
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modeling of aromatics formation in fuel-rich methane oxy-combustion with an automatically generated pressure-dependent mechanism†
Te-Chun
Chu
 a,
Zachary J.
Buras
a,
Patrick
Oßwald
b,
Mengjie
Liu
a,
Mark Jacob
Goldman
a and
William H.
Green
*a
a,
Zachary J.
Buras
a,
Patrick
Oßwald
b,
Mengjie
Liu
a,
Mark Jacob
Goldman
a and
William H.
Green
*a
aMassachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139, USA. E-mail: whgreen@mit.edu
bInstitute of Combustion Technology, German Aerospace Center (DLR), Pfaffenwaldring 38-40, D-70569 Stuttgart, Germany
First published on 7th December 2018
Abstract
With the rise in production of natural gas, there is increased interest in homogeneous partial oxidation (POX) to convert methane to syngas (CO + H2), ethene (C2H4) and acetylene (C2H2). In POX, polycyclic aromatic hydrocarbons (PAH) are important undesired byproducts. To improve the productivity of such POX processes, it is necessary to have an accurate chemical mechanism for methane-rich combustion including PAH. A new mechanism was created to capture the chemistry from C0 to C12, incorporating new information derived from recent quantum chemistry calculations, with help from the Reaction Mechanism Generator (RMG) software. For better estimation of kinetics and thermochemistry of aromatic species, including reactions through carbene intermediates, new reaction families and additional data from quantum chemistry calculations were added to RMG-database. Many of the rate coefficients in the new mechanism are significantly pressure-dependent at POX conditions. The new mechanism was validated against electron-ionization molecular beam mass spectrometry (EI-MBMS) data from a high-temperature flow reactor reported by Kohler et al. In this work quantification of additional species from those experiments is reported including phenylacetylene (C8H6), indene (C9H8), naphthalene (C10H8) and acenaphthylene (C12H8) at many temperatures for several feed compositions. Comparison of the experimental species concentration data and the new kinetic model is satisfactory; the new mechanism is generally more accurate than other published mechanisms. Moreover, because the new mechanism is composed of elementary chemical reaction steps instead of global fitted kinetics, pathway analysis of species could be investigated step-by-step to understand PAH formation. For methane-rich combustion, the most important routes to key aromatics are propargyl recombination for benzene, reactions of the propargyl radical with the phenyl radical for indene, and hydrogen abstraction acetylene addition (HACA) for naphthalene.
1. Introduction
With development of efficient production methods for shale gas in the early 2000s and the consequent fall in price, there has been increased interest in converting methane into chemicals.1 One direct method is non-catalytic partial oxidation (POX), which is particularly suitable for preparation of acetylene and ethylene.2 In POX, natural gas, mainly composed of methane (CH4), flows with a sub-stoichiometric amount of oxygen (O2) into a reactor. To better control and optimize POX, numerous experimental studies have been performed in flow reactors,3–7 shock tubes,8–11 premixed flames12–20 and inverse diffusion flames.21–24 Among these studies, in 2016 Kohler et al.3 performed experiments in a high-temperature flow reactor with quantitative speciation from mass spectrometry. That study provided a dependable consistent data set for validation of chemical kinetic models. Typically, temperatures in POX processes are around 1500–2000 K and a significant amount of soot has been observed in addition to the desired products.25–27Polycyclic aromatic hydrocarbons (PAH) have been identified as important precursors of soot in combustion, and their appearance is generally undesirable due to their contribution to air pollution28 and global warning.29 In addition, PAH formation can lead to clogging and coke formation in industrial applications. Soot is formed in four steps: first, species with the first aromatic ring are formed from small molecules; second, PAH are developed by ring growth pathways from the single ring; third, reactions involving PAH initiate and contribute to the growth of soot particles or coke deposits. The nascent soot or coke then reacts further, forming material with a high C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H ratio.30–32 In many studies, including this work, the dominant kinetics of gas-phase reactions and thermochemistry properties of the corresponding species are combined into a chemical mechanism that can be used to model various reaction conditions.
:H ratio.30–32 In many studies, including this work, the dominant kinetics of gas-phase reactions and thermochemistry properties of the corresponding species are combined into a chemical mechanism that can be used to model various reaction conditions.
There is plentiful literature detailing natural gas combustion mechanisms, including some studies on the formation of aromatic species. GRI-Mech 3.033 has been verified by experimental results including ignition delays, species profiles in shock tubes, laminar flame speeds, and species concentration profiles in a flow reactor. However, this mechanism was not intended for modeling fuel-rich situations forming aromatics, e.g. the largest species in GRI-Mech 3.0 has only 3 carbons. Several other popular methane mechanisms have been developed, including AramcoMech2.034–40 and FFCM-1,41 but like GRI-Mech, these are not intended for modeling systems forming substantial amounts of aromatics. USC-II was developed by Wang et al. on the basis of GRI-Mech 3.0 and other models. The focus was on chemistry of H2/CO/C1–C4 compounds; however, chemistry of first ring aromatic species, such as benzene, was considered as well.42 Using gas chromatography-mass spectrometry (GC-MS) to quantify PAH in a laminar flow reactor, Skjøth-Rasmussen et al. established a PAH mechanism up to pyrene.32 Though major species were predicted accurately, the volume fractions of soot predicted by the model were two to three orders of magnitude lower than the experimental values. The Blanquart mechanism43 and its subsequent version, the Narayanaswamy mechanism,44 investigated a wide range of hydrocarbon fuels from methane to iso-octane. In the Narayanaswamy mechanism, prediction of aromatics was validated by experiments initiated with toluene, ethylbenzene, styrene, xylene, and methylnaphthalene. Chernov et al.45 enhanced the Slavinskaya mechanism46 by incorporating PAH growth in methane-rich, ethylene and ethane co-flow flames, thereby improving the predictions of soot volume fractions. To the best of our knowledge, all the reported methane combustion mechanisms only used temperature-dependent Arrhenius kinetics for reactions relevant to PAH formation, and the pressure dependence of the rate coefficients (due to fall-off or chemical activation) was not considered. Under typical combustion conditions (e.g. 1800 K and 1 atm), many rate coefficients are in reality significantly dependent on both variables.47 Ignoring pressure dependence often leads to errors of more than a factor of two in the rate coefficients in this T,P range.
Reaction Mechanism Generator (RMG) is an open-source software package, which automatically constructs chemical mechanisms built on elementary chemical reaction steps.48 The reaction families defined in RMG and the tree structure of kinetic and thermodynamic data stored in RMG-database are the two bases for estimating the chemistry in RMG. Kinetic models are generated using a rate-based algorithm that excludes species estimated to have low rates of formation. CanTherm, a package included in RMG, can calculate phenomenological pressure-dependent rate coefficients directly from outputs of quantum chemistry codes, or from the species thermodynamic values in NASA polynomial format and reaction kinetics at the high-pressure limit,49 which allows RMG to generate pressure-dependent mechanisms. Furthermore, for predicting correct thermodynamic properties of PAH, estimation of polycyclic thermochemistry in RMG has been improved by two algorithms: similarity matching and bicyclic decomposition which extended the basic group additivity method.50 When RMG encounters cyclic molecules, the thermodynamic properties are estimated by the revised group additivity method instead of the basic one assuming each atom-based group is independent. The error in estimated thermodynamic properties of multi-ring species relevant to PAH formation was reduced by an order of magnitude.
Formation of two prototype aromatics, naphthalene and indene, exhibits the opportunity of PAH growth from the first aromatic ring in POX. Mebel et al.51 overviewed the theoretical studies on naphthalene and indene formation, and summarized ten pathways with potential energy surfaces (PES) reported in the literature. Elementary steps on each pathway were studied, and the reactivity of some routes are analogous to the formation of PAH larger than naphthalene and indene. Six pathways for naphthalene include:
(N1) Hydrogen abstraction acetylene addition (HACA)52,53 sequences,54,55
(N2) Addition of vinylacetylene to phenyl radical,51,56
(N3) Recombination of two cyclopentadienyl radicals and the reaction of cyclopentadienyl with cyclopentadiene,57–60
(N4) Reactions of propargyl radical with benzyl radical,61
(N5) Addition of 1,3-butadiene to phenyl radical,62
(N6) Conversion of indene or indenyl radical to naphthalene via methylation.63
Four pathways for indene include:
(I1) Reactions of phenyl radical with allene and propyne,63
(I2) Reactions of propargyl radical with benzene and phenyl radical,63
(I3) A reaction of benzyl radical with acetylene,64
(I4) Reactions of phenyl radical with propene and allyl radical.65
The high-pressure limit rates of elementary steps on these pathways either were provided in the Mebel et al. paper or could be calculated from reported molecular information in the cited papers.
This work used RMG to generate a new pressure-dependent mechanism, here named the Chu mechanism, specifically for methane-rich combustion with PAH growth obtained from elementary steps. The consideration of pressure dependence sets it apart from other mechanisms in the literature, and is expected to make the mechanism applicable across the wide range of pressures encountered in industrial processes and laboratory experiments. With existing experimental data up to benzene from Kohler et al.3 and additional quantification of larger aromatics in this work, simulated profiles using the Chu mechanism are validated. For important species, formation pathways are investigated to identify channels important for POX process development.
2. Experimental
The present study uses experimental data obtained from the DLR high temperature flow reactor.66 The experiments were performed targeting the conditions relevant to POX and to reforming processes relevant in gasification. The majority of the experimental results used herein were published previously.3 However, formation of aromatic species was not the primary target of the previous study. In this work, concentrations of additional species, including C5H4 (e.g. 1,3-pentadiyne), C8H6 (phenylacetylene), C9H8 (indene), C10H8 (naphthalene), and C12H8 (e.g. acenaphthylene), originating from the same measurement series have been evaluated to validate the new chemical kinetic model presented here.In brief, the flow reactor consists of an alumina tube (40 mm inner diameter) with a heated reaction segment of 100 cm out of the total length of 147 cm. The oven can provide temperatures up to 1900 K. Premixed gases (fuel and oxygen in 99.5% Ar) were fed at ambient temperature to the reactor using mass flow controllers. The reactor is operated at ambient pressure (960 ± 10 mbar). The reacting mixture passes through a known temperature profile imposed by the oven, and the composition at the reactor output is measured continuously using electron-ionization molecular beam mass spectrometry (EI-MBMS), with nominal 16 eV to try to minimize fragmentation of the parent ions. The oven passes through a distinct temperature decay ramp (1820 to 1080 K) during the experiments and species mole fractions at the reactor exit are recorded as a function of the oven temperature. Careful characterization of the system allows assignment of a distinct axial temperature profile for each oven temperature, which is afterwards used as input for kinetic modeling using the plug flow approximation.67 Due to the change in temperature, Reynolds numbers vary from 113–161, which assures the plug flow approximation; also, the residence time of gas ranges from 1.2–1.8 s.66 The experimental and numerical methods have been shown to give valid targets for development of kinetic mechanisms.67–70 A brief summary of the experimental conditions may be found in Table 1; the original publication gives more details.3
| Test case | I | II | III | IV | V |
|---|---|---|---|---|---|
| Fuel | CH4 | +CO2 | +CO, +CO2 | +C2H2 high | +C2H2 low |
| C/O | 0.625 | 0.556 | 0.571 | 0.667 | 0.634 |
| Ar [sccm] | 9950 | 9950 | 9950 | 9950 | 9950 |
| CH4 [sccm] | 25 | 25 | 25 | 20 | 25 |
| O2 [sccm] | 20 | 20 | 25 | 18 | 20.5 |
| CO [sccm] | 0 | 0 | 25 | 0 | 0 |
| CO2 [sccm] | 0 | 25 | 50 | 0 | 0 |
| C2H2 [sccm] | 0 | 0 | 0 | 2 | 0.5 |
For the present study, the full dataset was reanalyzed with respect to soot precursors and aromatic species. Species concentration profiles for the additional species mentioned above have been evaluated. Species assignment was performed on the determination of the exact mass, i.e. the elemental composition. The determination of the molecular structure is not feasible in the present experiment, and assignment was done solely from experience with isomer-resolving experiments.71,72 The assumed chemical structure is given in parentheses above. Quantification of those species follows the same approach as given in the original work. Due to blocking of the nozzle by particles, some data in Case I at low temperatures could not be quantified. For the rest of the missing data in other cases, species profiles were not given because the data were close to or lower than the detection limit. Since no cold gas calibration was performed for these species, the calibration relies on the relative ionization cross section (RICS) procedure to connect reference data measured with 70 V electrons to the current experiments using 16 V electrons, for estimation of the respective calibration factors. For the above-mentioned species, vinylacetylene was used as reference for the evaluation. It should be noted that the calibration relies on the chemical similarity of the considered species and increases the experimental uncertainty of the obtained absolute mole fraction significantly. Typically, deviations below a factor of two are observed, but deviations exceeding this value may occur in rare cases, in particular in cases where the mixture of isomers is highly uncertain. For the larger PAHs observed here, a larger uncertainty in the reported yields cannot be excluded since the similarity to the reference species (vinylacetylene) is not very high.
The absolute concentrations of the major species (CH4, O2, CO, CO2) are determined to within about 15% using direct mass spectrometry, where the measured mass spectrometry signals were converted into absolute concentrations using calibration procedures discussed in ref. 3. The experimental uncertainty in the absolute concentration of H2 obtained with these same calibration procedures is larger than for the other major species.3 We find that often a tighter error bar on the absolute H2 concentration can be obtained by using atom balances based on the more accurate values determined for the other major species, i.e. determine the absolute yield of H2 by difference. In this work, the measured relative signals of H2 as a function of temperature for each experiment were scaled to absolute H2 concentrations using the atom balance method, except for the experiment called Case III (the high CO/CO2 case) where the subtractions involved in the atom balance method introduce a high uncertainty. Although the error bars are usually smaller using the atom balance method than the H2 calibration method, in most cases the absolute concentrations inferred using the two methods agree within their error bars, so this is a modest correction to the H2 values reported in ref. 3. Full details on the atom balance methodology employed here and the resulting error bars on the absolute H2 concentrations are given in the ESI.†
3. Theoretical
To model the chemistry from small molecules to aromatic species containing up to 12 carbons in the DLR high temperature flow reactor, the new Chu mechanism with pressure dependence developed with RMG was applied to a plug flow reactor model with predefined temperature profiles in Chemical Workbench.73 The pressure-dependent mechanism consists of three main components: an RMG auto-generated mechanism to describe the chemistry from small molecules to the first aromatic ring, the chemistry for dominant pathways of forming naphthalene and indene, and the C12 chemistry from the reaction between naphthyl radicals and acetylene. The flow diagram for developing the Chu mechanism is shown in Fig. 1 and each part of the modeling process is discussed specifically in this section.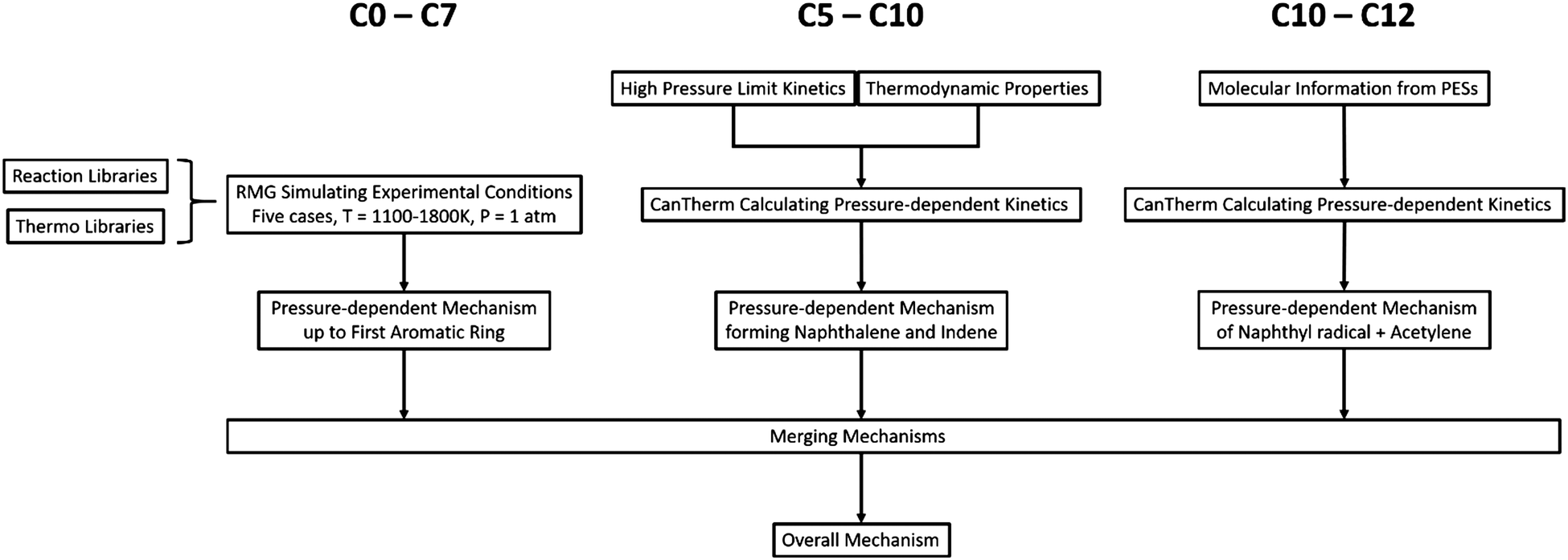 | ||
| Fig. 1 The flow diagram for developing the Chu mechanism. | ||
3.1 From small molecules to the first aromatic ring
In addition to teaching RMG new chemistry, there is another way to include important chemistry into the output mechanism: by creating kinetics/thermochemistry libraries, where data provided are prioritized in the mechanism generation. Due to the high sensitivity of the main processes to the small molecule chemistry, a reliable library of small molecule chemistry significantly improves the accuracy of the RMG-generated model's predictions for all the species, including the PAHs. FFCM-1 is a reaction model focusing on small hydrocarbons with chemistry from C0–C2. Its rate parameters have been optimized over a wide range of experimental data with careful consideration of uncertainties.41 The performance of FFCM-1 at our experimental conditions is validated, and the predictions of our model using FFCM-1 as a library agree well with the experimental measurements for small molecules; see Fig. S1 (ESI†). Therefore, FFCM-1 is applied as the primary library for kinetics and thermochemistry.
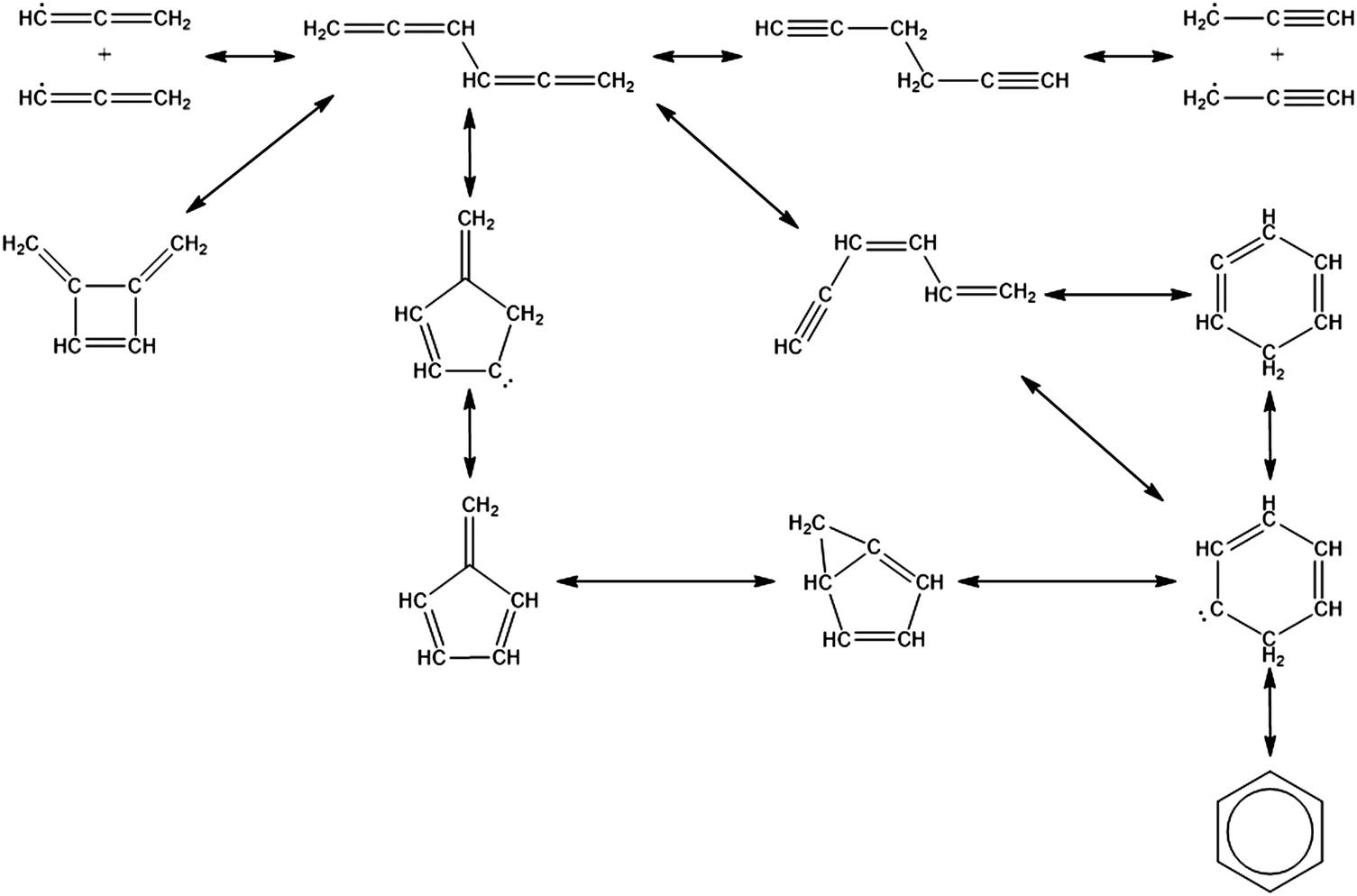 | ||
| Scheme 1 Elementary steps from propargyl radical recombination to fulvene and benzene. | ||
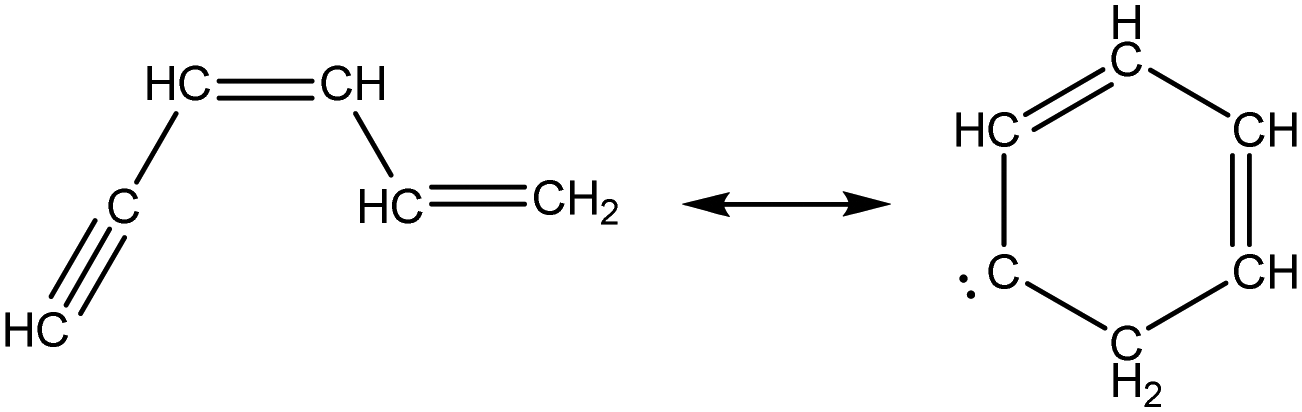
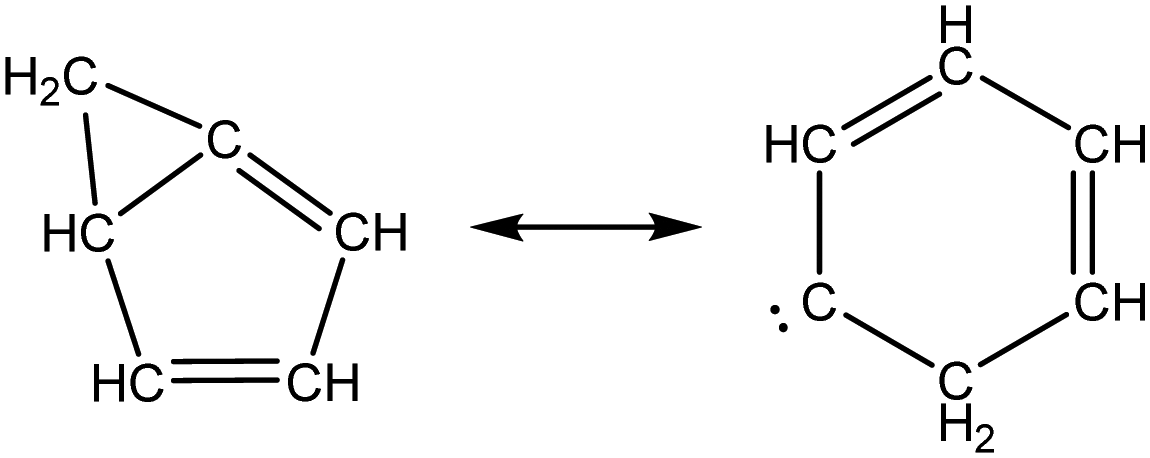
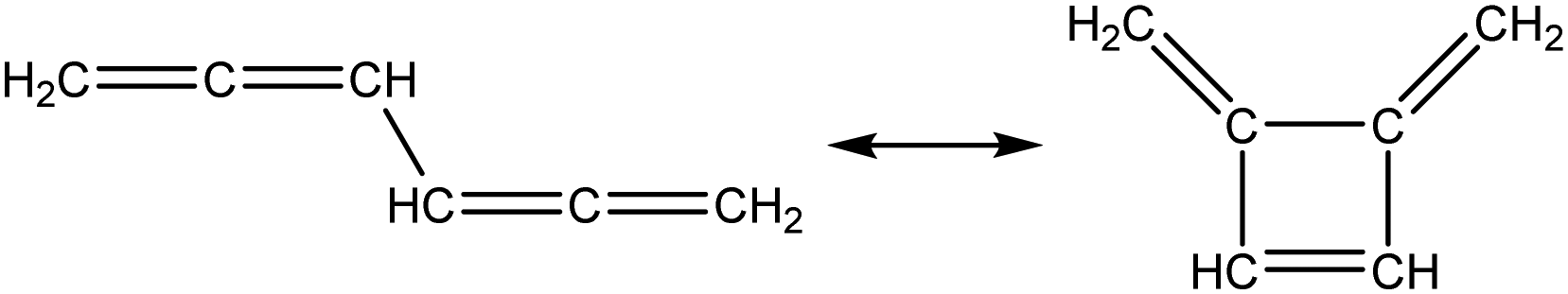
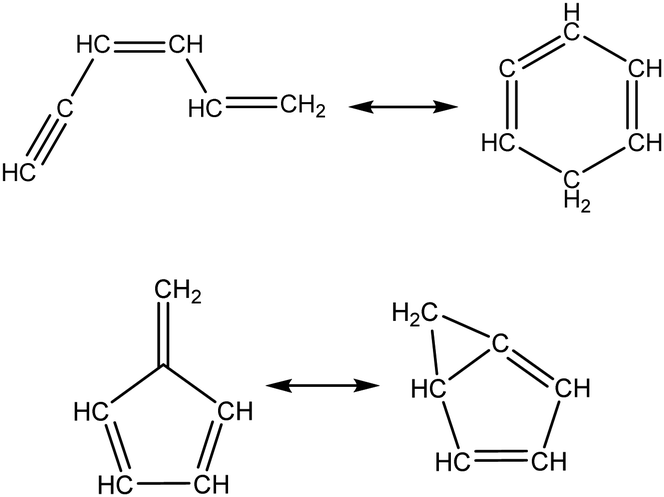
Once the capability to accurately estimate singlet carbene thermochemistry was added to RMG, attention could be given to the kinetics aspect of C3H3 recombination. Of the 13 elementary steps in Scheme 1, only two belonged to an existing reaction family in RMG (both “tail-to-tail” and “head-to-head” recombination belong to the R_Recombination family, whereas “tail-to-head” is not shown because its pathway to benzene formation has higher barriers reported by Miller et al.75); the kinetics of these three recombination steps were trained into RMG based on the calculation done by Georgievskii et al.76 The remaining 11 elementary reactions in Scheme 1 were grouped into eight new reaction families and added as training reactions to the new families in the RMG database, Table 2.
| Family | Name | Training reactions |
|---|---|---|

|
6_membered_central_C–C_shift |
|
|
Concerted_Intra_Diels_Alder_monocyclic_1,2_shiftH |
|
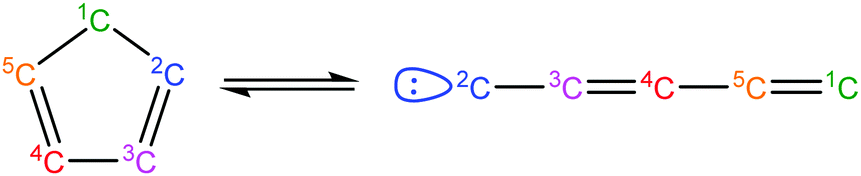
|
Cyclopentadiene_scission |
|

|
Intra_2+2_cycloaddition_Cd |
|
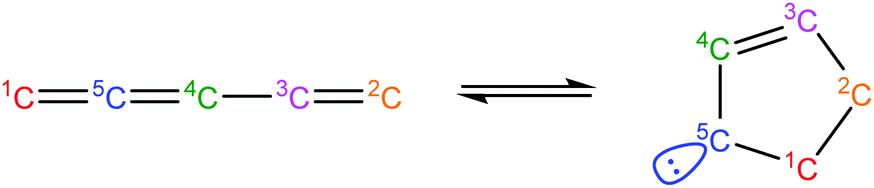
|
Intra_5_membered_conjugated_C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C_CC_addition C_CC_addition |
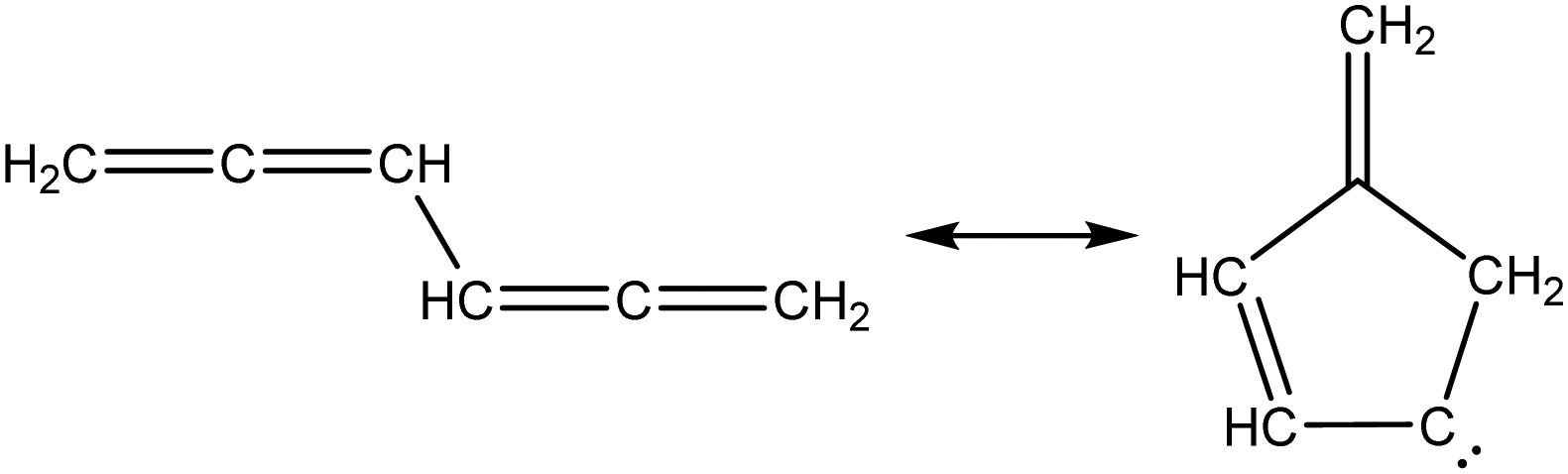
|
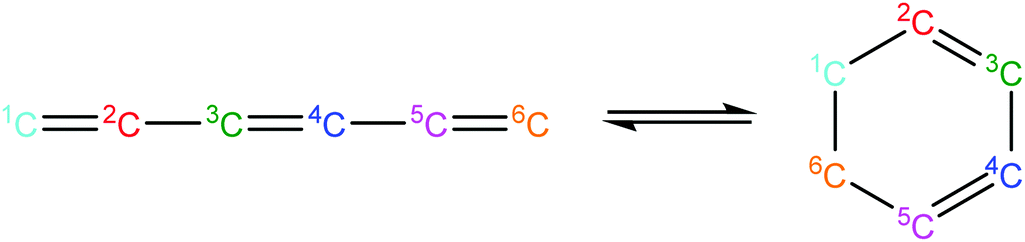
|
Intra_Diels_Alder_monocyclic |
|
|
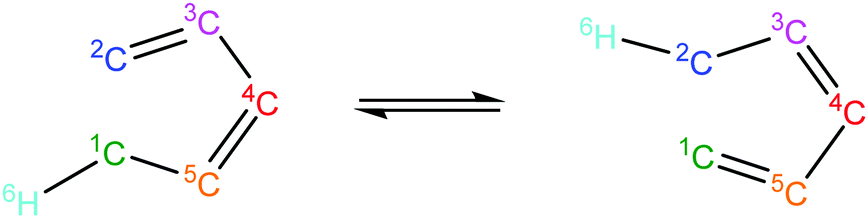
Intra_ene_reaction |
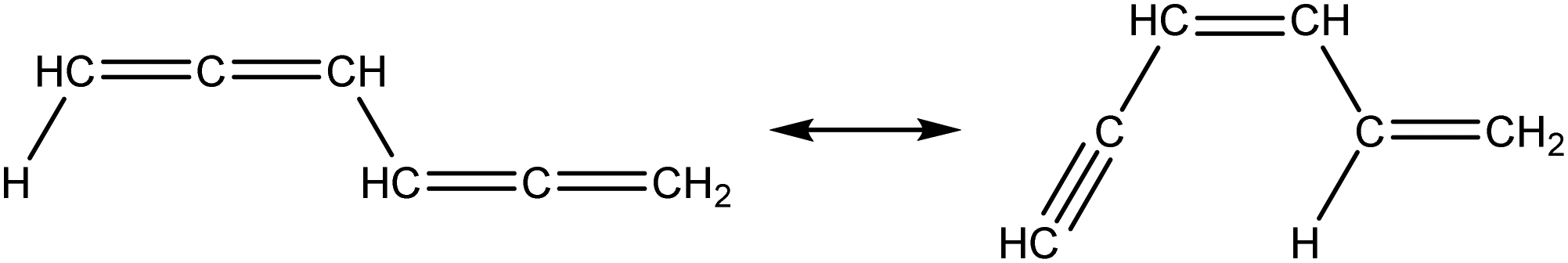
|

|
Singlet_Carbene_Intra_Disproportionation |
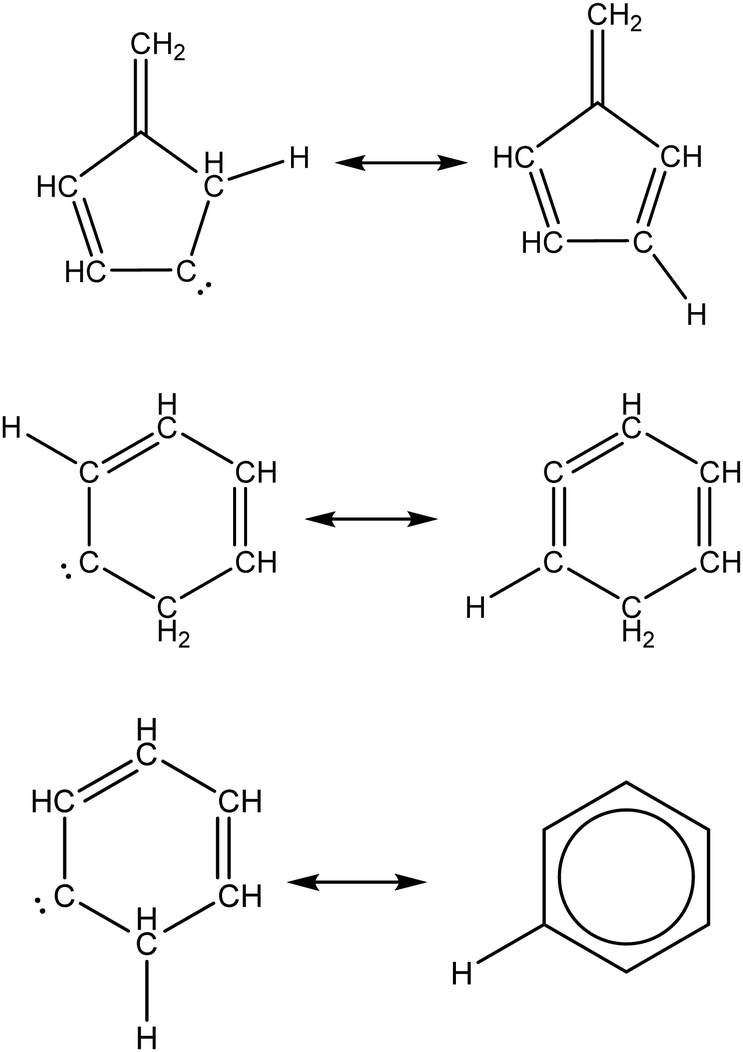
|
At this point, it is worthwhile to mention the advantage of “teaching” RMG new chemistry using the approach presented here (thermochemistry group corrections, reaction families and training reactions) as opposed to simply creating a thermochemistry/reaction library for the specific reactions. By incorporating new chemistry into the RMG database, rather than circumventing it via libraries, this knowledge will automatically be applied to all analogous systems, whereas a library will not.48 Using C3H3 recombination as an example, if one of the hydrogens in propargyl was replaced with a methyl group, RMG would be able to apply the new reaction families and thermo group corrections to find an analogous pathway to a substituted aromatic. The possibility of such pathways was proposed by Miller et al. when the original C3H3 recombination route to benzene was first fully elucidated,75 but to our knowledge this chemistry has not been explored, likely due to the overwhelming number of possible radical recombination pairs. RMG can facilitate this exploration, but only if it is aware of the relevant chemistry, as it is now.
Finally, as a check that all of the relevant aspects of the C3H3 recombination pathway to benzene were incorporated into the RMG database, RMG was used to reproduce the 1,5-hexadiyne pyrolysis experiments of Stein et al.79 (Fig. S3, ESI†). This experiment was used by Miller et al. to validate their predictions,75 which are also shown for comparison. The slight disagreement in the transition from a 3,4-methylenecyclobutene dominated product distribution to a fulvene dominated distribution is likely due to subtle thermochemistry differences, because the RMG predictions are relying on group additivity estimates for all species. Nonetheless, the agreement is satisfactory and provides confidence that RMG should now be able to fairly accurately predict benzene formation through C3H3 recombination, which has long been considered the dominant path to benzene and higher aromatics starting from only small molecules (like those in natural gas).
| Training reactions added | Sources of high pressure limit rates |
|---|---|
| a CBS-QB3 calculations performed with Gaussian03.90 | |
| C3H3 + O2 → HCCCH2OO | Hahn et al.81 |
| C3H3 + O2 → H2CCCHOO | Hahn et al.81 |
| C3H3 + H → CH3CCH | Harding et al.82 |
| C3H3 + H → CH2CCH2 | Harding et al.82 |
| C3H3 + CH3 → CH2CCHCH3 | Huang et al.83 |
| C3H3 + H2 → CH3CCH + H | Narendrapurapu et al.87 + CanTherm |
| C3H3 + H2 → CH2CCH2 + H | Narendrapurapu et al.87 + CanTherm |
| Elementary steps on C6H5 + O2 PES | Kislov et al.86 |
| CH2CCHCH3 + H → CHCCHCH3 + H2 | This worka |
| CH2CCHCH3 + CH3 → CHCCHCH3 + CH4 | This worka |
| CHCCH2CH3 + H → CHCCHCH3 + H2 | This worka |
| CHCCH2CH3 + CH3 → CHCCHCH3 + CH4 | This worka |
| CHCCHCH2 + H → CH2CCHCH2 | Ribeiro et al.88 |
| CHCCHCH2 + H → CHCCHCH3 | Ribeiro et al.88 |
| CHCCHCH2 + H → CHCCH2CH2 | Ribeiro et al.88 |
| CHCCCH + H → CHCHCCH | Klippenstein et al.89 |
| CHCCCH + H → CHCCCH2 | Klippenstein et al.89 |
H-abstraction from allene or propyne by a hydrogen atom also influences the formation of C3H3. Though no high-pressure limit rates of these two reactions have been reported before, detailed molecular information was given by Narendrapurapu et al. at the CCSDT(Q)/cc-pVXZ (X = D,T,Q,5,6)//CCSD(T)/cc-pVQZ level of theory.87 By using CanTherm, a tool for computing coefficients for chemical reactions using the results of quantum chemistry calculations, the desired rates were obtained and trained into RMG-database.48,49
The reactions of C4 unsaturated molecules showed up in sensitivity analysis, and a dominant pathway starts from C4H6, including 1,2-butadiene, 1,3-butadiene, and 1-butyne, to C4H4 (vinylacetylene), leading to the final least saturated species C4H2 (diacetylene). H-abstraction from the relatively more saturated species by H or CH3, followed by a β-scission reaction to remove a hydrogen atom, can increase the bond order by one and make species less saturated. The rate coefficients of these sensitive H-abstraction reactions were calculated in this work. The CBS-QB3 level of theory with Rigid Rotor Harmonic Oscillator (RRHO) + 1-D hindered-rotors was used to compute partition functions for both the stable molecules and transition states for these H-abstraction reactions, and the calculation of the rate coefficients was performed with CanTherm, including Eckart tunneling corrections. β-Scission reactions dissociating C4H5 into C4H4 and C4H3 into C4H2 were examined by Ribeiro et al.88 and Klippenstein et al.,89 respectively. The training reactions discussed in this section are summarized in Table 3.
3.2 From first to second aromatic ring
Once the mechanism for single aromatic ring formation is completed, the next step is to add the second ring formation chemistry with pressure dependence. Four of the six major pathways to naphthalene and all four of the pathways to indene reported by Mebel et al.51 follow straightforward 3-step processes: (1) single aromatic ring radicals (phenyl (C6H5) or benzyl (C7H7) radicals) attach to an unsaturated hydrocarbon; (2) the adduct radical cyclizes; (3) the bicyclic radical undergoes β-scission to form a stable bicyclic.For the PESs used in this part of the mechanism, pressure dependent kinetics were obtained using the approach of Allen et al.49 Thermochemistry properties, as NASA polynomials, and high-pressure rate constants, in the modified Arrhenius form, from the output of the RMG run were input into a modified form of CanTherm for generating pressure-dependent rates.48 This approach was used instead of calculating pressure-dependent rates directly using quantum chemistry output because some combined PESs came from more than one literature source using different levels of theory for quantum chemistry calculations. For example, the C10H9 PES is composed of reaction networks including C6H5C2H2 + C2H2 (G3(MP2,CC)//B3LYP/6-311G(d,p), Mebel et al.54), C6H4C2H3 + C2H2 (G3(MP2,CC)//B3LYP/6-311G(d,p), Mebel et al.54), C6H5 + C4H4 (G3(MP2,CC)//B3LYP/6-311G(d,p), Mebel et al.54), and C5H5 + C5H5-H (CBS-QB3//B3LYP/6-311G(2d,d,p), Long et al.60). Inconsistencies in species (e.g. energy and frequency) shared by multiple networks was resolved by merging these reaction networks from high-pressure limit rates and consistent thermochemistry properties estimated from RMG.
For each unimolecular product or reactant in the surface, the density of states, ρ(E) of the isomers is approximated by fitting functional groups to specific frequencies, using the method described by Allen et al.49 The remaining un-specified degrees of freedom, excluding translation and rotational modes, are fit to the heat capacity data of the NASA polynomials. The Lennard-Jones parameters, which were used to estimate collision rates, were estimated by group additivity. Microcanonical rate coefficients, k(E), were estimated from the high-pressure-limit rate coefficients, k(T), and ρ(E) using the inverse Laplace transform (ILT). For the modified Arrhenius form of k(T), there is an exact ILT formula for positive activation energies and n > −0.5. When the exact formula was not available or has numerical difficulties, the inverse Laplace was evaluated numerically. Pressure dependent rate coefficients were computed using the modified strong collision approximation with at least 250 grains spaced at less than 0.5 (kcal mol−1) apart. Collisional energy transfer rates were estimated using a single exponential model, with 〈ΔEdown〉 = (0.812 kcal mol−1) × (T/300 K)0.62 and Argon as a bath gas with ε = 561 K and σ = 4.46 Å.63 The rate coefficient k(T,P) was fit to PLOG format over a wide pressure range from 0.1–100 bar.
The first step to generating pressure-dependent kinetics of the ten pathways is to identify the PES that each pathway belongs to. There were multiple pathways sharing the same potential surface, and some species and elementary steps are in common. All the surfaces involved in the ten pathways are merged to be CxHy ([x,y] = [8,7], [9,7], [9,8], [9,9], [9,11], [10,7], [10,9], [10,10], [10,11]) PESs and the pressure-dependent rate coefficients are estimated from high-pressure limit rates and the thermochemistry properties of species on each PES using the method described above. Besides these reactions, critical H-abstraction reactions by H, CH3, and OH, which connect the different CxHy PESs, were incorporated from the rates calculated in literature or estimated by RMG. For example, H-abstraction from phenylacetylene to form C6H4C2H radicals is a critical step in the hydrogen-abstraction-acetylene-addition (HACA) pathway.
3.3 Beyond the second aromatic ring
HACA is often thought to be the dominant pathway leading to PAH formation in most combustion processes.52,53 Moreover, PAH radical reactions with acetylene have been predicted to have high exothermicities and low barriers. As a result, the reactions of naphthyl radicals with acetylene were assumed here to be the main pathway forming C12 species in this work.The PESs of 1-naphthyl (1-C10H7) and 2-naphthyl (2-C10H7) radicals reacting with acetylene were calculated by Kislov et al.93 and re-considered by Frenklach et al.94 Given the newest results at the G3(MP2,CC)//B3LYP/6-311G(d,p) level of theory, CanTherm computed pressure-dependent phenomenological rate coefficients leading to five C12H8 products from H-elimination channels, which are 1-ethynylnaphthalene, 2-ethynylnaphthalene, cyclobuta[a]naphthalene, cyclobuta[b]naphthalene, and acenaphthylene. Since the PESs were calculated at a consistent level of theory, CanTherm calculated ρ(E) and k(E) directly from the ab initio data, which gives a more accurate estimation of k(T,P) than the indirect ILT method discussed above. The complete Chu mechanism describing the methane-rich combustion from C0 to C12 with 480 species and 6789 reactions was developed by appending the first-to-second-ring part and the beyond-second-ring subset to the single aromatic ring RMG mechanism. The full mechanism is given in the ESI.†
3.4 Modeling of methane-rich oxy-combustion
As was done previously, the experiment was modeled as a plug flow reactor with predefined temperature profiles.3,66,67 The assumptions made in the modeling are: (1) no axial diffusion, (2) no radial gradients, and (3) uniform axial velocity through the reactor. Therefore, the residence time (τ) can be directly transformed to the spatial position (x) in the plug flow reactor model. The software Chemical Workbench (version: 4.2.21387) allows users to import a large number (235 in this work) of temperature profiles, and conveniently perform calculations for each T(x).73 For all the temperature profiles, the gas temperature at 1 cm intervals in the reactor was given by a correlation to the respective oven temperature from a scaling procedure based on experimentally determined centerline temperature profiles.67 To make a fair comparison with the experimental measurement by EI-MBMS and consider the limitation of EI-MBMS on distinguishing isomers, the mole fractions of important isomers predicted by the model were lumped at the end of the reactor and the lumped concentration was used in the model vs. data comparison. In Table S2 (ESI†), the names and structures of the important isomers in the Chu mechanism are listed.Inlet flow conditions for the five test cases were simulated using the parameters in Table 1. In addition to the new Chu mechanism, two literature mechanisms including PAH were chosen for kinetic simulations and comparison with the experimental data: the Narayanaswamy mechanism44 and the Chernov mechanism.45 The Narayanaswamy mechanism was developed based on a mechanism from Blanquart et al.,43 and the combined mechanism included reactions of various substituted aromatic compounds, and PAH formation up to four aromatic rings. Understanding the physical and chemical properties of the surrogate fuel is the main target of the Narayanaswamy mechanism, which was validated against plug flow reactor data, ignition delay times, species profiles measured in shock tube experiments, and laminar burning velocities. The Chernov mechanism was enhanced from the Slavinskaya mechanism, which was developed for a laminar co-flow non-premixed ethylene–air diffusion flame,46 to accommodate PAH formation in methane, ethylene and ethane co-flow flames. In prior modeling work for a turbulent high pressure partial oxidation (HP POX) pilot-scale reactor, the Chernov mechanism was found to be semi-accurate.95
Pathway analysis was done in Chemical Workbench at the oven temperature corresponding to peak formation of the species of interest, which is the most characteristic condition of the species formation.73 The pathway analysis was illustrated based on integrated fluxes through the entire length of the reactor, and the species in the pathway analysis schemes were mapped out from the results in Chemical Workbench. Rate of production (ROP) analysis was performed at the point reaching the highest gas temperature at the oven temperature used in the pathway analysis, and the values presented are the percentages of reactants converted into products.
4. Results & discussion
4.1 Major species profiles
Mole fractions of major species for all five test cases are reported in Fig. 2, obtained from experiment and from model calculations using the literature mechanisms of Narayanaswamy44 and Chernov,45 and the mechanism developed in this work. For each case, the reactants CH4 and O2 are presented in the plot next to the plot of the major products CO, CO2, and H2; the y-axis scale is kept identical for comparison. In all of the experiments, consumption of CH4 and O2 was very small below 1250 K. Between 1250 and 1400 K, prompt decay of reactants was seen with a corresponding increase of products CO and H2. Above 1400 K, the mole fraction of O2 at the reactor exit became negligible, while CH4 was not completely consumed due to the non-stoichiometric equivalence ratio. The products CO and H2, on the other hand, gradually build up with increasing oven temperature until reaching a maximum at the highest oven temperature. The other major product, CO2 behaved similarly to CO and H2 in Case I, IV, and V. In Case II and III where CO2 was a reactant, its concentration remained stable below 1400 K but dropped off as temperature increased from 1400 K to 1800 K.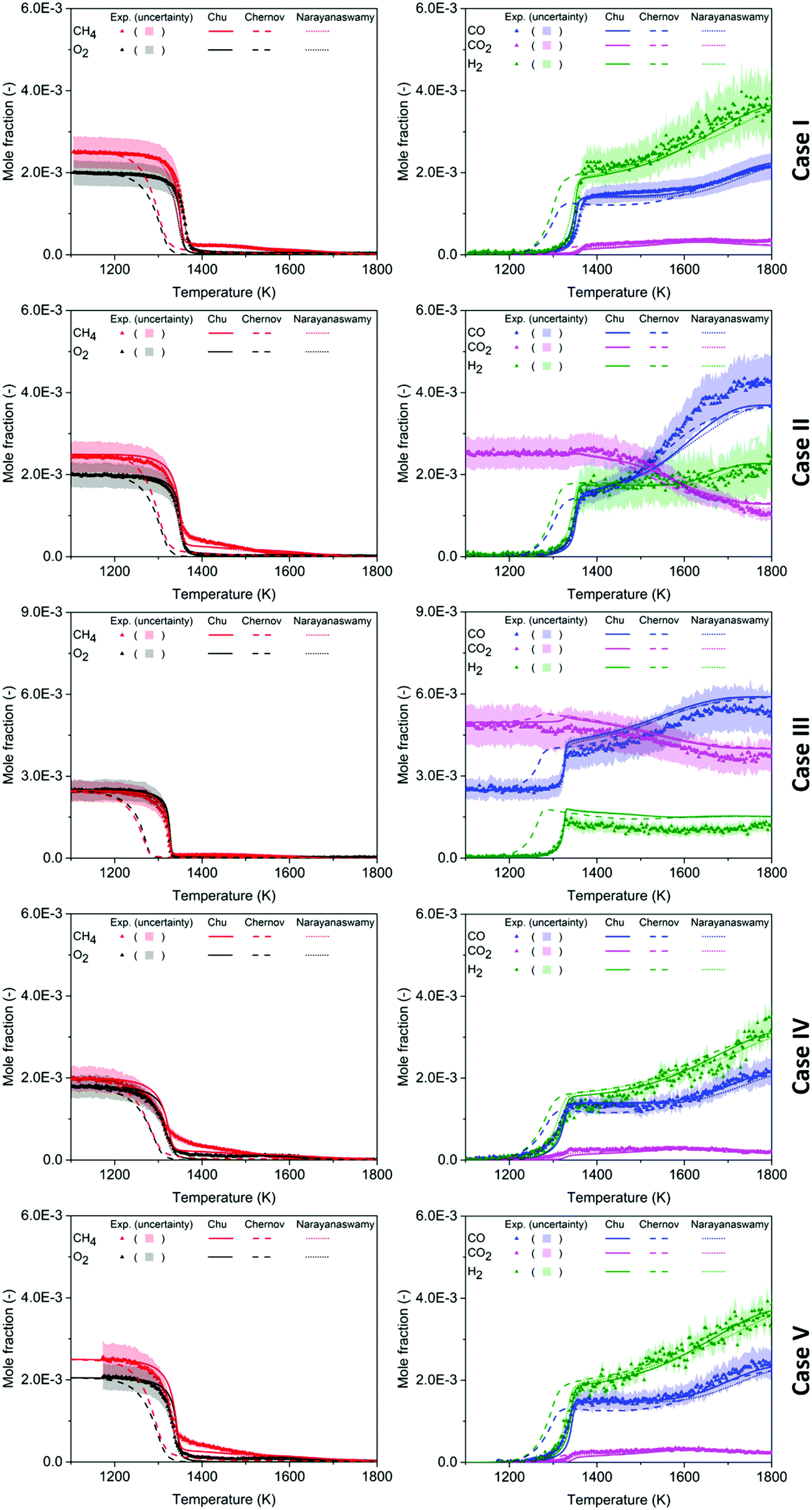 | ||
| Fig. 2 Mole fraction profiles of major species as a function of oven temperature for five test cases, measurements (symbols and uncertainty band) and predictions of three models (lines). | ||
In general, the predictions made by the Chu and the Narayanaswamy mechanisms match the experimental measurements on the major species better than the Chernov mechanism. As mentioned in the theoretical Section 3.1.1, the chemistry of species from C0–C2 in the Chu mechanism is mainly captured by the FFCM-1 model. Therefore, the mole fraction profiles of major species predicted by this mechanism behave similarly to the Case I results of the FFCM-1 model shown in Fig. S1 (ESI†). Once noticeable difference between them is the mole fraction of H2 from 1350 K to 1500 K, where the Chu mechanism gives mole fractions ∼10% lower than FFCM-1. Additional reactions in the current mechanism, e.g. H-abstraction between H2 and larger species not included in FFCM-1, provide more channels for H2 consumption.
Overall, the Chu mechanism predicts the major species concentrations close to or within the experimental uncertainty range (CH4, O2, CO, CO2: 15% using calibrations,3 and the slightly larger error bars on H2 mostly determined using the atom balance method, see ESI†) throughout the whole temperature range. The main discrepancies are between the measured and predicted concentration of CH4 around 1400 K in Cases II, IV, and V, and in the absolute yield of H2 in Case III, see Fig. 2. We do not know the origin of the CH4 discrepancy at that T range in those particular cases; it is curious that the model and all the other CH4 measurements are in excellent agreement. We were not able to use our preferred method (see ESI†) to convert the measured H2 signals to absolute mole fractions in Case III, so those H2 data points were determined using a different procedure than all the other H2 data. We suspect that the Case III experimental error bars on H2 are too optimistic.
The predicted and measured ignition temperatures, defined as the inflection point of the CH4 mole fraction vs. temperature plot, are summarized in Table 4. The Chu and the Narayanaswamy mechanisms both predict the ignition temperature in all test cases within 15 K of the experimental value; on the other hand, the Chernov mechanism estimates the ignition occurs at a lower temperature by 60–70 K. Kohler et al.3 have identified the rate coefficients of CH3 + O2 → CH2O + OH assumed in the Chernov mechanism as accounting for the discrepancy. The rate coefficients of CH3 + O2 → CH2O + OH in the Chu and the Narayanaswamy mechanisms differ by less than a factor of two within the temperature range of the experiment, while the Chernov value is an order of magnitude higher than the value used in the current mechanism (from FFCM-1). This reaction converts the relatively unreactive CH3 radical into the much more reactive OH radical.
4.2 C2 and oxygenated species profiles
To understand the formation of PAHs, the pathways giving rise to intermediates between major products and aromatic species were investigated in the mechanism and validated by the experiment. Here we focus on Cases I, II and IV. The experimental data of Case III were affected by background signals in EI-MBMS, especially for aromatic species with low mole fractions in absolute scale, so the comparison with model predictions is hard to access. Compared to Case V, Case IV has a more extreme amount of C2H2 added to the inlet flow; thus, Case IV is more representative of the effect of adding C2H2. The experimental and predicted mole fraction profiles for Case III and Case V are reported in Fig. S5–S7 (ESI†).Mole fraction profiles of C2H6, C2H4, C2H2 for Cases I, II and IV are shown in Fig. 3. The mole fractions of C2H6 and C2H4 increase rapidly near 1250 K and decrease suddenly after reaching maxima at ∼1350 K, close to the ignition temperature. According to all three models, formation of C2H6 predominantly comes from recombination of CH3 radicals, and the rate coefficient in the Chu model is from the FFCM-1 model. The destruction of C2H6 is mainly caused by H-abstraction by H and OH radicals, generating C2H5 radicals. C2H5 radicals undergo β-scission producing C2H4 and H radicals, which is the leading pathway to C2H4 formation. The route involving H-abstraction followed by β-scission of the resulting radical also leads to consumption of C2H4 and formation of C2H2. In addition to elementary steps, one direct concerted pressure-dependent pathway in the Chu mechanism, C2H4 + M → C2H2 + H2 + M, from FFCM-1, also leads to consumption of C2H4. The observed decreasing saturation of C2 species from low to high temperature is expected, it follows the T-dependence of the chemical equilibria. The predicted maximum mole fractions of C2 species by this model fall within the 20% uncertainty of the experimental data.
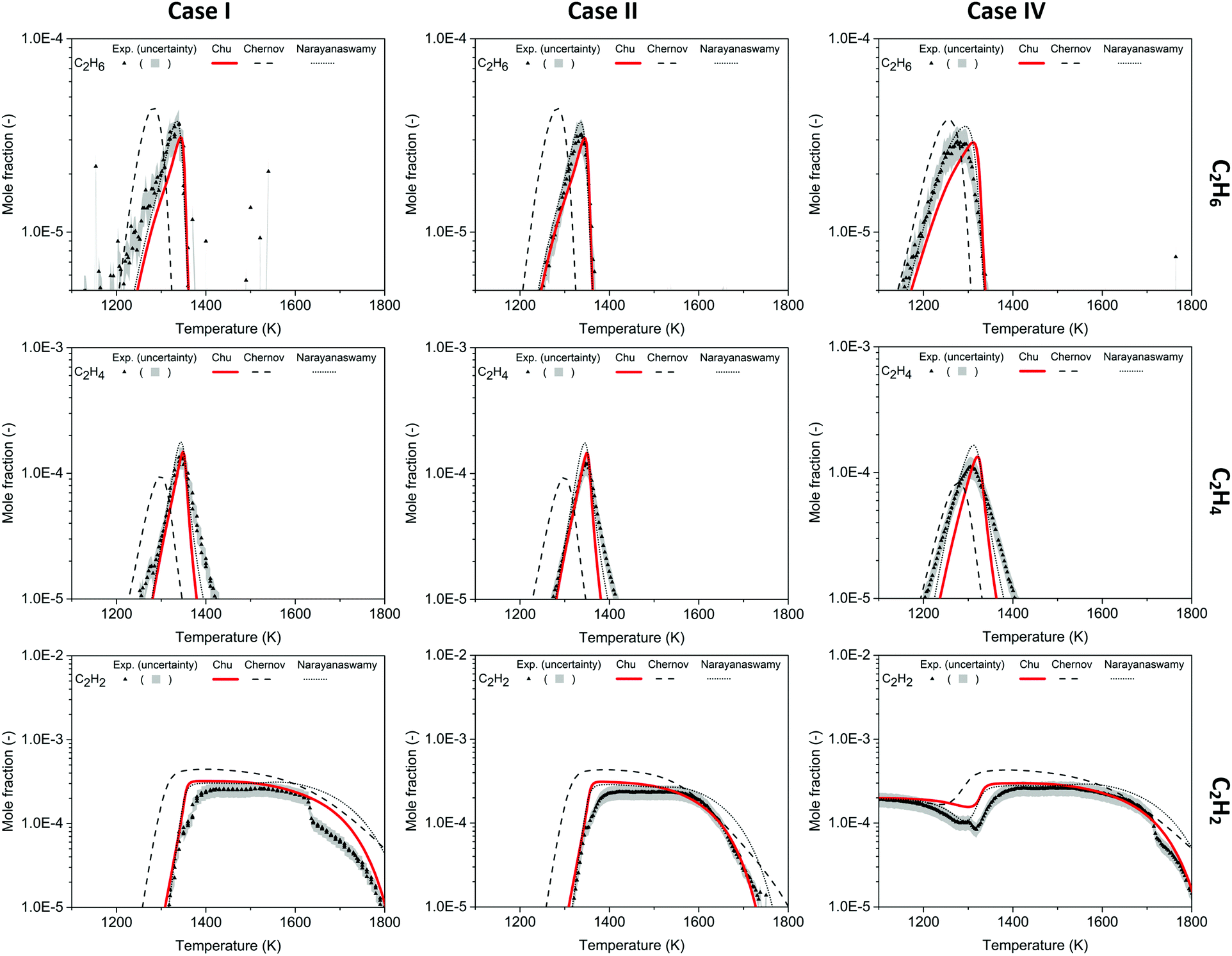 | ||
| Fig. 3 Mole fraction profiles of C2H6, C2H4, C2H2 as a function of oven temperature for Case I, II and IV, measurements (symbols and uncertainty band) and predictions of three models (lines). | ||
At 1600 K, the mole fraction of C2H2 starts to decrease in the experiment, and Kohler et al.3 stated that the sharp drop in C2H2 concentration observed in the experiment was not predicted by any mechanism used in their work, which were the Chernov,45 GRI-Mech 3.033 and USC-II42 mechanisms. Although the Chu mechanism does not perfectly match the C2H2 dip in Case I, it is able to better predict mole fractions compared to other mechanisms for T > 1600 K, and the model-experiment agreement is even better in Case II and Case IV. A pathway analysis using the current mechanism indicates that C2H2 is mostly consumed via OH + C2H2 → CH2CO (ethenone) + H at high temperature in POX reactors, and ethenone (also called ketene) would further react with H radical to form CO and CH3. Accurate prediction of C2H2 yields at high temperature is critical for the POX process, since C2H2 is a valuable product.
The oxygenated species methanal (CH2O, aka formaldehyde) and CH2CO are included in the Chu mechanism and quantified in the experiment. Because the RICS calibration method was used for CH2O and CH2CO quantification, an uncertainty factor of 2–4 is expected for those experimental data. In addition, the bimodality of CH2CO profiles may result from the instabilities of the experimental system instead of the chemistry.3 The mole fraction profiles of CH2O and CH2CO are shown in Fig. S8 (ESI†), and the current mechanism agrees well with the experimental quantification, except for missing the bimodality in Case IV. The reaction CH3 + O2 → CH2O + OH is a key reaction not only in the ignition of CH4, but also in the formation of CH2O in the POX process. The Chu and the Narayanaswamy mechanisms both predict the CH2O profiles within the experimental uncertainty, whereas the Chernov mechanism, which uses a very different rate coefficient for this reaction, overestimates the CH2O mole fraction by a factor of 4–6.
4.3 Unsaturated C3–C5 species profiles
As temperature increases, a trend of growing size in the molecules is seen in this methane-rich combustion study. Among species with the same number of carbons, more saturated species normally appear first at low temperature, and the appearance of C4 species follows this trend. Starting with the most saturated C4 species quantified in the experiment – C4H6, it is produced near the ignition temperature and four important isomers were found in the Chu model: 1-butyne, 2-butyne, 1,2-butadiene, and 1,3-butadiene. Vinylacetylene (C4H4) forms at higher temperature and reaches its maximum concentration at ∼1380 K in Cases I and II and ∼1270 in Case IV. Finally, the most unsaturated C4 species, butadiyne (aka diacetylene, C4H2), becomes dominant among all the intermediates above 1500 K, with an order of magnitude greater concentration than the maximum of C4H6 and C4H4 in Fig. 4.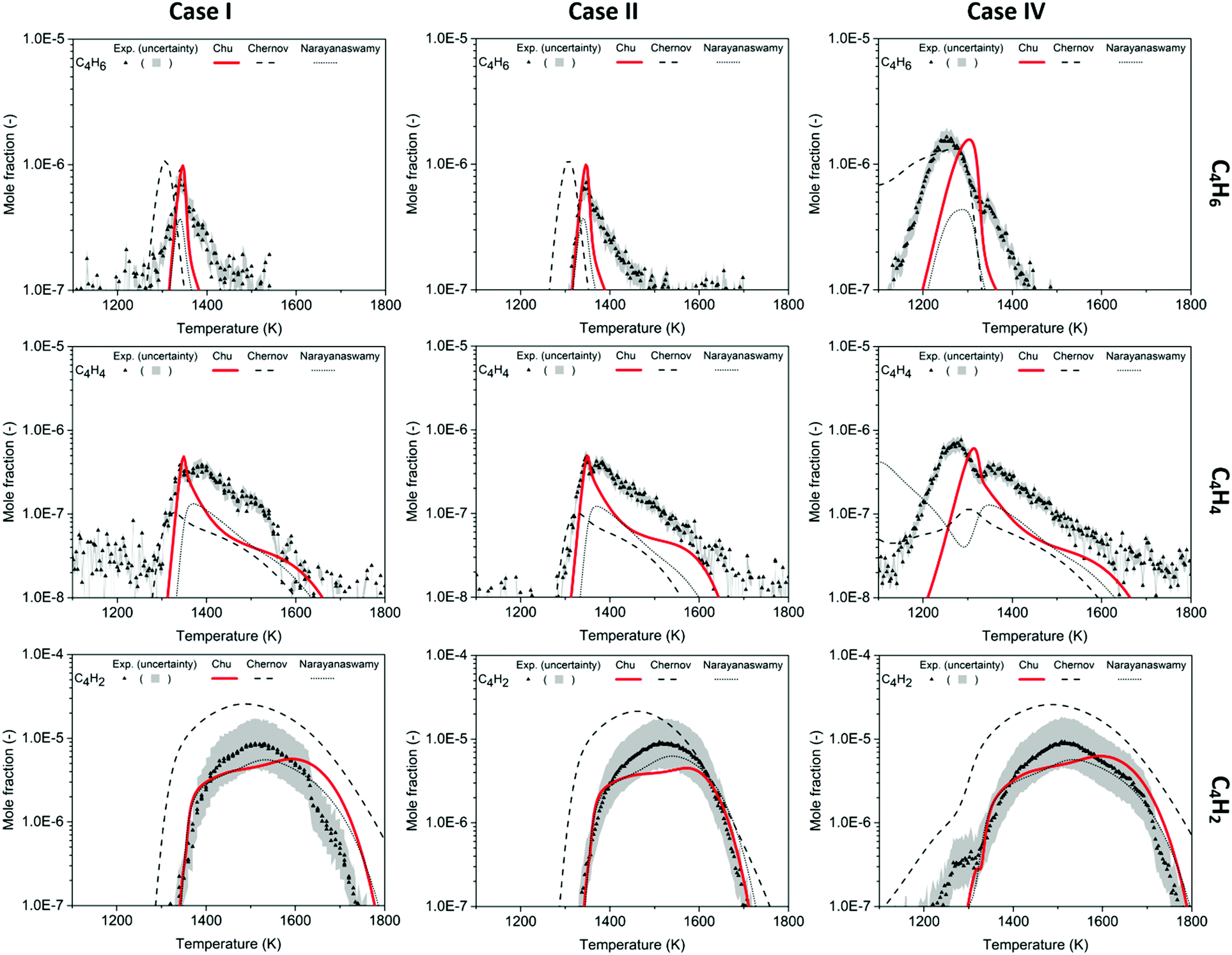 | ||
| Fig. 4 Mole fraction profiles of C4H6, C4H4, C4H2 as a function of oven temperature for Case I, II and IV, measurements (symbols and uncertainty band) and predictions of three models (lines). | ||
In the mole fraction profiles of C4H6, the experimental concentration peaks at 1340 K in Case I, 1347 K in Case II, and 1253 K in Case IV. Although the experimental quantification of C4H6 used 1,3-butadiene to determine the ionization cross section, there should be other isomers that contribute to the measured C4H6 signals. In Fig. 4, the sum of C4H6 mole fractions predicted in this work is compared to the experimental profile, assuming all isomers have the same response factor in the mass spectrometer. Both the magnitude and the temperature dependence are predicted fairly well by the Chu model (and more accurately than by the other mechanisms). 1,3-Butadiene is predicted as the most dominant C4H6 isomer, and its formation mainly comes from recombination of CH3 and propargyl radicals, see Scheme 2. 2-Butyne has different relative contributions to C4H6 isomers in Case I, II and IV, and its formation is summarized below:
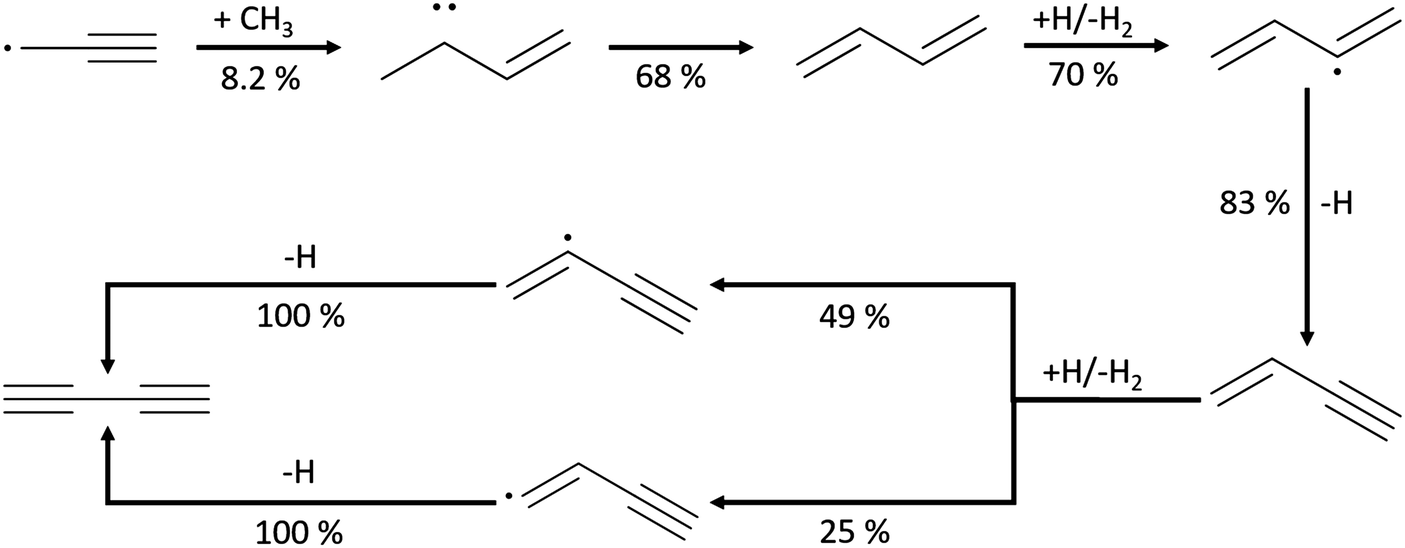 | ||
| Scheme 2 Leading pathways of diacetylene (C4H2) formation found in the Chu mechanism and the ROP analysis at the oven temperature (1592 K) maximizing C4H2 concentration in Case 1. | ||
• Case I (only CH4 and O2) and Case II (CH4 + O2 + CO2)
– 2-Butyne is <5% of lumped concentration of C4H6 isomers
– General pathway: H-facilitated isomerization of 1,2-butadiene and 1,3-butadiene; chemically activated channel of the recombination of propargyl radicals and CH3
• Case IV (CH4 + O2 + C2H2)
– Doubled relative abundance of 2-butyne, ∼10%
– Additional pathway: initiated by CH3 addition to C2H2 (CH3 + C2H2 → propyne (C3H4) + H), followed by a second CH3 addition and β-scission (CH3 + propyne (C3H4) → 2-butyne (C4H6) + H)
The vinylacetylene (C4H4) mole fraction rises rapidly as the reactor is heated above 1300 K in Cases I and II, and the rise is captured by the Chu mechanism. On the other hand, Case IV exhibits bimodality with a minor peak at 1271 K which is not predicted by the Chu mechanism. For all cases shown in Fig. 4, the maximum mole fraction of C4H4 is predicted by the current mechanism within the measurement uncertainty, even though the shape of the yield vs. temperature profile is not predicted as well. Similar to the desaturation sequence of C2 species, vinylacetylene is primarily generated by desaturation of C4H6 isomers in the Chu mechanism, e.g. by H-abstraction from 1,3-butadiene followed by β-scission. Moreover, this work shows a good agreement with the experiments that the concentration of C4H6 and C4H4 is very small at low temperature (before ignition). In contrast, in Case IV the Chernov mechanism and the Narayanaswamy mechanism predict fairly large mole fractions of some of these species at low temperatures.
As the least saturated C4 species with stable thermochemical properties, diacetylene (C4H2) was found to be one of the most abundant species in the experiment. Its mole fraction peaks at T ∼ 1500 K, and, considering the uncertainty of the experiment, the temperature dependence was successfully predicted by the Chu mechanism in Fig. 4. As expected, a desaturation pathway is found where C4H4 loses two hydrogens to produce C4H2, which is presented in Scheme 2. Starting from recombination of propargyl radical with CH3, a thermalized intermediate is formed before formation of 1,3-butadiene. From the portion of the C4H6 PES found by RMG, this thermalized intermediate gives the only elementary step connecting to 1,3-butadiene. This indicates that the channel via thermalized carbene is predicted by RMG to be faster than the direct well-skipping channel C3H3 + CH3(+ M) → C4H6(+ M) at the given conditions. 1,3-Butadiene undergoes H-abstraction by H, CH3, and OH radicals, before β-scission to form vinylacetylene. For H-abstraction from vinylacetylene, there are two pathways, and the one generating i-C4H3 with resonance stabilization is favored. Finally, β-scissions of both n-C4H3 and i-C4H3 form C4H2.
In addition to C4 species, there were unsaturated C3 and C5 species observed in the experiment, which are propene (C3H6) and C5H4 with mole fraction profiles shown in Fig. S9 (ESI†). The experiment shows the maximum concentration of C3H6 is near the ignition temperature with 1.9 ppm in Case I, 1.4 ppm in Case II and 1.1 ppm in Case IV. The predictions of the Chu mechanism and the Chernov mechanism are in decent agreement with experiment, but the Narayanaswamy mechanism overestimates the C3H6 concentration by more than a factor of four. In this work, the chemically-activated reaction CH3 + C2H4 produces C3H6 with the co-product H atom. Two isomers of C5H4 have notable concentrations: 1,3-pentadiyne (major) and 1,2-pentadien-4-yne (minor). In the temperature range from 1400–1600 K, C5H4 is observed experimentally with a noticeable amount, and the Chu mechanism predicts the mole fraction accurately. In addition, the tiny spike in C5H4 concentration observed in Case II and IV (see Fig. S9, ESI†) is correctly predicted.
4.4 Propargyl radicals and single ring aromatic species profiles
Propargyl radical (C3H3), a resonance-stabilized radical (RSR), is known to be important to benzene formation in flames and combustion.96 Due to its low reactivity toward O281 and the absence of easy H-loss channels, C3H3 accumulates in the reactor to a non-negligible amount, where the maximum measured concentration is 0.8 ppm in Case I, 0.7 ppm in Case II, and 1.3 ppm in Case II, shown in Fig. 5. In the previous Köhler et al. work,3 the USC-II and Chernov mechanisms were found to disagree with the experiment on the temperature dependence of C3H3 relative to C6H6.3 The observed early growth of C3H3 is captured by the Chu model in Cases I and II, but the concentration between 1350–1550 K is still underestimated.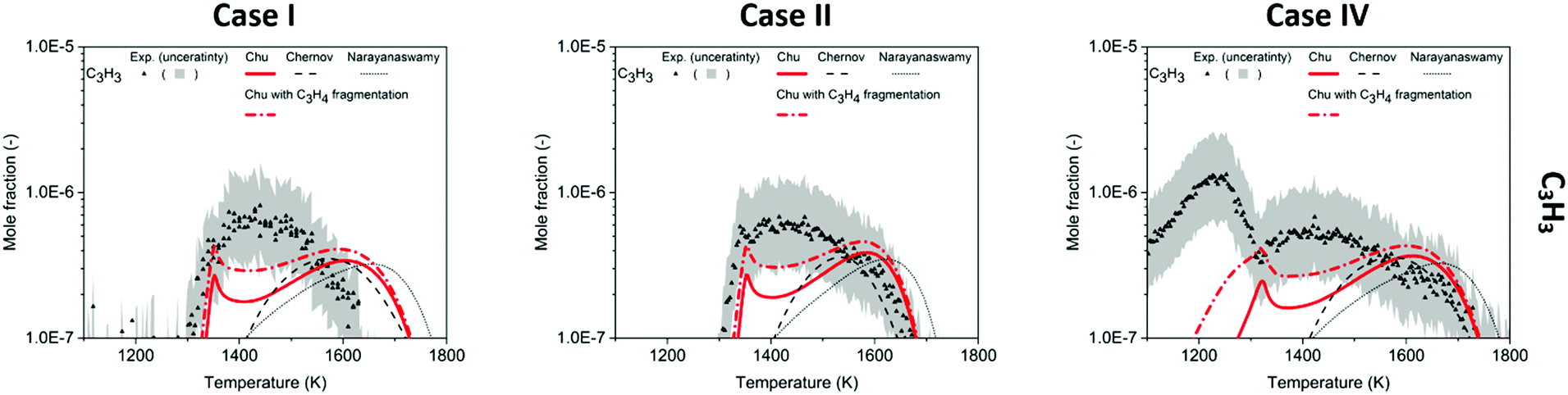 | ||
| Fig. 5 Mole fraction profiles of C3H3 and total m/z = 39 signal as a function of oven temperature for Case I, II and IV, measurements (symbols and uncertainty band) and predictions of three models (lines). Contribution of m/z = 39 fragments from C3H4 species are additionally shown and significantly improves the model predictions at lower temperatures. | ||
A large amount of m/z = 39 signal is measured in the experiment near 1250 K in Case IV, but none of the mechanisms predict high concentrations of any species with mass 39 amu at that condition. The inconsistency between the experiment and the model in Case IV may be caused by fragmentation of propyne and allene under electron-impact ionization. The ionization energy used in the experiment was 16 eV, which is significantly higher than the ionization energy of these species, i.e. 10.36 eV for propyne and 9.69 eV for allene.97 This extra energy causes the ions to fragment into several peaks on the mass spectrum; in this case, the peak m/z = 40 is the parent peak for both propyne and allene, and one child peak m/z = 39 overlaps with the signal arising from C3H3 formed in the oxidation process. In this work, the fragmentation ratios (signals(m/z = 39)/signals(m/z = 40)) were measured from cold gas samples of pure propyne and pure allene, see Fig. S10 (ESI†). At 16 eV, 13.4% of propyne and 8.5% of allene fragment into C3H3+. However, using this fragmentation pattern to correct the experimental profiles is not possible since the parent ion (C3H4) signal was not separated from the diluent argon in the original work. Instead, the calibration factors determined for propyne and allene were quantified in this work (see ESI†), and the calibration factor for propargyl radicals quantified (original work) by Köhler et al.3 has been used to calculate the contribution of C3H4 fragments to the C3H3 mole fraction profile based on the simulated profiles. Comparing the calibration factors in the Köhler et al. work3 and this work might not be ideal because there are more than three years between the two measurements; however, these data are the only available information. Including the effect of C3H4 fragmentation, the bimodal distribution of C3H3 in Case IV becomes distinct in the Chu model, Fig. 5, despite the imperfect temperature dependence. Also, the modeled data match the experimental measurement better in Case I and II, validating the Chu prediction of C3H3 with consideration of fragmentation.
For quantification of benzene (C6H6), the RICS calibration method was used, and a factor of two uncertainty is expected. The shape of C6H6 mole fraction profile from the Chu model agrees well with the trend in the experiment in Fig. 6. The simplest methane-rich condition in Case I shows the maximum to be 2.4 ppm experimentally, and 3.1 ppm in the model; with the addition of CO2 in Case II, the peak is 2.5 ppm in the experiment and 3.5 ppm in the model. For Case IV, the additional C2H2 makes the different models reveal their different treatments of acetylene consumption and shows the biggest deviations from the experimental results.3 The experimental bimodality (0.73 ppm and 2.4 ppm) is captured by the current model (0.96 ppm and 3.5 ppm), and compared to the Narayanaswamy mechanisms, the profile shapes and the maximum peak of C6H6 evaluated in this work are closer to the experimental results. At high temperatures, the Chu mechanism overestimates C6H6, likely because RMG is missing or incorrectly estimating some chemistry related to benzene/phenyl consumption, which will need to be studied and trained into RMG-database in the future.
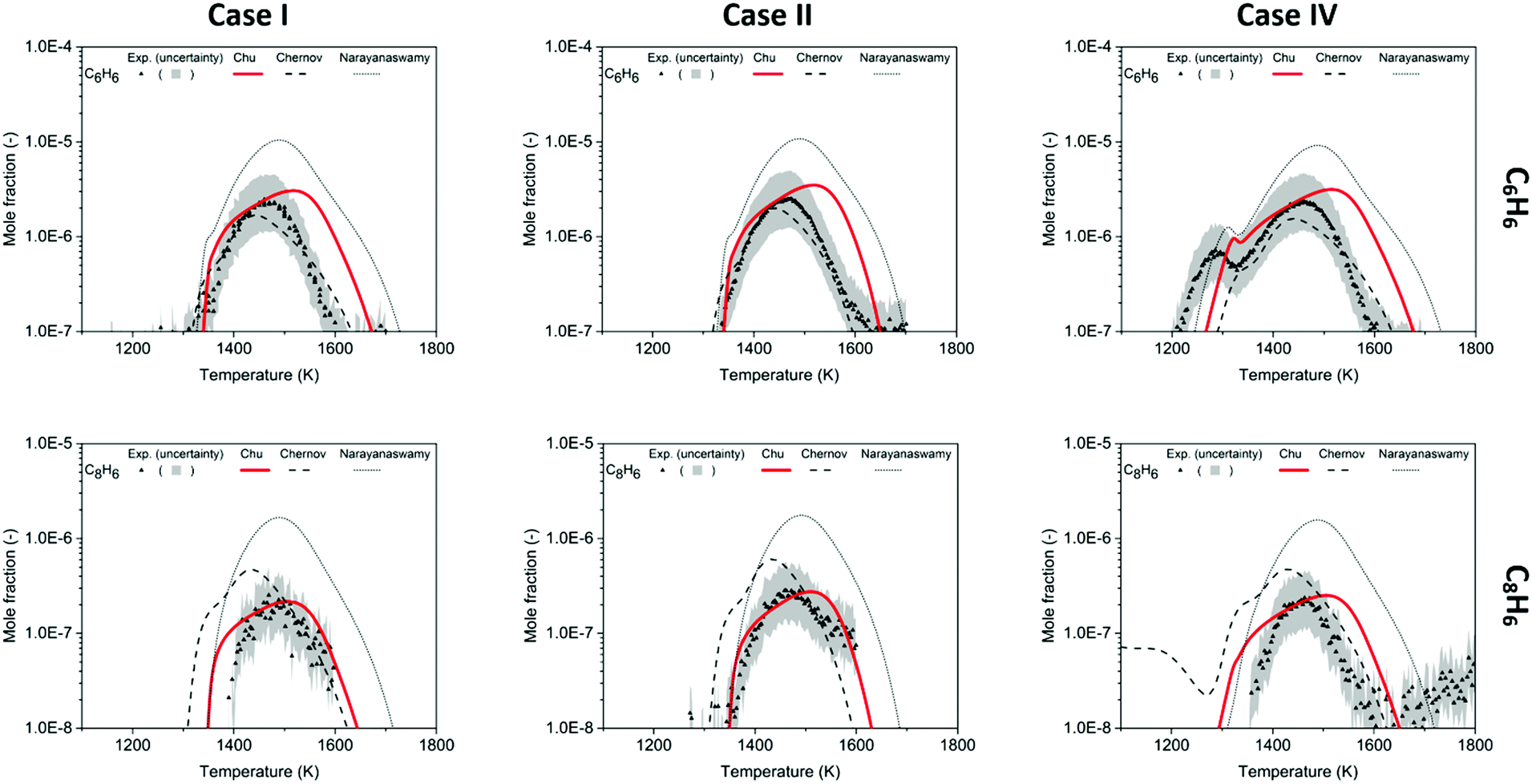 | ||
| Fig. 6 Mole fraction profiles of C6H6 and C8H6 as a function of oven temperature for Case I, II and IV, measurements (symbols) and model predictions (lines). The gray band indicates experimental uncertainty. | ||
To understand aromatics formation in methane-rich combustion, pathway analysis of benzene was performed at the oven temperature (1517 K for Case I) reaching maximum concentration. Not surprisingly, the propargyl recombination pathway added to RMG-database contributed most to benzene formation. In Scheme 3, the leading pathway (solid line) starting from propargyl radicals has a well-skipping channel forming thermalized hexa-1,3-dien-5-yne, which itself reacts by another well-skipping pressure-dependent reaction path to form benzene. There are also some minor pathways found in the analysis of benzene formation. First, “tail-to-tail” and “head-to-head” recombination generate hexa-1,2,4,5-tetraene and hexa-1,5-diyne, respectively. These recombination products can react along the major pathway or form fulvene, the second most stable species on C6H6 PES. Due to the large barriers from fulvene to benzene, RMG found this process was dominated by the H-facilitated pathway initiated by H radicals attacking fulvene, where each of the additions had an activation energy of less than 5 kcal mol−1. Subsequent reactions on the C6H7 surface lead to benzene + H formation through a well-skipping reaction.
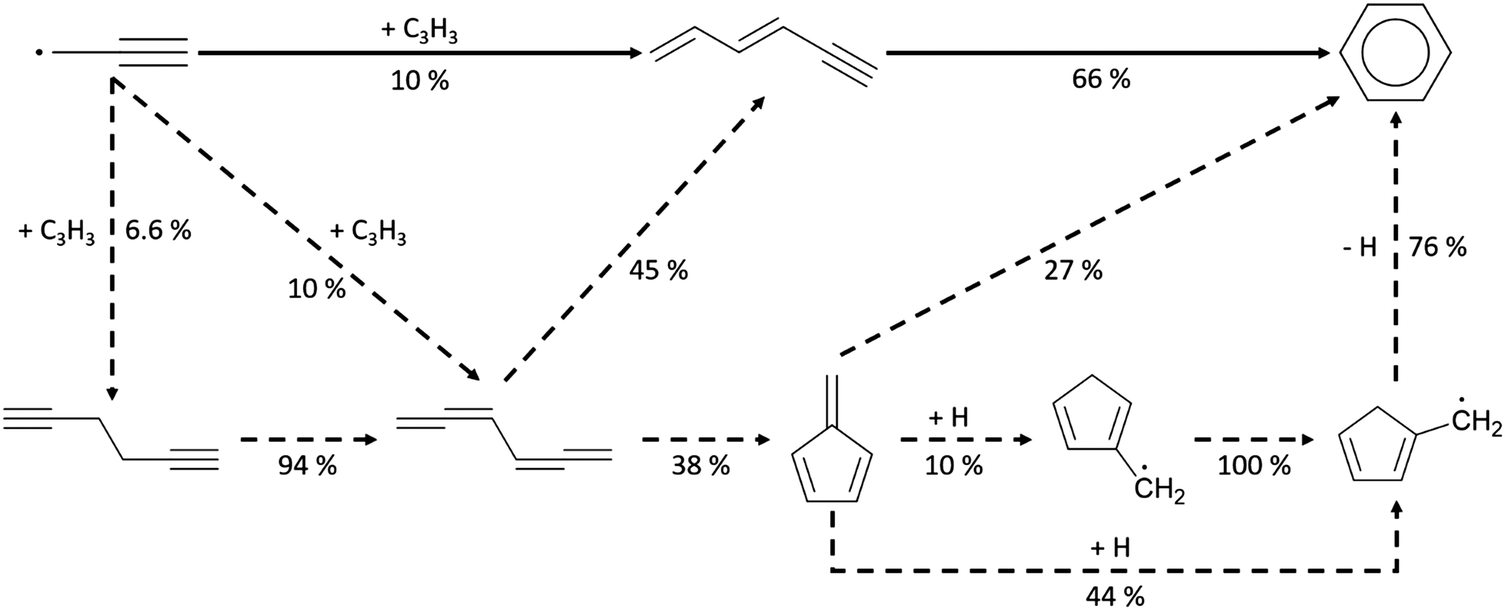 | ||
| Scheme 3 Leading pathways of benzene formation from propargyl radicals found in the Chu mechanism, and the ROP analysis at the oven temperature (1517 K) maximizing C6H6 concentration in Case 1 (major pathway – solid line, minor pathways – dash line). | ||
In these experiments, phenylacetylene (C8H6) was detected to be the most abundant among the species larger than benzene. Similarly, Gudiyella et al. studied high temperature pyrolysis (HTP) of natural gas validated by pilot plant data and found that C8H6 prevailed in aromatics in the high temperature pyrolysis reactor network. The first reason is its thermochemical stability, and its lack of H-loss channel; the second is the high concentration of C2H2, which readily reacts with phenyl radicals via the HACA pathway.55 In Fig. 6, the peaks of C8H6 are ∼10% of the amplitude of peaks of C6H6. The Chu model estimates the mole fraction of C8H6 within the experimental uncertainty and accurately predicts the profile from 1400 K to 1600 K. For the chemistry beyond first aromatic ring in the Chu mechanism, all the reactions were appended by the method discussed above. Since HACA is the only pathway to C8H6 in the model, the satisfying match between the experimental data and the model prediction validates the dominance of HACA generating C8H6 in methane-rich combustion.
4.5 Polycyclic aromatic hydrocarbons (PAHs) profiles
The experimental data of C9H8, C10H8 and C12H8 provide direct evidence of PAH formation from combustion of rich methane and serve as important validation data. For the experimental data of C9H8 shown in Fig. 7, only part of the profiles could be obtained because of a blocked nozzle in Case I and signals lower than the detection limit in Case II. However, the available profiles indicate that C9H8 mole fractions reach up to ∼30 ppb at 1500 K in Cases I and IV. Because the background signals reach the same order of magnitude as the C9H8 signals, the real uncertainty of the experimental data is expected to be larger than the area plotted in Fig. 7. Therefore, the predictions including the profile shape made by the Chu and Narayanaswamy mechanisms seem satisfactory. The pathway analysis of C9H8 formation in Scheme 4a indicates that recombination of phenyl and propargyl radicals is the dominant path. Propa-1,2-dienylbenzene is produced from the initial recombination step, and a hydrogen atom attacks the center carbon on the chain to form 1-phenylpropen-3-yl radical. To accomplish the second ring formation, a radical on the terminal carbon attacks the benzene ring at the ortho position as the ring closure step, and a final β-scission generates indene.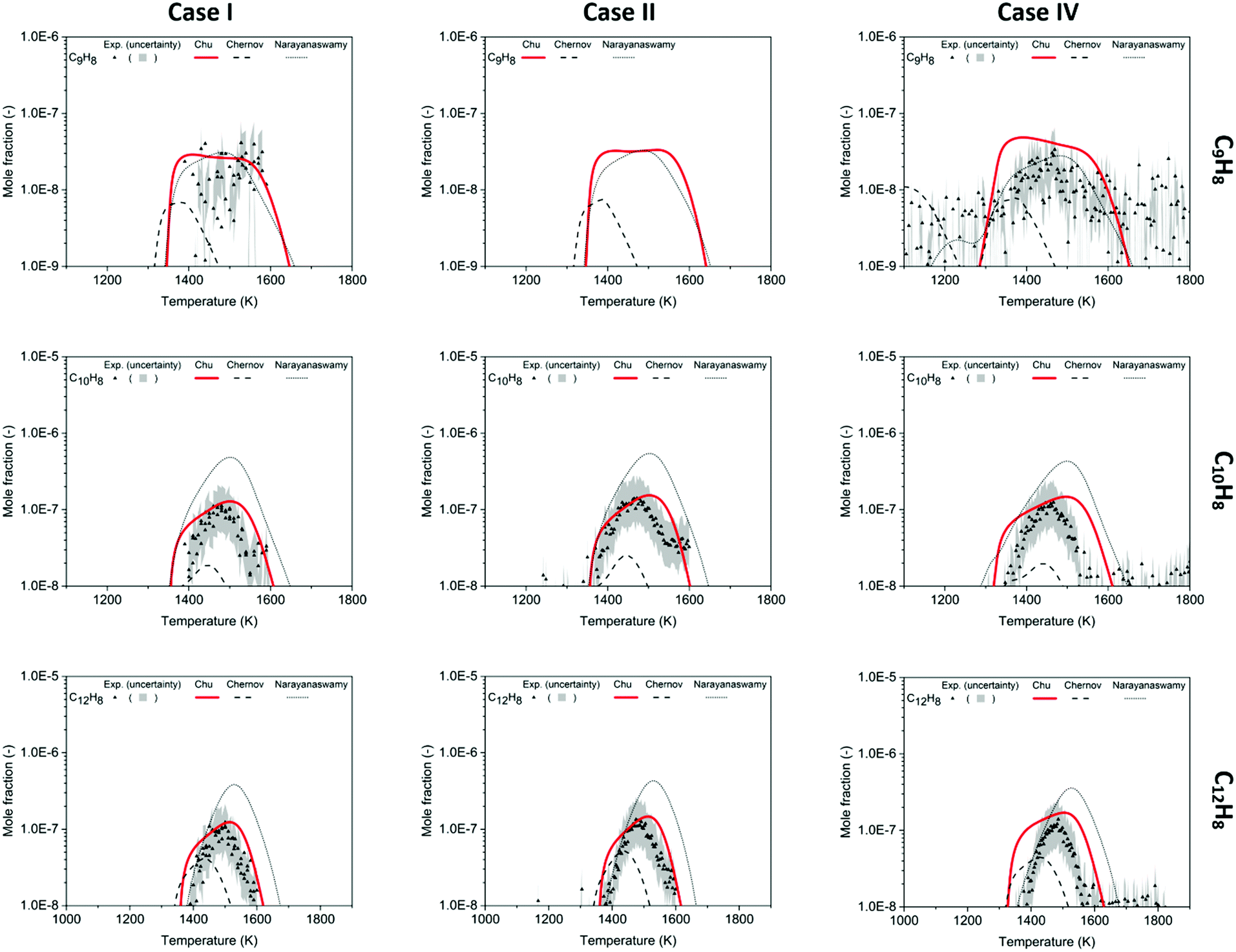 | ||
| Fig. 7 Mole fraction profiles of C9H8, C10H8, C12H8 as a function of oven temperature for Case I, II and IV, measurements (symbols and uncertainty band) and predictions of three models (lines). In the Case II experiment, the C9H8 signal was hard to discern above the noise. | ||
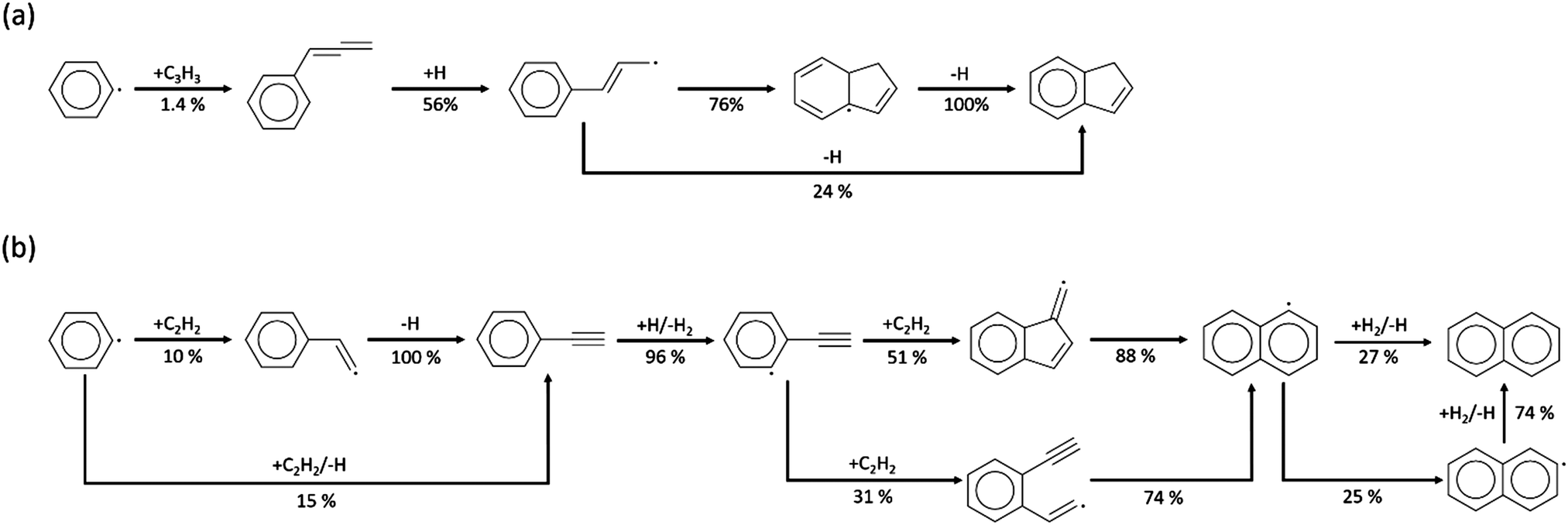 | ||
| Scheme 4 Pathway analysis of (a) indene and (b) naphthalene formation in the Chu model and the ROP analysis at the oven temperature (1415 K for indene and 1502 K for naphthalene) maximizing species concentration in Case 1. | ||
Compared to C9H8, C10H8 was found to have higher concentrations at the conditions studied in this work with naphthalene as the most dominant isomer. The experiment shows a clear peak of C10H8 of 110 ppb at 1471 K in Case I, 140 ppb at 1468 K in Case II and 120 ppb at 1466 K in Case IV, shown in Fig. 7. The maximum concentration of naphthalene, the most stable aromatic species with two rings, is ∼5% of the maximum concentration of benzene. The oven temperature maximizing C10H8 is ∼30 K higher in the Chu model than the experiment; however, the model estimates the maximum concentration with the smallest deviation from the experimental data versus the other two mechanisms.
The HACA pathway is identified as the leading pathway to naphthalene formation among the six pathways summarized by Mebel et al.51 There are three different HACA routes to naphthalene: Frenklach's original HACA route (with phenylacetylene formation), the Bittner–Howard HACA route (with C10H9 isomer formation) and the modified Frenklach route (with styrene formation).54 From the pathway analysis shown in Scheme 4b, Frenklach's original HACA route is the most important one with the appended H-abstraction reactions in the Chu mechanism initiating the second acetylene addition by generating radicals on the ortho position of phenylacetylene. In addition, the importance of pressure dependence in this mechanism is emphasized by the chemically-activated channels in the pathway analysis. The first C2H2 addition in Scheme 4b has two channels: (1) the first elementary step forms the thermalized C8H7 adduct, then the β-scission product, phenylacetylene + H, is generated by the second elementary step; (2) a chemically activated channel characterized in the pressure-dependent network directly produces phenylacetylene + H from phenyl radical + C2H2. In the ROP analysis at the oven temperature maximizing C10H8 concentration in Case I, the chemically-activated channel accounts for 15% of the consumption of phenyl radical, which was more important than the two-step thermalized channel (10%).
The largest quantified species in this work is C12H8 which is calibrated as acenaphthylene. In the experimental data, the maximum mole fraction of C12H8 is 110 ppb at 1491 K in Case I, 130 ppb at 1490 K in Case II and 120 ppb at 1487 K in Case IV. The amount of C12H8 formed is similar to C10H8 with the peaks appearing ∼20 K higher. There are five C12H8 isomers in the Chu mechanism, and acenaphthylene, the stable product produced following the reaction 1-naphthyl radical + C2H2, has the highest concentration in the model. The second dominant isomer is 2-ethynylnaphthalene, which is the product of 2-naphthyl radicals + C2H2. Comparing the modeling results with the experimental data in Fig. 7, the Chu mechanism has an accurate prediction similar to the performance of other aromatics in terms of temperature dependence and maximum concentration. Given the satisfactory predictions made in this study up to C12 species (three-ring aromatics), future methane-rich mechanism development will focus on chemistry of larger PAHs by training RMG with new kinetics and thermochemical data.
5. Conclusions
The Chu mechanism with pressure dependence including aromatics formation for methane-rich combustion has been developed using RMG. It captures detailed chemistry from small molecules to three-ring aromatics by using the reaction library feature of RMG, training RMG-database with reactions relevant to first aromatic ring formation, and calculating pressure-dependent rates using CanTherm for naphthalene, indene, and acenaphthylene formation. Most importantly, all of the kinetics and thermochemical parameters are taken as is from RMG-database estimates or calculations based on reported PESs without any optimization or fitting to the experimental data.The species mole fraction dataset from the high-temperature flow reactor methane-rich combustion experiments has been augmented by quantifying additional species including PAHs. In the model simulation using the Chu mechanism, major species, C2 chemicals, and oxygenated species profiles are predicted similarly to FFCM-1. At high temperatures (>1600 K), the Chu mechanism does not overestimate C2H2 compared to the Chernov and Narayanaswamy mechanisms. For unsaturated C3–C5 stable molecules, the lumped mole fraction profiles estimated by the Chu model matched decently with experiment, although low absolute concentration and presence of indistinguishable isomers in the experiment made analysis harder. Detailed pathways have been identified for C4 species desaturation, where H-abstraction and β-scission steps convert 1,3-butadiene to vinylacetylene and ultimately diacetylene. The new mechanism accurately captures the overall profile and maximum concentration of aromatic species including benzene, phenylacetylene, indene, naphthalene, and acenaphthylene. The dominant pathways were propargyl self-recombination for benzene, phenyl plus propargyl reactions for indene, and Frenklach's original HACA route for naphthalene. Future mechanism development should focus on pathways to larger PAHs.
In summary, the Chu mechanism developed in this work has been successfully validated using methane-rich combustion experiments and is recommended for future similar modeling projects. The incorporation of high-accuracy detailed aromatic chemistry based on quantum chemical calculations into RMG-database is expected to significantly improve the fidelity of future RMG models and will allow future RMG users to build aromatic formation mechanisms automatically.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support for this research by SABIC, and the collaboration with Dr. Soumya Gudiyella.Notes and references
- M. Inman, Nature, 2014, 516, 28–30 CrossRef CAS PubMed.
- P. Nikolaus, W. Rudolf, W. Rudolf and F. Werner, United States Patent Office, 1966.
- M. Köhler, P. Oßwald, H. Xu, T. Kathrotia, C. Hasse and U. Riedel, Chem. Eng. Sci., 2016, 139, 249–260 CrossRef.
- S. Han, J. Park, S. Song and K. M. Chun, Int. J. Hydrogen Energy, 2010, 35, 8762–8771 CrossRef CAS.
- R. C. Lund, J. J. Geest and G. Peter, Int. J. Chem. Kinet., 2008, 40, 778–807 CrossRef.
- B. Lemke, C. Roodhouse, N. Glumac and H. Krier, Int. J. Hydrogen Energy, 2005, 30, 893–902 CrossRef CAS.
- A. A. Konnov, J. N. Zhu, J. H. Bromly and D.-K. Zhang, Combust. Sci. Technol., 2004, 176, 1093–1116 CrossRef CAS.
- Y. Levy, E. Olchanski, V. Sherbaum, V. Erenburg and A. Burcat, J. Propul. Power, 2006, 22, 669–676 CrossRef CAS.
- E. L. Petersen, D. F. Davidson and R. K. Hanson, Combust. Flame, 1999, 117, 272–290 CrossRef CAS.
- D. B. Olson and W. C. Gardiner, Combust. Flame, 1978, 32, 151–161 CrossRef CAS.
- A. Lifshitz, K. Scheller, A. Burcat and G. B. Skinner, Combust. Flame, 1971, 16, 311–321 CrossRef CAS.
- H. Watanabe, F. Arai and K. Okazaki, Combust. Flame, 2013, 160, 2375–2385 CrossRef CAS.
- C. H. Smith, D. I. Pineda and J. L. Ellzey, Combust. Flame, 2013, 160, 557–564 CrossRef CAS.
- A. Turbiez, A. El Bakali, J. F. Pauwels, A. Rida and P. Meunier, Fuel, 2004, 83, 933–941 CrossRef CAS.
- S. G. Davis and C. K. Law, Combust. Sci. Technol., 1998, 140, 427–449 CrossRef CAS.
- M. Musick, P. J. Van Tiggelen and J. Vandooren, Combust. Flame, 1996, 105, 433–450 CrossRef CAS.
- M. J. Castaldi, A. M. Vincitore and S. M. Senkan, Combust. Sci. Technol., 1995, 107, 1–19 CrossRef CAS.
- C. J. Langley and A. R. Burgess, Proc. R. Soc. London, Ser. A, 1989, 421, 259–278 CrossRef CAS.
- F. N. Egolfopoulos, P. Cho and C. K. Law, Combust. Flame, 1989, 76, 375–391 CrossRef CAS.
- J. H. Bechtel, R. J. Blint, C. J. Dasch and D. A. Weinberger, Combust. Flame, 1981, 42, 197–213 CrossRef CAS.
- B. Garten, F. Hunger, D. Messig, B. Stelzner, D. Trimis and C. Hasse, Int. J. Therm. Sci., 2015, 87, 68–84 CrossRef CAS.
- B. Stelzner, F. Hunger, S. Voss, J. Keller, C. Hasse and D. Trimis, Proc. Combust. Inst., 2013, 34, 1045–1055 CrossRef CAS.
- B. Stelzner, F. Hunger, A. Laugwitz, M. Gräbner, S. Voss, K. Uebel, M. Schurz, R. Schimpke, S. Weise, S. Krzack, D. Trimis, C. Hasse and B. Meyer, Fuel Process. Technol., 2013, 110, 33–45 CrossRef CAS.
- F. Hunger, B. Stelzner, D. Trimis and C. Hasse, Flow, Turbul. Combust., 2013, 90, 833–857 CrossRef CAS.
- A. Keller, R. Kovacs and K. H. Homann, Phys. Chem. Chem. Phys., 2000, 2, 1667–1675 RSC.
- R. J. Berger and G. B. Marin, Ind. Eng. Chem. Res., 1999, 38, 2582–2592 CrossRef CAS.
- S. Gudiyella, Z. J. Buras, T.-C. Chu, I. Lengyel, S. Pannala and W. H. Green, Ind. Eng. Chem. Res., 2018, 57, 7404–7420 CrossRef CAS.
- Q. Di, L. Dai and Y. Wang, JAMA, 2017, 318, 2446–2456 CrossRef PubMed.
- D. Shindell, J. C. I. Kuylenstierna, E. Vignati, R. van Dingenen, M. Amann, Z. Klimont, S. C. Anenberg, N. Muller, G. Janssens-Maenhout, F. Raes, J. Schwartz, G. Faluvegi, L. Pozzoli, K. Kupiainen, L. Höglund-Isaksson, L. Emberson, D. Streets, V. Ramanathan, K. Hicks, N. T. K. Oanh, G. Milly, M. Williams, V. Demkine and D. Fowler, Science, 2012, 335, 183–189 CrossRef CAS PubMed.
- M. Frenklach and H. Wang, Symp. (Int.) Combust., [Proc.], 1991, 23, 1559–1566 CrossRef.
- C. A. Schuetz and M. Frenklach, Proc. Combust. Inst., 2002, 29, 2307–2314 CrossRef CAS.
- M. S. Skjøth-Rasmussen, P. Glarborg, M. Østberg, J. T. Johannessen, H. Livbjerg, A. D. Jensen and T. S. Christensen, Combust. Flame, 2004, 136, 91–128 CrossRef.
- G. P. Smith, D. M. Golden, M. Frenklach, N. W. Moriarty, B. Eiteneer, M. Goldenberg, C. T. Bowman, R. K. Hanson, S. Song, W. C. Gardiner, V. V. Lissianski Jr. and Z. Qin, GRI-Mech 3.0, http://www.me.berkeley.edu/gri_mech/.
- Y. Li, C.-W. Zhou, K. P. Somers, K. Zhang and H. J. Curran, Proc. Combust. Inst., 2017, 36, 403–411 CrossRef CAS.
- C.-W. Zhou, Y. Li, E. O'Connor, K. P. Somers, S. Thion, C. Keesee, O. Mathieu, E. L. Petersen, T. A. DeVerter, M. A. Oehlschlaeger, G. Kukkadapu, C.-J. Sung, M. Alrefae, F. Khaled, A. Farooq, P. Dirrenberger, P.-A. Glaude, F. Battin-Leclerc, J. Santner, Y. Ju, T. Held, F. M. Haas, F. L. Dryer and H. J. Curran, Combust. Flame, 2016, 167, 353–379 CrossRef CAS.
- U. Burke, W. K. Metcalfe, S. M. Burke, K. A. Heufer, P. Dagaut and H. J. Curran, Combust. Flame, 2016, 165, 125–136 CrossRef CAS.
- S. M. Burke, U. Burke, R. Mc Donagh, O. Mathieu, I. Osorio, C. Keesee, A. Morones, E. L. Petersen, W. Wang, T. A. DeVerter, M. A. Oehlschlaeger, B. Rhodes, R. K. Hanson, D. F. Davidson, B. W. Weber, C.-J. Sung, J. Santner, Y. Ju, F. M. Haas, F. L. Dryer, E. N. Volkov, E. J. K. Nilsson, A. A. Konnov, M. Alrefae, F. Khaled, A. Farooq, P. Dirrenberger, P.-A. Glaude, F. Battin-Leclerc and H. J. Curran, Combust. Flame, 2015, 162, 296–314 CrossRef CAS.
- S. M. Burke, W. Metcalfe, O. Herbinet, F. Battin-Leclerc, F. M. Haas, J. Santner, F. L. Dryer and H. J. Curran, Combust. Flame, 2014, 161, 2765–2784 CrossRef CAS.
- A. Kéromnès, W. K. Metcalfe, K. A. Heufer, N. Donohoe, A. K. Das, C.-J. Sung, J. Herzler, C. Naumann, P. Griebel, O. Mathieu, M. C. Krejci, E. L. Petersen, W. J. Pitz and H. J. Curran, Combust. Flame, 2013, 160, 995–1011 CrossRef.
- W. K. Metcalfe, S. M. Burke, S. S. Ahmed and H. J. Curran, Int. J. Chem. Kinet., 2013, 45, 638–675 CrossRef CAS.
- G. P. Smith, Y. Tao and H. Wang, Foundational Fuel Chemistry Model Version 1.0 (FFCM-1), 2016, http://nanoenergy.stanford.edu/ffcm1.
- H. Wang, X. You, A. V. Joshi, S. G. Davis, A. Laskin, F. Egolfopoulos and C. K. Law, USC Mech Version II. High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds, http://ignis.usc.edu/USC_Mech_II.htm.
- G. Blanquart, P. Pepiot-Desjardins and H. Pitsch, Combust. Flame, 2009, 156, 588–607 CrossRef CAS.
- K. Narayanaswamy, G. Blanquart and H. Pitsch, Combust. Flame, 2010, 157, 1879–1898 CrossRef CAS.
- V. Chernov, M. J. Thomson, S. B. Dworkin, N. A. Slavinskaya and U. Riedel, Combust. Flame, 2014, 161, 592–601 CrossRef CAS.
- N. A. Slavinskaya and P. Frank, Combust. Flame, 2009, 156, 1705–1722 CrossRef CAS.
- B. M. Wong, D. M. Matheu and W. H. Green, J. Phys. Chem. A, 2003, 107, 6206–6211 CrossRef CAS.
- C. W. Gao, J. W. Allen, W. H. Green and R. H. West, Comput. Phys. Commun., 2016, 203, 212–225 CrossRef CAS.
- J. W. Allen, C. F. Goldsmith and W. H. Green, Phys. Chem. Chem. Phys., 2012, 14, 1131–1155 RSC.
- K. Han, A. Jamal, C. A. Grambow, Z. J. Buras and W. H. Green, Int. J. Chem. Kinet., 2018, 50, 294–303 CrossRef CAS.
- A. M. Mebel, A. Landera and R. I. Kaiser, J. Phys. Chem. A, 2017, 121, 901–926 CrossRef CAS PubMed.
- M. Frenklach, Phys. Chem. Chem. Phys., 2002, 4, 2028–2037 RSC.
- H. Richter and J. B. Howard, Prog. Energy Combust. Sci., 2000, 26, 565–608 CrossRef CAS.
- A. M. Mebel, Y. Georgievskii, A. W. Jasper and S. J. Klippenstein, Proc. Combust. Inst., 2017, 36, 919–926 CrossRef CAS.
- V. V. Kislov, N. I. Islamova, A. M. Kolker, S. H. Lin and A. M. Mebel, J. Chem. Theory Comput., 2005, 1, 908–924 CrossRef CAS PubMed.
- J. Aguilera-Iparraguirre and W. Klopper, J. Chem. Theory Comput., 2007, 3, 139–145 CrossRef CAS PubMed.
- A. M. Mebel and V. V. Kislov, J. Phys. Chem. A, 2009, 113, 9825–9833 CrossRef CAS PubMed.
- V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2008, 112, 700–716 CrossRef CAS.
- A. J. Vervust, M. R. Djokic, S. S. Merchant, H.-H. Carstensen, A. E. Long, G. B. Marin, W. H. Green and K. M. Van Geem, Energy Fuels, 2018, 32, 3920–3934 CrossRef CAS.
- A. E. Long, S. S. Merchant, A. G. Vandeputte, H.-H. Carstensen, A. J. Vervust, G. B. Marin, K. M. Van Geem and W. H. Green, Combust. Flame, 2018, 187, 247–256 CrossRef CAS.
- A. Matsugi and A. Miyoshi, Int. J. Chem. Kinet., 2012, 44, 206–218 CrossRef CAS.
- H. Ismail, J. Park, B. M. Wong, W. H. Green and M. C. Lin, Proc. Combust. Inst., 2005, 30, 1049–1056 CrossRef.
- A. M. Mebel, Y. Georgievskii, A. W. Jasper and S. J. Klippenstein, Faraday Discuss., 2016, 195, 637–670 RSC.
- V. V. Kislov and A. M. Mebel, J. Phys. Chem. A, 2007, 111, 3922–3931 CrossRef CAS.
- Z. J. Buras, T.-C. Chu, A. Jamal, N. W. Yee, J. E. Middaugh and W. H. Green, Phys. Chem. Chem. Phys., 2018, 20, 13191–13214 RSC.
- P. Oßwald and M. Köhler, Rev. Sci. Instrum., 2015, 86, 105–109 CrossRef PubMed.
- T. Kathrotia, C. Naumann, P. Oßwald, M. Köhler and U. Riedel, Combust. Flame, 2017, 179, 172–184 CrossRef CAS.
- T. Kathrotia, P. Oßwald, M. Köhler, N. A. Slavinskaya and U. Riedel, Combust. Flame, 2018, 194, 426–438 CrossRef CAS.
- M. Köhler, T. Kathrotia, P. Oßwald, M. L. Fischer-Tammer, K. Moshammer and U. Riedel, Combust. Flame, 2015, 162, 3197–3209 CrossRef.
- P. Oßwald, R. Whitside, J. Schäffer and M. Köhler, Fuel, 2017, 187, 43–50 CrossRef.
- P. Oßwald, P. Hemberger, T. Bierkandt, E. Akyildiz, M. Köhler, A. Bodi, T. Gerber and T. Kasper, Rev. Sci. Instrum., 2014, 85, 025101 CrossRef PubMed.
- D. Krüger, P. Oßwald, M. Köhler, P. Hemberger, T. Bierkandt, Y. Karakaya and T. Kasper, Combust. Flame, 2018, 191, 343–352 CrossRef.
- Chemical Workbench, Kintech Lab, 2016, 4.1.18493.
- A. Fahr and A. Nayak, Int. J. Chem. Kinet., 2000, 32, 118–124 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 7783–7799 CrossRef CAS.
- Y. Georgievskii, J. A. Miller and S. J. Klippenstein, Phys. Chem. Chem. Phys., 2007, 9, 4259–4268 RSC.
- S. W. Benson, Thermochemical kinetics: methods for the estimation of thermochemical data and rate parameters, Wiley, 1968 Search PubMed.
- H. L. Woodcock, D. Moran, B. R. Brooks, P. v. R. Schleyer and H. F. Schaefer, J. Am. Chem. Soc., 2007, 129, 3763–3770 CrossRef CAS PubMed.
- S. E. Stein, J. A. Walker, M. M. Suryan and A. Fahr, Symp. (Int.) Combust., [Proc.], 1991, 23, 85–90 CrossRef.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 2680–2692 CrossRef CAS.
- D. K. Hahn, S. J. Klippenstein and J. A. Miller, Faraday Discuss., 2002, 119, 79–100 RSC.
- L. B. Harding, S. J. Klippenstein and Y. Georgievskii, J. Phys. Chem. A, 2007, 111, 3789–3801 CrossRef CAS PubMed.
- C. Huang, B. Yang and F. Zhang, Combust. Flame, 2017, 184, 167–175 CrossRef CAS.
- M. J. Fadden, C. Barckholtz and C. M. Hadad, J. Phys. Chem. A, 2000, 104, 3004–3011 CrossRef CAS.
- I. V. Tokmakov, G.-S. Kim, V. V. Kislov, A. M. Mebel and M. C. Lin, J. Phys. Chem. A, 2005, 109, 6114–6127 CrossRef CAS PubMed.
- V. V. Kislov, R. I. Singh, D. E. Edwards, A. M. Mebel and M. Frenklach, Proc. Combust. Inst., 2015, 35, 1861–1869 CrossRef CAS.
- B. S. Narendrapurapu, A. C. Simmonett, H. F. Schaefer, J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2011, 115, 14209–14214 CrossRef CAS PubMed.
- J. M. Ribeiro and A. M. Mebel, Phys. Chem. Chem. Phys., 2017, 19, 14543–14554 RSC.
- S. J. Klippenstein and J. A. Miller, J. Phys. Chem. A, 2005, 109, 4285–4295 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN03, 2003 Search PubMed.
- S. J. Klippenstein, L. B. Harding and Y. Georgievskii, Proc. Combust. Inst., 2007, 31, 221–229 CrossRef.
- S.-H. Li, J.-J. Guo, R. Li, F. Wang and X.-Y. Li, J. Phys. Chem. A, 2016, 120, 3424–3432 CrossRef CAS PubMed.
- V. V. Kislov, A. I. Sadovnikov and A. M. Mebel, J. Phys. Chem. A, 2013, 117, 4794–4816 CrossRef CAS PubMed.
- M. Frenklach, R. I. Singh and A. M. Mebel, Proc. Combust. Inst., 2018 DOI:10.1016/j.proci.2018.05.068.
- M. Vascellari, H. Xu, S. Hartl, F. Hunger and C. Hasse, Chem. Eng. Sci., 2015, 134, 694–707 CrossRef CAS.
- J. A. Miller and C. F. Melius, Combust. Flame, 1992, 91, 21–39 CrossRef CAS.
- T. A. Cool, K. Nakajima, T. A. Mostefaoui, F. Qi, A. McIlroy, P. R. Westmoreland, M. E. Law, L. Poisson, D. S. Peterka and M. Ahmed, J. Chem. Phys., 2003, 119, 8356–8365 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp06097e |
| This journal is © the Owner Societies 2019 |