
Rate exchange rather than relaxation controls structural recovery
Birte
Riechers
 * and
Ranko
Richert
* and
Ranko
Richert
School of Molecular Sciences, Arizona State University, Tempe, AZ 85287, USA. E-mail: briecher@asu.edu
First published on 26th September 2018
Abstract
We observe structural recovery after an electric field step by probing the dielectric loss profile near its maximum, which displays a field-induced shift towards lower frequencies. These dynamics display time aging-time superposition (TaTS) for the majority of relaxation modes, thus implying homogeneous recovery dynamics. Although assumed by generally accepted models, the same modes can not be responsible for structural relaxation and for structural recovery, as the former is heterogeneous and the latter is homogeneous regarding the nature of the dynamics. This conflict is resolved by proposing that structural recovery is governed by rate exchange, a process that refers to the homogeneous fluctuations of rate constants in equilibrium and restores ergodicity more slowly than the relaxation observed as a simple correlation decay. This recognition has wide-ranging consequences on how aging and nonlinear dynamics such as scanning calorimetry should be modeled.
Introduction
The structural relaxation of liquids is often characterized by responses to small perturbations whose amplitudes ensure that the system remains in equilibrium.1 The situation is more complicated if systems depart from equilibrium, either as a result of changing temperature or pressure, or by any perturbation leading to a departure from linear response.2 An example known as physical aging is the slow evolution of the configurational state towards equilibrium at a temperature T after a quench from an equilibrium state to a temperature T < Tg, where Tg denotes the glass transition temperature. Tool and Narayanaswamy described the discrepancy between the equilibrium structure at temperature T and actual structure by means of a fictive temperature Tf > T,3,4 implying that the relaxation timescale τ depends on both T and Tf until equilibrium is restored by a process denoted physical aging or structural recovery.2 Quantitative modeling of such non-equilibrium dynamics requires a detailed understanding of the processes that restore equilibrium.Widely used approaches to the kinetics that restore equilibrium are the Tool–Narayanaswamy–Moynihan (TNM), the Kovacs–Aklonis–Hutchinson–Ramos (KAHR), and related models,2,5,6 which make two important assumptions: (i) the deviation from equilibrium can be captured by a single parameter (Tf), and (ii) relaxation dynamics control structural recovery. A consequence of a single fictive temperature is that the susceptibility will not change shape in the process of physical aging, which is referred to as time-aging time-superposition (TaTS). Assuming that recovery is entirely controlled by relaxation dynamics facilitates that relaxation data obtained from linear response experiments serve as input to aging models.
After recognizing the heterogeneous nature of structural relaxation in the linear regime,7,8 fast and slow relaxation modes are considered largely independent. As a result, the dispersive nature of the α-relaxation that is observed by numerous techniques is viewed as a superposition of (locally) exponential processes. Assuming that these relaxation modes control physical aging and other nonlinear processes suggests that fast modes would recover equilibrium more quickly than others.9,10 Clearly, such a model is inconsistent with the commonly observed feature of TaTS, as the relaxation mode spectrum would narrow in the course of aging if structural recovery were as heterogeneous as relaxation dynamics.11–13 The conundrum is that the observation of TaTS implies homogeneous structural recovery, while relaxation dynamics are consistently found to be heterogeneous. Possible solutions are that either TaTS is not valid for the entire relaxation spectrum, or structural recovery should not be modeled on the basis of relaxation modes.
The typical frequency-domain probing of physical aging induced by a temperature step is restricted to high frequencies relative to the peak frequency, thus covering only a few percent of the fastest relaxation modes contributing to the overall response.14 This is mainly due to the necessity of averaging over several periods of an oscillatory probing field for the purpose of accurately identifying the state of the system. Most such measurements are also restricted to slow aging in order to allow for sufficient thermal equilibration prior to the onset of aging,15 but faster equipment performance has been reported.16 As a representative example, the work of Lunkenheimer et al.15 provides detailed dielectric loss data for a range of frequencies recorded in the course of physical aging of various materials. Here, the lowest test frequency is about 3 decades higher than the loss peak frequency, νmax, so that only the fastest ∼3% of the contributions to the spectrum are observed regarding their aging behavior.14 In other words, only the small fraction of modes that reside in the high-frequency flank have been subject to aging experiments, while the recovery dynamics of the bulk of the modes remained obscure. Thus, despite the number of reports on aging,15–19 the validity of TaTS has not been demonstrated for the majority of modes, which reside near the most probable relaxation frequency. A decisive conclusion on the validity of TaTS for the entire spectrum requires observing structural recovery of modes near the loss peak.
In this work, evidence is provided for TaTS to apply not only to the few fastest, but to the majority of modes. The homogeneous nature of structural recovery is then rationalized by the notion that physical aging is controlled by rate exchange rather than relaxation processes, with rate exchange referring to the fluctuations of time constants that restore ergodicity on a time scale (τex) slower than relaxation dynamics (τα). We report time-resolved dielectric measurements of physical aging at frequencies ranging from νmax to about 100 × νmax, thus probing more than 95% of relaxation modes. Instead of employing a temperature change, a high electric bias-field is applied to drive the sample away from its prior equilibrium state within microseconds.20 The structural recovery is then measured by a small amplitude oscillatory field at different frequencies. Due to averaging over the results of thousands of field transitions,20 this approach facilitates aging measurement with a time resolution of a single period (1/ν) and thus using test frequencies near the peak loss frequency.
Experimental
The compounds of this study, glycerol (GLY, 99.5%, Aldrich) and 2-methyltetrahydrofuran (MTHF, 99%, stabilizer-free, Acros), were used as received. All measurements are based on a capacitor cell consisting of two spring-loaded stainless steel disks separated by a Teflon ring of 10 μm nominal thickness and 14 mm inner diameter.21 The cell was mounted onto the cold-stage of a He-refrigerator cryostat (Leybold RDK 6-320), with temperature controlled by a Lakeshore Model 340 using DT-470-CU sensors. Low-field (E < 1.4 kV cm−1) dielectric measurements were performed using a gain/phase analyzer (Solartron SI-1260) equipped with a transimpedance amplifier (Mestec DM-1360).The aim of the electric field applied to the sample is twofold: (i) a large amplitude bias field is applied to shift the state of the sample away from its previous equilibrium position, thereby initiating a structural recovery process, analogous to the role of a temperature down jump in the case of physical aging; (ii) a small amplitude oscillatory signal is applied to measure the dielectric loss ε′′ at the selected frequency ω. The role of the subsequent data analysis is to evaluate the change of ε′′(ω) for each period following the bias field step. A simplified schematic depiction of this field protocol is shown in Fig. 1, with the zero field interval between two field bursts (axis break) being long compared with the time the high field is applied. To achieve signal averaging, the field burst is applied 5000 times and the average result for the voltage and current traces are recorded.
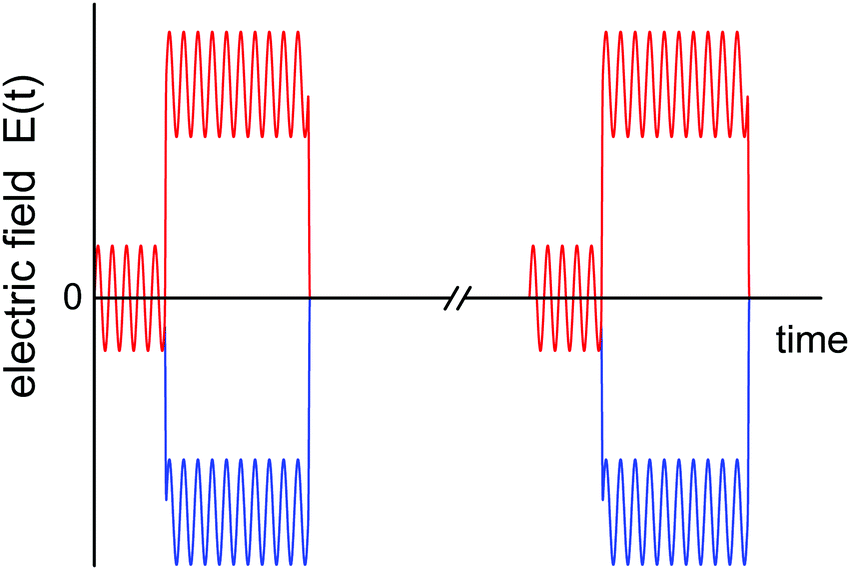 | ||
| Fig. 1 Schematic representation of the field protocol used for the present experiment. The oscillatory component is used to obtain a value for ε′′(ω) for each period, and each of the 5000 high amplitude bias field steps (only two shown here) induces a structural recovery process that is monitored with a time resolution of one period, i.e., 2π/ω. Measurements are repeated for a range of frequencies ω to cover the dielectric loss spectrum. | ||
These time-resolved high-field measurements were realized by an arbitrary waveform generator (Stanford Research Systems DS-345) with high voltage amplifier (Trek PZD 700). The waveforms consisted of an integer number of periods with a phase shift, V(t) = V0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(ωt − π/4), with a dc-bias voltage VB superimposed only after a certain number of periods without dc-bias, see Fig. 1. The voltage ratio was VB/V0 = 4 for measurements on GLY and VB/V0 = 9 for MTHF. The dc-bias transition was realized as a linear ramp with duration 0.1/ν and it defines t = 0 at its center. Due to the phase-shift, the dc-bias step is completed within the second half of the last ‘low-field’ period, increasing the time resolution relative to previous experiments.20,22,23 Due to the limitation to 16300 points defining a waveform, some experiments were repeated at a lower point density per period to allow for a larger number of periods, thereby extending the time window to about 18 ms. In such cases, the results for both high and low point densities are overlaid in the graphs, and no systematic differences are detected. Waveform signals were repeated at a rate of 1–2 Hz with the high-field duty cycle kept below 15% to ensure that field induced effects had subsided prior to the next field step. Voltage and current signals were recorded by a digitizing oscilloscope (Nicolet Sigma 100). All results are based on the average of two measurements of opposing bias-polarity, +VB and −VB (red and blue trace in Fig. 1, respectively), which eliminates the direct polarization response to the bias-step.22 A Fourier analysis of each period (i.e., between times t = 2π(n + 1/8)/ω and 2π(n + 9/8)/ω for integer n) of the resulting signals (voltage V, current I) yielded amplitude (AV, AI) and phase (φV, φI) values, from which the dielectric loss ε′′ was determined for every period via22
sin(ωt − π/4), with a dc-bias voltage VB superimposed only after a certain number of periods without dc-bias, see Fig. 1. The voltage ratio was VB/V0 = 4 for measurements on GLY and VB/V0 = 9 for MTHF. The dc-bias transition was realized as a linear ramp with duration 0.1/ν and it defines t = 0 at its center. Due to the phase-shift, the dc-bias step is completed within the second half of the last ‘low-field’ period, increasing the time resolution relative to previous experiments.20,22,23 Due to the limitation to 16300 points defining a waveform, some experiments were repeated at a lower point density per period to allow for a larger number of periods, thereby extending the time window to about 18 ms. In such cases, the results for both high and low point densities are overlaid in the graphs, and no systematic differences are detected. Waveform signals were repeated at a rate of 1–2 Hz with the high-field duty cycle kept below 15% to ensure that field induced effects had subsided prior to the next field step. Voltage and current signals were recorded by a digitizing oscilloscope (Nicolet Sigma 100). All results are based on the average of two measurements of opposing bias-polarity, +VB and −VB (red and blue trace in Fig. 1, respectively), which eliminates the direct polarization response to the bias-step.22 A Fourier analysis of each period (i.e., between times t = 2π(n + 1/8)/ω and 2π(n + 9/8)/ω for integer n) of the resulting signals (voltage V, current I) yielded amplitude (AV, AI) and phase (φV, φI) values, from which the dielectric loss ε′′ was determined for every period via22
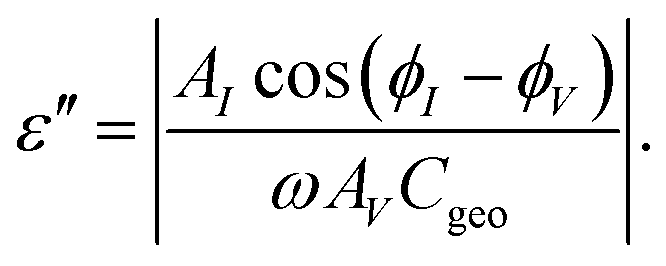 | (1) |
Results
Low-field dielectric loss data of glycerol are compiled in Fig. 2, comparing linear response impedance data taken before (ε′′loinitial) and after (ε′′lofinal) all high-field measurements on that sample had been performed. The difference in loss amplitude between ε′′loinitial and ε′′lofinal is very small, indicating that all field-induced changes are practically reversible. Also included in Fig. 2 are ε′′lo results from the time-resolved technique, derived from the periods without bias field (t < 0). These values are taken at the frequencies indicated by arrows and agree favorably with the ordinary impedance data, validating that field induced changes have faded prior to each of the thousands of subsequent field steps. To characterize the primary relaxation process, ε′′loinitial was fitted by the sum of a Havriliak–Negami (HN) and a Cole–Davidson (CD) function,24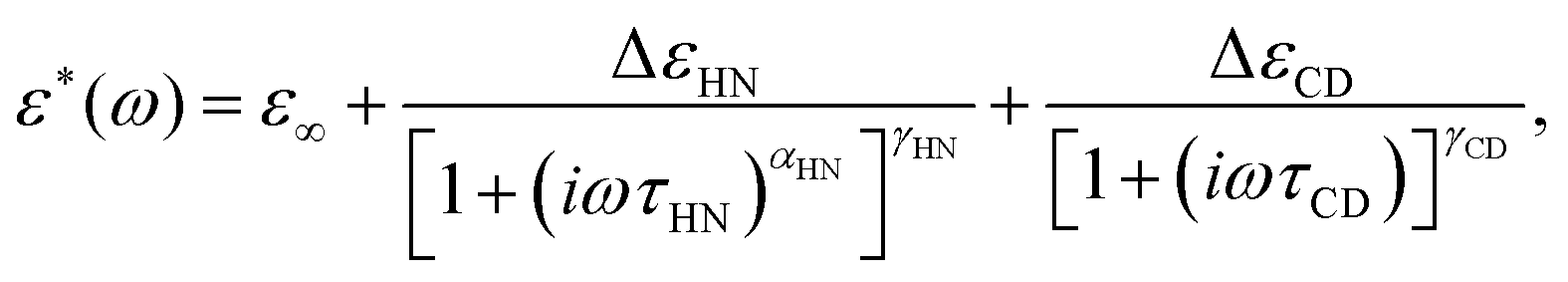 | (2) |
logε′′/∂logν at the test frequencies, the values are otherwise inconsequential for the remainder of the analysis.
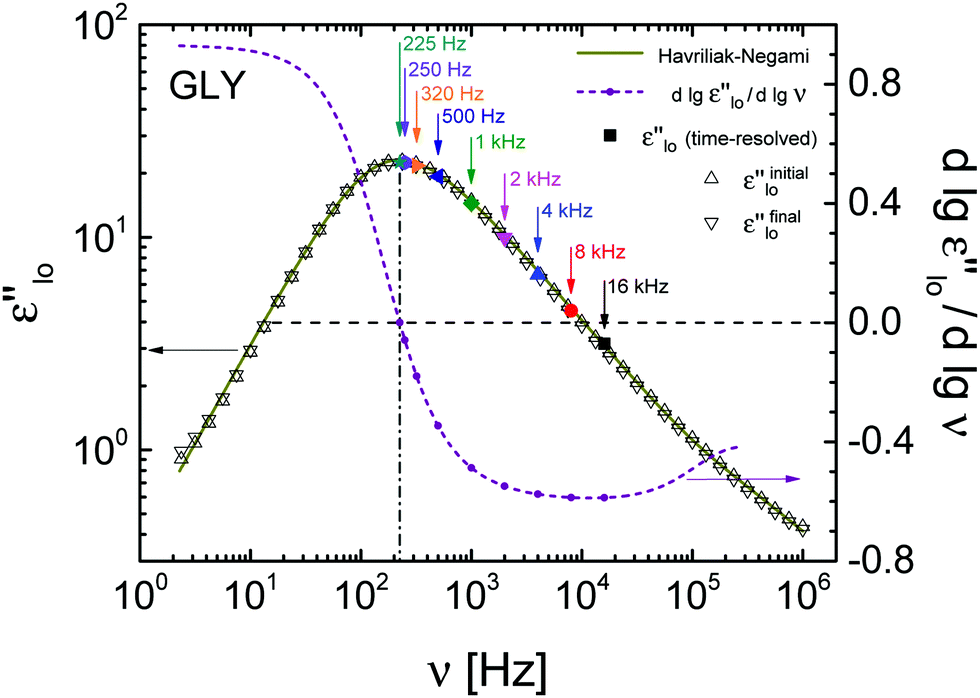 | ||
| Fig. 2 Experimental results (open symbols) of the low-field dielectric loss spectrum of glycerol and its derivative (dashed line) at T = 211 K. The solid line is a fit to the data using eqn (2). Solid symbols represent the low-field (t < 0) response of time-resolved measurements. | ||
To reveal the relative change in ε′′ that arises from the field-induced perturbation at t = 0, Fig. 3 quantifies this nonlinear effect by (ε′′hi − ε′′lo)/ε′′lo, with ε′′lo and ε′′hi referring to data for t < 0 and t > 0, respectively. When the dc-bias field is applied, an instantaneous positive signal arises at t = 0, followed by a decay towards a plateau for t > 0. The initial change is influenced by a ‘heating’-like contribution due to the energy absorbed from the field step, which can be calculated on the basis of a well-tested model,22,25 see the inset of Fig. 3. The parameters used in this calculation are the dc-field of EB = 196 kV cm−1, the activation parameter ∂lnτ/∂T = −0.339 K−1 based on Vogel–Fulcher–Tammann (VFT) fit results,26 and the configurational contribution of the excess heat capacity ΔCcfg = fcΔCexc = 0.8 × 1.245 J K−1 cm−3.20 Clearly, this ‘heating’ effect on (ε′′hi − ε′′lo)/ε′′lo decays more rapidly than the slower transition towards a negative level, which displays frequency dependent values ranging from −0.75% to −1.5%.
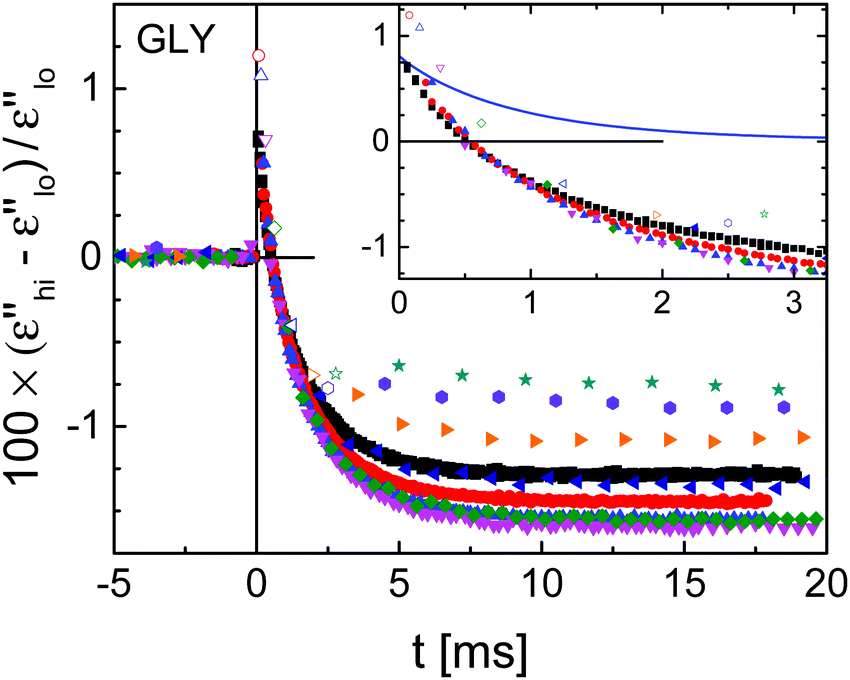 | ||
| Fig. 3 Experimental results for glycerol at T = 211 K (symbols) of the field-induced time-resolved relative change in dielectric loss for nine different frequencies as indicated in Fig. 2. Superposition with a dc-bias field of EB = 196 kV cm−1 starts at t = 0 s. The inset shows details of the signal just after applying the dc-bias field along with the calculated ‘heating’ contribution (line). | ||
According to previous observations on glycerol subject to high static electric fields,20 the depression of ε′′hi for ν > νmax is largely due to a shift of the loss spectrum to lower frequencies, equivalent to longer relaxation times as in a temperature down jump. In order to translate the changes observed for ε′′ into a shift of the spectrum along the frequency axis, the saturation corrected values of (ε′′hi − ε′′lo)/ε′′lo ≈ Δlnε′′ are divided by the derivative ∂logε′′/∂logν, see Fig. 2.27,28 The variation of the plateau values with frequency is considerably smaller for the resulting Δlnν traces than for Δlnε′′ itself. The average (250 Hz ≤ ν ≤ 16 kHz) steady state ‘horizontal shift’ amounts to Δlnν = −0.014 at a field of EB = 196 kV cm−1, consistent with a decrease in fictive temperature.25
From the raw results for the field induced relative changes of the dielectric loss in Fig. 3, it is obvious that the decay time constants of these traces show little change even though the probing frequency changes by a factor of 70. Clearly, the long-time plateau levels change with the probe frequency, which is mostly a matter of the change in slope, ∂logε′′/∂logν. In order to obtain a clearer picture of the structural recovery dynamics, the curves have been corrected for the energy absorption effect and normalized to a steady state level of −1, as depicted in Fig. 4 with traces offset vertically for clarity. To facilitate a better comparison, a frequency invariant fit function is added for each curve. The choice for that decay function is motivated below.
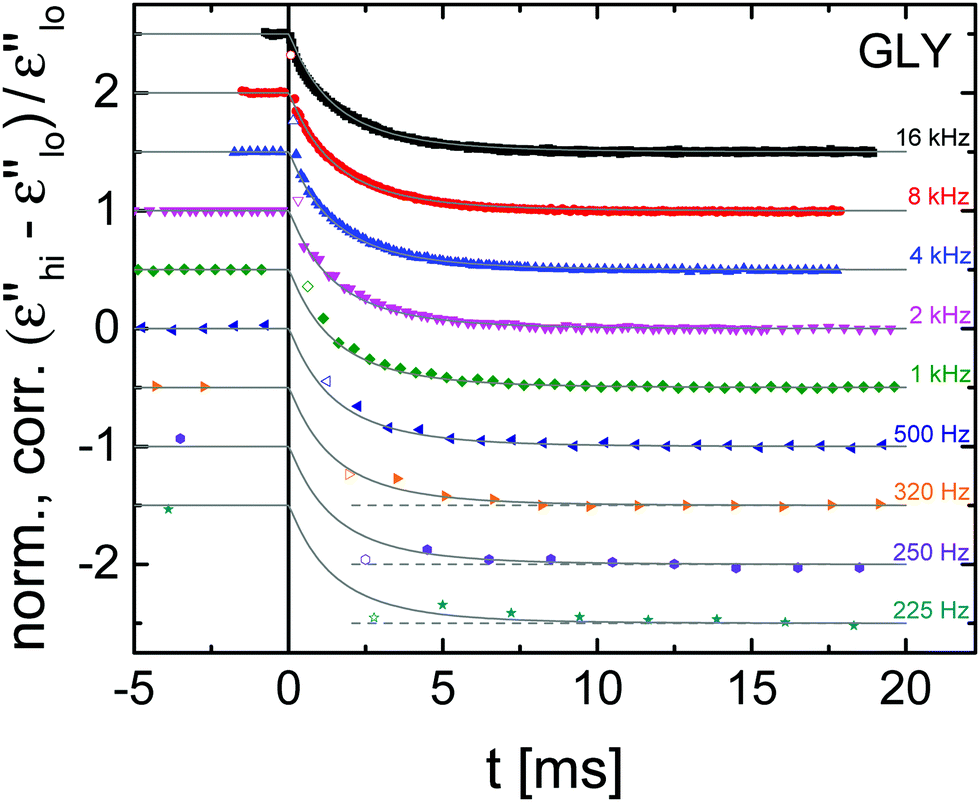 | ||
| Fig. 4 Experimental results (symbols) of the time-resolved relative change in dielectric loss for glycerol at T = 211 K after ‘heating’ correction, normalization to negative unity, and vertical stacking for clarity. The high-field amplitude is EB = 196 kV cm−1 for times t ≥ 0. ϕ2(t)-fits (gray lines) sharing the same parameters for all frequencies serve as guides. | ||
As with many glass-forming liquids, the polarization step-response of glycerol is well represented by the Kohlrausch–Williams–Watts (KWW) function.1,29,30 The parameters for such a KWW fit, ϕ(t) = ϕ0{1 − exp[−(t/τKWW)β]}, can be deduced from the above HN-fit results.31 This translation yields τKWW = 0.58 ms and β = 0.65, but ϕ(t) is not expected to match the decays of Fig. 4.22 To understand this, one needs to recognize that (ε′′hi − ε′′lo)/ε′′lo is not proportional to polarization P, but rather to P2.25 To account for this quadratic dependence, we follow earlier practice22,23,25 and describe the nonlinear response with ϕ2(t) = ϕ0{1 − exp[−(t/τadj)β]}2, which matches the data with an adjusted time constant τadj = 1.4 × τKWW = 0.81 ms. As can be observed in Fig. 4, the agreement between experimental results and the ϕ2(t) function is independent of the probing frequency, clearly demonstrating that structural recovery is frequency invariant within the present range. Given the limited time resolution for the lower frequencies, the time-dependence revealed by these traces depends critically on the initial data point of each curve. To visualize the impact of those first data points, a dashed horizontal line at the steady state level serves as a guide. Although the value of every first period of a data set is offset from the trend of the remaining data, the clear conclusion from Fig. 4 is that no frequency dependence can be detected regarding the dynamics of structural recovery.
To be able to compare the results of glycerol with a second organic glass-former, the time-dependent high-field response of MTHF was measured at T = 97 K. The time-resolved data were evaluated as described for glycerol with parameters for the ‘heating’ correction of EB = 253 kV cm−1, ∂lnτ/∂T = −1.22 K−1, and ΔCcfg = fcΔCexc = 0.6 × 0.97 J K−1 cm−3. Plateau values of the relative nonlinear contribution of MTHF follow a comparable trend as for glycerol and result in an average frequency shift Δlnν = −0.025 at a field of EB = 253 V cm−1. The normalization of (ε′′hi − ε′′lo)/ε′′lo measured at various frequencies collapses all data sets to a master curve, as shown in Fig. 5. Again, a common timescale of structural recovery dominates the evolution of the data for all probe frequencies. To derive time constants from these decays, a ϕ2(t)-function using τKWW and β was calculated. The low-field data shown in the inset of Fig. 5 provided the necessary HN parameters: ΔεHN = 14.8, log10(τHN/s) = −2.75, αHN = 0.94, γHN = 0.54, and ΔεCD = 0. The corresponding peak loss is positioned at νmax = 135 Hz. Matching the data could not to be obtained by adjusting solely τKWW, so that the ϕ2(t)-function plotted in Fig. 5 is based on the adjusted parameters τadj = 3 × τKWW = 2.03 ms and βadj = 1.6 × β = 0.8. The main result for MTHF is that, again, the structural recovery process is frequency invariant and slower than the dielectric relaxation. For the time-scale ratio, τadj/τKWW, single frequency measurements on other glass-formers have found 1.7 for glycerol,22 about 1.5 for cresolphthalein dimethylether,27 and 2.0 for N-methyl-ε-caprolactam,23 all similar to the present findings of 1.4 for GLY and 3.0 for MTHF.
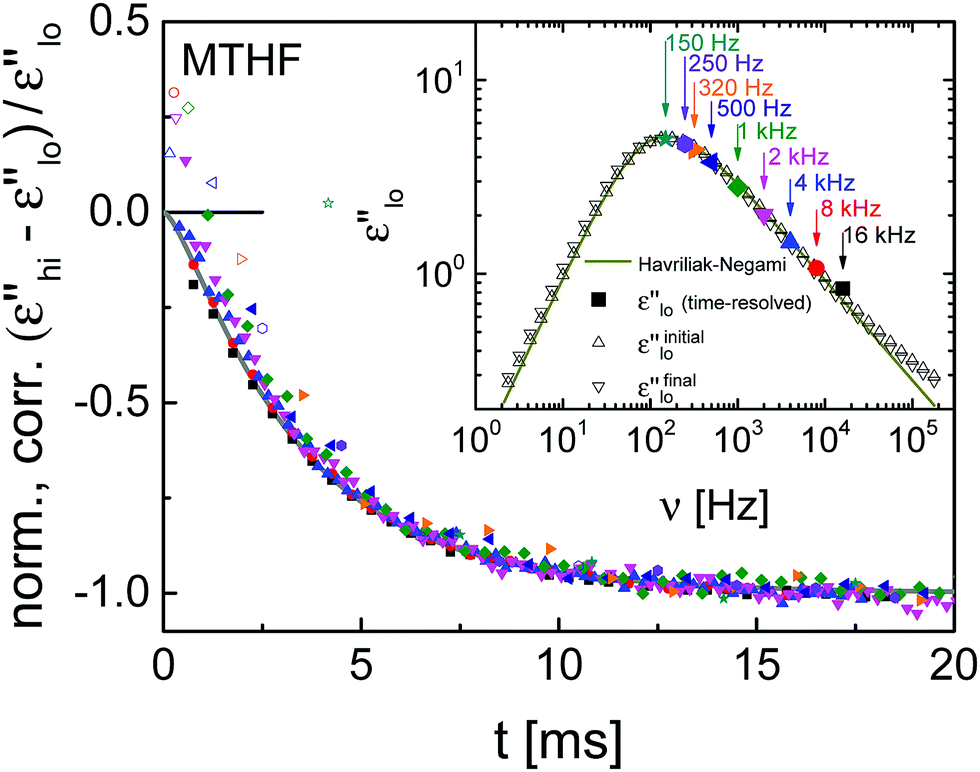 | ||
| Fig. 5 Experimental results (symbols) of the time-resolved relative change in dielectric loss for MTHF at T = 97 K after ‘heating’ correction and normalization to negative unity, along with a ϕ2(t)-fit (gray line). The field is EB = 253 kV cm−1. The inset shows experimental results (open symbols) of the corresponding low-field dielectric loss spectrum, the solid line is a fit using eqn (2). Solid symbols correspond to the low-field (t < 0) response of time-resolved measurements. | ||
In both cases, GLY and MTHF, the probe frequencies span the two decades from the loss peak position to the onset of the excess wing, i.e., in which the recovery dynamics of the α-peak dominates over possibly different dynamics originating from a secondary process. Based on the HN-parameters, the probability density of relaxation times, g(lnτ), has been determined for GLY and for MTHF.14 Accounting for the present frequency range located near the loss peak, νmax to 100νmax, integrating g(lnτ) from ln(τlo) = −ln(2π100νmax) to ln(τhi) = −ln(2πνmax) reveals that 55% of all relaxation modes have their peak located within the frequency regime probed presently. For glycerol, most modes with peak-positions at frequencies below the experimental range also contribute to our measurements, as the remaining width of the spectrum is only about half the width (wD/2) of a Debye-peak. Therefore, using ln(τhi) = −ln(2πνmax) + wD/2, it is reasonable to assume that about 95% of relaxation modes are involved in the recovery dynamics measurements of this work.
Discussion
An important aspect of this discussion is the clear separation of what is referred to as structural relaxation and structural recovery. In what follows, we associate relaxation dynamics with responses to perturbations within the linear regime, where the response is a true reflection of the equilibrium fluctuations of the system in its present state.32 By contrast, structural recovery refers to the process of the system approaching a new equilibrium state, i.e., a process along which relaxation parameters such as amplitudes and time constants (or fictive temperatures) are bound to change.2 Consequently, structural recovery involves nonlinear responses. A common observation of structural recovery is the physical aging that sets in after a temperature quench, where time constants increase as the system approaches the new equilibrium state.16 Although exceptions can be found,33 the majority of physical aging experiments display TaTS,15–19i.e., a homogeneous recovery scenario in which a single fictive temperature is sufficient to define the transient non-equilibrium state. As a result of the typical experimental approach to physical aging found in the literature, the validity of this statement has been confined to the small fraction of the fastest relaxation modes out of the entire relaxation time distribution.The present observations outlined above demonstrate that fast and slow relaxation modes display identical dynamics regarding structural recovery, implying that this process of ‘physical aging’ in response to an electric field step is consistent with TaTS, a homogeneous nature of structural recovery, and a single fictive temperature. By virtue of the technique of this study, this notion is found to hold for the majority of modes that contribute to the overall approach to equilibrium, not only the few fastest ones. Such a structural recovery behavior is distinct from what is known about structural relaxation in supercooled liquids, which is heterogeneous in the sense that fast and slow contributions to the response act independently.7,8 Clearly, this calls into question the common assumption that structural recovery can be modeled directly on the basis of the relaxation behavior that is derived from linear response experiments, which is the basis for the widely used models such as TNM or KAHR.5,6 Within the KAHR approach, the TaTS feature is introduced via an ad hoc assumption: while the model starts with the concept of heterogeneous recovery dynamics and a spectrum of fictive temperatures, a subsequent step reduces the system to a single fictive temperature and thus homogeneous recovery dynamics, with no explanation other than stating: “although this approach is mathematically attractive, it appears physically unreasonable since the time scale of molecular motions must depend on the overall state of a system rather than, independently, on the particular value of each individual ordering parameter”.6 At the time, i.e. prior to 1979, relaxation dynamics were assumed homogeneous. Given the later recognition of the heterogeneous nature of relaxation dynamics,34 a more valid justification of TaTS is required.
To further illustrate the situation, we focus on dielectric polarization as an example, but analogous arguments will hold for other techniques. Low-field dielectric relaxation measurements provide polarization decay data under conditions such that time constants do not change in the course of the relaxation process (linear response). However, changes of the time constants are relevant in physical aging or structural recovery,2 any experiments driving the system beyond linear response,8 and rate exchange.35 Rate exchange refers to the final slow process that restores ergodicity by fluctuations of time constants (τ) on the time scale of τex, a process that is required to restore identity of ensemble and time averages in a heterogeneous system.7,8 As such, rate exchange itself is a dynamically homogeneous process. Because rate exchange preserves the polarization state, dielectric relaxation experiments will not sense rate exchange as such and thus can not reveal the time scale of rate exchange. Nuclear magnetic resonance experiments can determine τex directly by observing the equilibration of a sub-ensemble of relaxation modes,36 resulting in τex values that are somewhat longer than the average time of structural relaxation, τα.7,8 Many studies on the ratio τex/τα report values near 2, including those for GLY and MTHF.37 The ratio τex/τα could be material specific, and values outside the 2–3 range have been reported on the basis of other techniques.7,8,33
With rate exchange representing fluctuations of τ's in equilibrium and structural recovery being directed changes of τ's in response to a perturbation, it seems reasonable to connect the two by a fluctuation dissipation relation (for small perturbations).32,38 Consequently, structural recovery should be viewed as governed by rate exchange dynamics. Consistent with this picture, the present field-induced recovery experiments observe dynamics closer to τex than to τα. Therefore, rate exchange dynamics should serve as the proper input to TNM-type models of aging, thus predicting somewhat slower aging dynamics and inherently yielding TaTS with a single fictive temperature. A deeper insight into rate exchange will improve our understanding of structural recovery, and vice versa.
An alternative approach to the difference between relaxation and recovery process rests on the following realization: when the dielectric relaxation process is practically complete, the structure associated with the heterogeneity of fast and slow modes persists until rate exchange has restored ergodicity. Relative to common usage, a better definition of true structural relaxation may be the process of restoring ergodicity. Thus, the dielectric relaxation decays faster than such a true structural relaxation, which can not be measured by simple correlation decay experiments. In this language, true structural relaxation would be the same as structural recovery.
Summary and conclusions
Using a technique that facilitates measuring structural recovery for frequencies near the loss peak maximum, it is found that TaTS is valid not only for the small fraction of the fastest contributions to the α-process, but for practically the entire spectrum. This identifies structural recovery as a homogeneous process, i.e., fast and slow contributions to the overall relaxation approach equilibrium with a common time dependence. As a result, a single fictive temperature is capable of delineating the non-equilibrium state of the system. A consequence of structural recovery being homogeneous is that structural relaxation can not be governed by the same modes, as the latter is heterogeneous. A possible solution to this problem is to associate structural recovery with rate exchange rather than relaxation dynamics. As the final process restoring ergodicity of a heterogeneously relaxing system, rate exchange is bound to be homogeneous. Moreover, structural recovery and rate exchange refer to the process of time constants adjusting towards the new equilibrium state and time constants fluctuating in equilibrium, respectively. For not too large perturbations, recovery and rate exchange may thus be viewed as linked by fluctuation–dissipation relations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fruitful discussions with D. Matyushov are gratefully acknowledged. This work was supported by the National Science Foundation under Grant No. CHE-1564663.References
- C. A. Angell, K. L. Ngai, G. B. McKenna, P. F. McMillan and S. W. Martin, J. Appl. Phys., 2000, 88, 3113 CrossRef CAS.
- I. M. Hodge, J. Non-Cryst. Solids, 1994, 169, 211 CrossRef CAS.
- A. Tool and C. Eichlin, J. Am. Ceram. Soc., 1931, 14, 276 CrossRef CAS; A. Tool, J. Am. Ceram. Soc., 1946, 29, 240 CrossRef.
- S. Narayanaswamy and R. Gordon, J. Am. Ceram. Soc., 1970, 53, 380 CrossRef CAS; O. Narayanaswamy, J. Am. Ceram. Soc., 1971, 54, 491 CrossRef.
- C. T. Moynihan, P. B. Macedo, C. J. Montrose, P. K. Gupta, M. A. DeBolt, J. F. Dill, B. E. Dom, P. W. Drake, A. J. Easteal, P. B. Elterman, R. P. Moeller, H. Sasabe and J. A. Wilder, Ann. N. Y. Acad. Sci., 1976, 279, 15 CrossRef CAS.
- A. J. Kovacs, J. J. Aklonis, J. M. Hutchinson and A. R. Ramos, J. Polym. Sci., Polym. Phys. Ed., 1979, 17, 1097 CrossRef CAS.
- M. D. Ediger, Annu. Rev. Phys. Chem., 2000, 51, 99 CrossRef CAS PubMed.
- R. Richert, J. Phys.: Condens. Matter, 2002, 14, R703 CrossRef CAS.
- R. Richert, J. Chem. Phys., 2011, 134, 144501 CrossRef PubMed.
- S. K. S. Mazinani and R. Richert, J. Chem. Phys., 2012, 136, 174515 CrossRef PubMed.
- G. Diezemann, J. Chem. Phys., 2005, 123, 204510 CrossRef PubMed.
- I. Saika-Voivod and F. Sciortino, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2004, 70, 041202 CrossRef PubMed.
- R. Richert, Phys. Rev. Lett., 2010, 104, 085702 CrossRef.
- W. Huang and R. Richert, J. Chem. Phys., 2009, 130, 194509 CrossRef.
- P. Lunkenheimer, R. Wehn, U. Schneider and A. Loidl, Phys. Rev. Lett., 2005, 95, 055702 CrossRef CAS PubMed.
- T. Hecksher, N. B. Olsen, K. Niss and J. C. Dyre, J. Chem. Phys., 2010, 133, 174514 CrossRef PubMed.
- E. Schlosser and A. Schönhals, Polymer, 1991, 32, 2135 CrossRef CAS.
- R. L. Leheny and S. R. Nagel, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 5154 CrossRef CAS.
- M. Wübbenhorst and J. van Turnhout, J. Non-Cryst. Solids, 2002, 305, 40 CrossRef.
- S. Samanta and R. Richert, J. Chem. Phys., 2015, 142, 044504 CrossRef PubMed.
- K. Adrjanowicz, M. Paluch and R. Richert, Phys. Chem. Chem. Phys., 2018, 20, 925 RSC.
- A. R. Young-Gonzales, S. Samanta and R. Richert, J. Chem. Phys., 2015, 143, 104504 CrossRef PubMed.
- S. Samanta and R. Richert, J. Phys. Chem. B, 2016, 120, 7737 CrossRef CAS PubMed.
- S. Havriliak and S. Negami, Polymer, 1967, 8, 161 CrossRef CAS.
- R. Richert, J. Phys.: Condens. Matter, 2017, 29, 363001 CrossRef PubMed.
- F. Stickel, PhD thesis, Mainz University, Shaker, Aachen, 1995.
- S. Samanta, O. Yamamuro and R. Richert, Thermochim. Acta, 2016, 636, 57 CrossRef CAS.
- B. Riechers, K. Samwer and R. Richert, J. Chem. Phys., 2015, 142, 154504 CrossRef PubMed.
- R. Kohlrausch, Pogg. Ann. Phys. Chem., 1854, 91, 179 CrossRef.
- G. Williams and D. C. Watts, Trans. Faraday Soc., 1970, 66, 80 RSC.
- R. Richert, Adv. Chem. Phys., 2014, 156, 101 CrossRef.
- R. Kubo, J. Phys. Soc. Jpn., 1957, 12, 570 CrossRef.
- O. Gulbiten, J. C. Mauro and P. Lucas, J. Chem. Phys., 2013, 138, 244504 CrossRef PubMed.
- K. Schmidt-Rohr and H. W. Spiess, Phys. Rev. Lett., 1991, 66, 3020 CrossRef CAS PubMed.
- A. Heuer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1997, 56, 730 CrossRef CAS.
- A. Heuer, M. Wilhelm, H. Zimmermann and H. W. Spiess, Phys. Rev. Lett., 1995, 75, 2851 CrossRef CAS PubMed.
- F. Qi, T. El Goresy, R. Böhmer, A. Döβ, G. Diezemann, G. Hinze, H. Sillescu, T. Blochowicz, C. Gainaru, E. Rössler and H. Zimmermann, J. Chem. Phys., 2003, 118, 7431 CrossRef CAS.
- S. Samanta and R. Richert, J. Chem. Phys., 2014, 140, 054503 CrossRef PubMed.
| This journal is © the Owner Societies 2019 |