | 1.448 | 1.449 | 1.447 | |
| TSM | N–C | 1.237 | 1.234 | 1.240 |
| C–B | 1.614 | 1.615 | 1.613 | |
| B–H | 1.279 | 1.275 | 1.284 | |
| N–H | 1.558 | 1.577 | 1.540 | |
| C–C | 1.452 | 1.453 | 1.450 |
| 0 | 30 | 50 | 80 | 100 | 200 | 300 | |
|---|---|---|---|---|---|---|---|
| μ | 4.56 | 3.71 | 3.14 | 2.29 | 1.71 | −1.19 | −4.27 |
| e (N) | −0.29 | −0.27 | −0.25 | −0.23 | −0.23 | −0.20 | −0.17 |
| e (−CPh) | 0.29 | 0.27 | 0.25 | 0.23 | 0.23 | 0.20 | 0.17 |
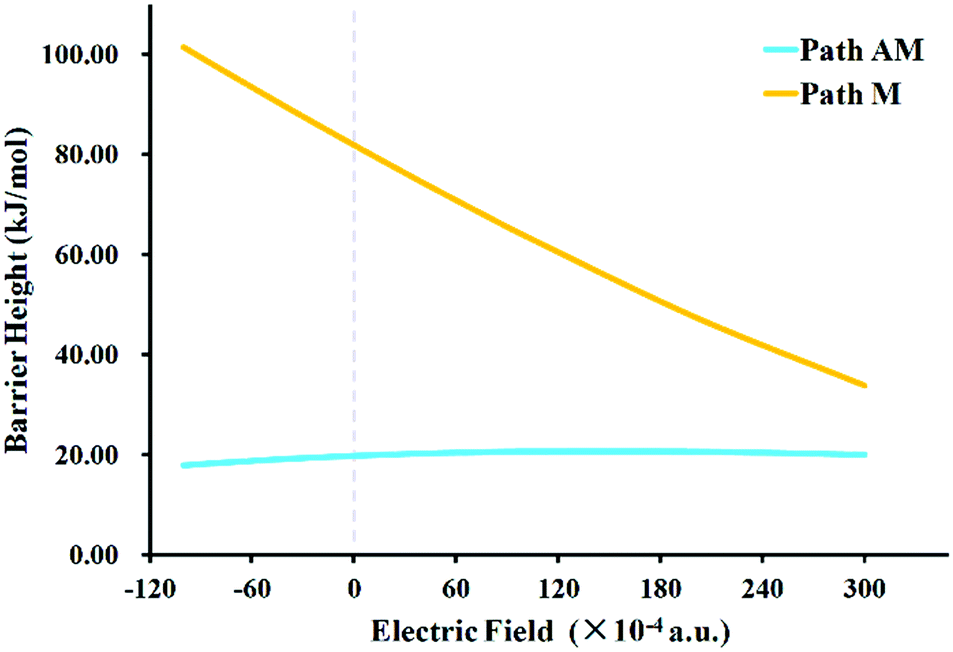 | ||
| Fig. 4 Variations of barrier heights for path AM and path M with changing EEF (orange line: path M; light blue line: path AM). | ||
In summary, the effect of an EEF on the hydroboration of benzonitrile was firstly studied in this work. The exploration of the reaction in the gas phase was found to have two possible pathways, path AM and path M, of which path AM is the more favorable channel by virtue of its lower barrier height. IRC analysis and Mayer bond orders were calculated to prove which bonds were formed (N–B and C–H for path AM, N–H and C–B for path M) and impaired/ruptured (NC/B–H). Furthermore, an ELF isosurface was used to visualize this changing process.
Intriguingly, as the EEF was applied for hydroboration, the structures of the stationary points along the potential energy surfaces changed a little. Similar to the result for the aminoborane complex,21 the polarization of the N–B bond was the main factor that caused the variations of the N–B distance. The EEF induced changes in the charges of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, and further a change in the dipole moment, which was the root cause of the effect on the reactions caused by the EEF. The barrier height of path M lowered gradually with increasing EEF along +FZ, and that for path AM increased. Thus, path M became more favorable in these conditions than in a vacuum. Moreover, path M might compete with path AM and even overcome it if the EEF becomes large enough. The solvent and its synergistic effects were investigated with the EEF; the study indicated that solvents C6H6, THF and DMSO are unfavourable for these hydroboration reactions. This study demonstrates theoretically that an EEF could accelerate/inhibit the hydroboration of benzonitrile, and control its selectivity.
N bond, and further a change in the dipole moment, which was the root cause of the effect on the reactions caused by the EEF. The barrier height of path M lowered gradually with increasing EEF along +FZ, and that for path AM increased. Thus, path M became more favorable in these conditions than in a vacuum. Moreover, path M might compete with path AM and even overcome it if the EEF becomes large enough. The solvent and its synergistic effects were investigated with the EEF; the study indicated that solvents C6H6, THF and DMSO are unfavourable for these hydroboration reactions. This study demonstrates theoretically that an EEF could accelerate/inhibit the hydroboration of benzonitrile, and control its selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the National Science Foundation of China (NSFC 21473026) and the Science and Technology Development Planning of Jilin Province (20140101046JC), and H.-L. X. acknowledges support from a project funded by the China Postdoctoral Science Foundation (2014M560227) and the Fundamental Research Funds for the Central Universities 2412018ZD008.References
- (a) R. M. Mikhailov, Organoboron Compounds in Organic Synthesis, Harwood Academic, 1984 Search PubMed; (b) H. C. Brown, Boranes in Organic Chemistry, Cornell University Press, Ithaca, 1972 Search PubMed; (c) P. Chen, R. A. Lalancette and F. Jäkle, Angew. Chem., Int. Ed., 2012, 51, 7994 ( Angew. Chem. , 2012 , 124 , 8118 ) CrossRef CAS PubMed; (d) Y. L. Rao, H. Amarne and S. Wang, Coord. Chem. Rev., 2012, 256, 759 CrossRef CAS; (e) W. Luo, P. G. Campbell, L. N. Zakharov and S. Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326 CrossRef CAS PubMed.
- (a) H. C. Brown, Hydroboration, Benjamin, W. A., New York, 1962 Search PubMed; (b) H. C. Brown and C. F. Lane, J. Am. Chem. Soc., 1970, 92, 6660 CrossRef CAS; (c) J. V. B. Kanth and H. C. Brown, J. Org. Chem., 2001, 66, 5359 CrossRef CAS PubMed.
- (a) C. C. Zhang, M. Zhou, S. W. Liu, B. J. Wang, Z. P. Mao, H. Xu, Y. Zhong, L. P. Zhang, B. Xu and X. F. Sui, Carbohydr. Polym., 2018, 191, 17 CrossRef CAS PubMed; (b) M. F. Pang, C. J. Wu, X. W. Zhuang, F. J. Zhang, M. C. Su, Q. X. Tong, C. H. Tung and W. G. Wang, Organometallics, 2018, 37, 1462 CrossRef CAS; (c) Y. L. Wu, C. K. Shan, J. X. Ying, J. Su, J. Zhu, L. L. Liu and Y. Y. Zhao, Green Chem., 2017, 19, 4169 RSC.
- (a) V. L. Weidner, C. J. Barger, M. Delferro, T. L. Lohr and T. J. Marks, ACS Catal., 2017, 7, 1244 CrossRef CAS; (b) D. Mukherjee, A. Ellern and A. D. Sadow, Chem. Sci., 2014, 5, 959 RSC.
- (a) L. A. Oro, D. Carmona and J. M. Fraile, in Metal-Catalysis in Industrial Organic Processes, ed. Chiusoli, G. P. and Maitlis, P. M., RSC Publishing, London, 2006, pp. 79–113 Search PubMed; (b) N. N. Zhou, X. A. Yuan, Y. Zhao, J. Xie and C. J. Zhu, Angew. Chem., 2018, 130, 4054 CrossRef; (c) D. T. Yang, S. K. Mellerup, X. Wang, J. S. Lu and S. N. Wang, Angew. Chem., Int. Ed., 2015, 54, 5498 CrossRef CAS PubMed; (d) C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628 RSC.
- J. Seyden-Penne, Reductions by Alumino and Borohydrides in Organic Synthesis, Wiley-VCH, New York, 2nd edn, 1997 Search PubMed.
- (a) M. M. Midland and A. Kazubski, J. Org. Chem., 1992, 57, 2953 CrossRef CAS; (b) M. J. S. Dewar, G. J. Gleicher and B. P. Robinson, J. Am. Chem. Soc., 1964, 86, 5698 CrossRef CAS.
- (a) R. Arévalo, C. M. Vogels, G. A. MacNeil, L. Riera, J. Pérez and S. A. Westcott, Dalton Trans., 2017, 46, 7750 RSC; (b) D. Mukherjee, S. Shirase, T. P. Spaniol, K. Mashima and J. Okuda, Chem. Commun., 2016, 52, 13155 RSC; (c) C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628 RSC; (d) J. R. Lawson, L. C. Wilkins and R. L. Melen, Chem. – Eur. J., 2017, 23, 10997 CrossRef CAS PubMed.
- (a) S. Shaik, S. P. de Visser and D. Kumar, J. Am. Chem. Soc., 2004, 126, 11746 CrossRef CAS PubMed; (b) H. Hirao, H. Chen, M. A. Carvajal, Y. Wang and S. Shaik, J. Am. Chem. Soc., 2008, 130, 3319 CrossRef CAS PubMed; (c) R. Meir, H. Chen, W. Z. Lai and S. Shaik, Chem. Phys. Chem., 2010, 11, 301 CrossRef CAS PubMed; (d) S. Shaik, D. Mandal and R. Ramanan, Nat. Chem., 2016, 8, 1091 CrossRef CAS PubMed.
- (a) M. Alemani, M. V. Peters, S. Hecht, K. H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 126, 14446 CrossRef PubMed; (b) A. C. Aragonès, N. L. Haworth, N. Darwish, S. Ciampi, N. J. Bloomfield, G. G. Wallace, I. Diez-Perez and M. L. Coote, Nature, 2016, 531, 88 CrossRef PubMed; (c) A. C. Aragonés, N. Darwish, S. Ciampi, F. Sanz, J. J. Gooding and I. Díez-Perez, Nat. Commun., 2017, 8, 15056 CrossRef PubMed.
- (a) B. Xu and N. J. Tao, Science, 2003, 301, 1221 CrossRef CAS PubMed; (b) W. Haiss, R. J. Nichols, H. van. Zalinge, S. J. Higgins, D. Bethella and D. J. Schiffrin, Phys. Chem. Chem. Phys., 2004, 6, 4330 RSC; (c) W. Haiss, C.-S. Wang, I. Grace, A. S. Batsanov, D. J. Schiffrin, S. J. Higgins, M. R. Bryce, C. J. Lambert and R. J. Nichols., Nature, 2006, 5, 995 CrossRef CAS PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1992, 96, 2155 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1992, 97, 9173 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (d) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (e) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (f) J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- A. Y. Khalimon, P. Farha, L. G. Kuzminab and G. I. Nikonov, Chem. Commun., 2012, 48, 455 RSC.
- (a) J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808 CrossRef CAS PubMed; (b) A. Y. Khalimon, P. M. Farhaa and G. I. Nikonov, Dalton Trans., 2015, 44, 18945 RSC.
- C. Gonzales and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef.
- (a) I. Mayer, Chem. Phys. Lett., 1983, 97, 270 CrossRef CAS; (b) I. Mayer, J. Comput. Chem., 2007, 28, 204 CrossRef CAS PubMed.
- (a) A. Savin, R. Nesper, S. Wengert and T. F. Fässler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS; (b) T. Lu and F. W. Chen, Acta Phys.-Chim. Sin., 2011, 27, 2786 CAS.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- T. Lu and F. W. Chen, J. Theor. Comput. Chem., 2012, 11, 163 CrossRef CAS.
- G. Merino, V. I. Bakhmutov and A. Vela, J. Phys. Chem. A, 2002, 106, 8491 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06704j |
| This journal is © the Owner Societies 2019 |