
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the impact of competing intra- and intermolecular triplet-state quenching on photobleaching and photoswitching kinetics of organic fluorophores†
Jochem H.
Smit
 a,
Jasper H. M.
van der Velde
a,
Jingyi
Huang
b,
Vanessa
Trauschke‡
a,
Sarah S.
Henrikus§
a,
Si
Chen
a,
Nikolaos
Eleftheriadis
a,
Eliza M.
Warszawik
b,
Andreas
Herrmann
bc and
Thorben
Cordes
*ad
a,
Jasper H. M.
van der Velde
a,
Jingyi
Huang
b,
Vanessa
Trauschke‡
a,
Sarah S.
Henrikus§
a,
Si
Chen
a,
Nikolaos
Eleftheriadis
a,
Eliza M.
Warszawik
b,
Andreas
Herrmann
bc and
Thorben
Cordes
*ad
aMolecular Microscopy Research Group, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
bDepartment of Polymer Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
cDWI – Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52056 Aachen, Germany
dPhysical and Synthetic Biology, Faculty of Biology, Ludwig-Maximilians-Universität München, Großhadernerstr. 2-4, 82152 Planegg-Martinsried, Germany. E-mail: cordes@bio.lmu.de
First published on 30th November 2018
Abstract
While buffer cocktails remain the most commonly used method for photostabilization and photoswitching of fluorescent markers, intramolecular triplet-state quenchers emerge as an alternative strategy to impart fluorophores with ‘self-healing’ or even functional properties such as photoswitching. In this contribution, we evaluated combinations of both approaches and show that inter- and intramolecular triplet-state quenching processes compete with each other. We find that although the rate of triplet-state quenching is additive, the photostability is limited by the faster pathway. Often intramolecular processes dominate the photophysical situation for combinations of covalently-linked and solution-based photostabilizers and photoswitching agents. Furthermore we show that intramolecular photostabilizers can protect fluorophores from reversible off-switching events caused by solution-additives, which was previously misinterpreted as photobleaching. Our studies also provide practical guidance for usage of photostabilizer–dye conjugates for STORM-type super-resolution microscopy permitting the exploitation of their improved photophysics for increased spatio-temporal resolution. Finally, we provide evidence that the biochemical environment, e.g., proximity of aromatic amino-acids such as tryptophan, reduces the photostabilization efficiency of commonly used buffer cocktails. Not only have our results important implications for a deeper mechanistic understanding of self-healing dyes, but they will provide a general framework to select label positions for optimal and reproducible photostability or photoswitching kinetics in different biochemical environments.
1. Introduction
Non-fluorescent dark-states such as the triplet or radical states have been identified as major sources of photobleaching in (organic) fluorophores due to their relatively long lifetime and high chemical reactivity.1–3 Besides their tendency to directly undergo irreversible chemical reactions to non-fluorescent products (photobleaching), they can be involved in the generation of other transient and chemically reactive species. Notably, these dark-states can be used for localization-based super-resolution imaging (e.g., in techniques abbreviated STORM4,5etc.) in case that the photoswitching kinetics can be controlled. To optimize signal strength and quality, quencher molecules, herein denoted as photostabilizers, are used to deplete triplet- or radical-states. Common quenching mechanisms for high photostability are triplet–triplet annihilation via molecular oxygen,6–8 dexter-type triplet energy transfer (cyclooctatetraene (COT),9–11 diphenylhexatriene (DPHT)12) or photo-induced electron transfer (nitrophenyl-compounds,11 Trolox (TX),1,13 methylviologen (MV), ascorbic acid (AA)2,14). Whereas other damage pathways such as multi-photon absorption processes and bleaching from singlet states are known to occur,15 the primary strategy for photostabilization relies on suppressing triplet- and radical-state formation as well as removal of molecular oxygen14 to avoid formation of reactive oxygen species (ROS6–8,16,17).All approaches depend upon collisions or short-range interactions between photostabilizer and reactive fluorophore intermediates. These are generated by either addition of the photostabilizer to the imaging buffer at millimolar concentrations2,3,11,13 to allow diffusional intermolecular quenching (Fig. 1a), or via covalent linkage, i.e., to create high local concentrations of the photostabilizer (Fig. 1b).18–25
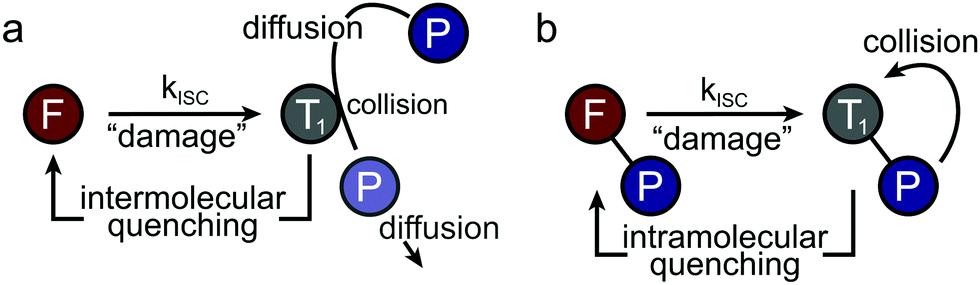 | ||
| Fig. 1 Methods to create collisions and to allow short-range interactions between photostabilizer and reactive fluorophore state (here triplet-state T1) using intermolecular diffusional quenching (a) or intramolecular processes (b). In both cases the long-lived reactive triplet-state collides with the photostabilizer to restore the fluorophore's singlet ground state. | ||
Currently, intermolecular photostabilization (Fig. 1a) with buffer cocktails is the most flexible approach to stabilize various classes of organic fluorophores in different biochemical environments.1–3,9–15,26–30 This approach is widely applied in fluorescence microscopy and the antioxidant TX,13 COT9–11 or combinations of redox-active compounds (“ROXS”2) have become the additives of choice.
Intramolecular photostabilization via triplet-state quenchers (Fig. 1b) was suggested as a mechanism of enhancing dye laser technologies in the 1980s.31,32 These papers lack, however, clear experimental evidence of triplet-state quenching that relates to improved performance of the used dye constructs.31,32 The approach was then utilized by the Blanchard lab18 in 2012 for fluorescence microscopy and was further developed in later years.19–25 The method can improve photophysical parameters whenever buffer additives would not be tolerated due to toxicity or interference with biological structure and function. Furthermore, intramolecular photostabilization is an effective option for live-cell imaging24 or in cases where buffer additives remain ineffective due to the lack of collisions between photostabilizer and an “enclosed” fluorophore. In spite of their promise, self-healing dyes have been less frequently used in part because of challenges in bioconjugation and less optimal photostabilization compared to solution additives especially when using redox-based stabilizers.23
Recently, their applicability for super-resolution microscopy has been suggested. Previous studies by our group demonstrated that self-healing dyes allow for STED-type imaging in fixed mammalian cells with a higher number of accessible successive images due to an increased photostability.23 It is, however, unclear whether self-healing dyes can be used for STORM imaging via fluorophore blinking (ON/OFF switching). Another related, yet unanswered question is whether inter- and intramolecular photostabilization can be used in combination33 to increase the overall photostabilization efficiency or the range of fluorophores that can be stabilized simultaneously. To address these questions, we studied the effect of commonly used solution-based healers (TX, COT) on a number of commonly-used fluorophores and their photostabilizer-conjugates, using an aromatic nitro moiety, TX and COT. Our results suggest that either inter- or intramolecular triplet-state quenching dominate the photophysical processes of self-healing dyes, a fact that prohibits a useful combination of both approaches. These findings also prompted us to characterize the interaction of a photoswitching buffer additive used for STORM-type super-resolution imaging, tris(2-carboxyethyl)phosphine (TCEP) and cysteamine (MEA), with Cy5-conjugates with NPA, TX and COT. Similar to previous findings, our data shows that the intramolecular photostabilizer outcompetes intermolecular pathways. TCEP and MEA induce reversible off-switching, a process that can be used for superior STORM imaging, but the observed “apparent photobleaching” can also lead to misinterpretation of photostability values whenever these compounds are used in buffer systems. These results suggest an additional role for intramolecular triplet-state quenchers; rather than inferring photobleaching, they most likely act as protective agents for reversible off-switching reactions. Ultimately, we link our findings, i.e., a non-additive behaviour of inter- and intramolecular photostabilization effects to interactions of fluorophores with natural aromatic amino-acids and show that tryptophan residues significantly reduce diffusion-based photostabilization effects.
2. Results
The focus of previous work on self-healing fluorophores18,21–25 has been on the optimization of photostabilization efficiency,22,24 the development of versatile bioconjugation strategies,18,21,23 and to benchmark the method for different fluorophore types23,25 or environmental conditions.18,22 Here, we conducted experiments in which photostabilizers were present in solution and linked to the fluorophore simultaneously (Fig. 2), in an attempt to determine whether inter- and intramolecular photostabilization effects can be additive.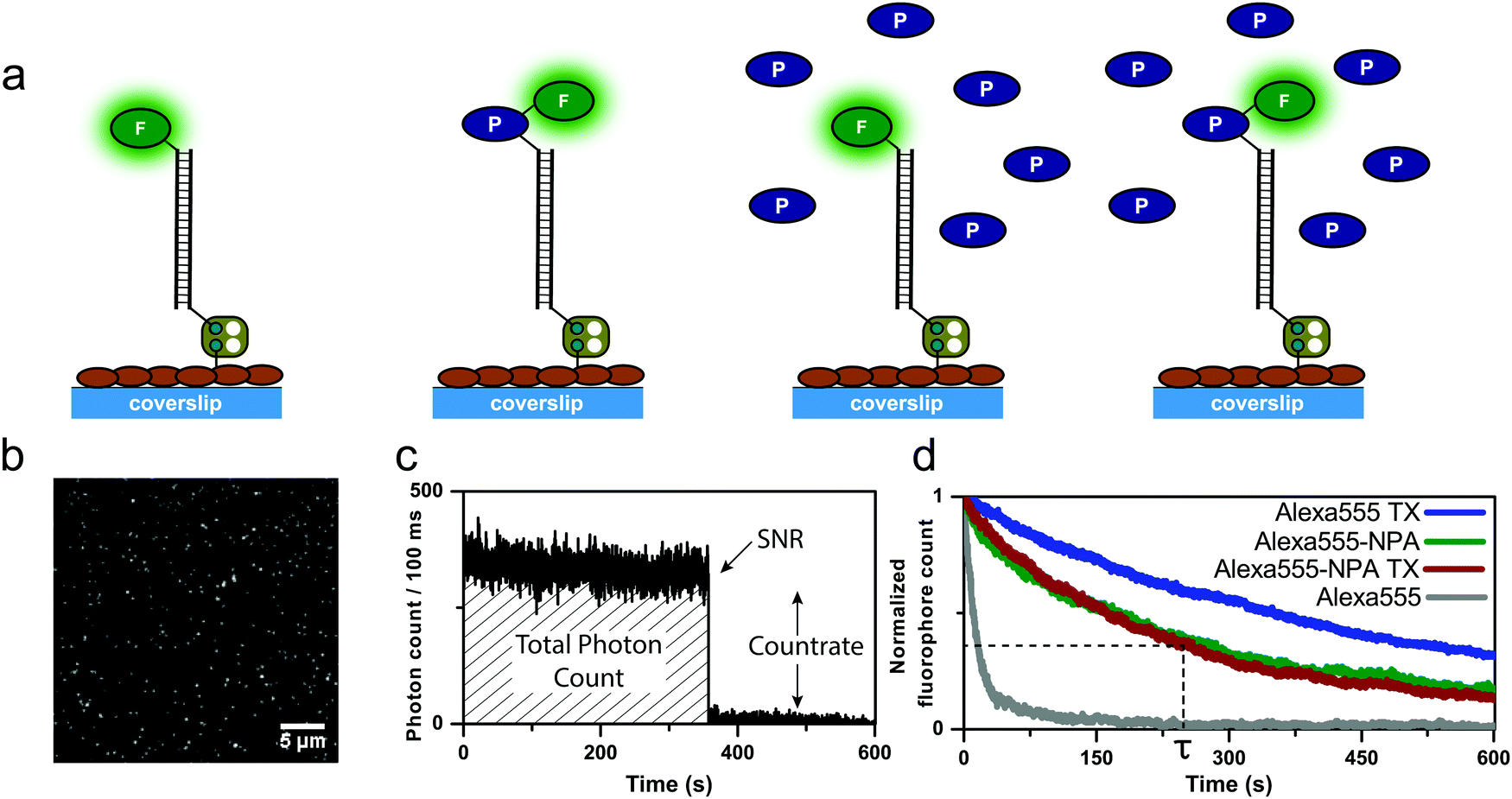 | ||
| Fig. 2 Experimental strategy and design for the characterization of the photophysical parameters of DNA-linked dyes and their photostabilizer conjugates. Different buffer systems were tested in the absence of oxygen using an enzymatic oxygen removal systems based on glucose-oxidase and catalase (GOX). (a) Schematics of immobilization and experimental conditions. Representative single-molecule data sets of (b) TIRF images of individual fluorophores, (c) fluorescent transients of individual fluorophores which were used to extract photophysical parameters. (d) Normalized plots of fluorophore counts in each time frame providing quantitative information on photobleaching kinetics. | ||
To this end, fluorescent dyes and their photostabilizer–dye conjugates were immobilized on a streptavidin-functionalized coverslip via double-stranded DNA-scaffolds as described previously23 (Fig. 2a). Total internal reflection fluorescence (TIRF) microscopy was employed to quantify the photophysical properties of different permutations of inter- and intramolecular photostabilization (Fig. 2a) in a deoxygenated environment. Quantitative photophysical parameters were obtained from single emitters (Fig. 2b–d) identified in TIRF images. Detailed analysis of their fluorescent transients allowed us to extract the count-rate and signal-to-noise ratio (SNR). The number of fluorescent emitters over time (Fig. 2d) was fitted to an exponential decay which gave the mean photobleaching lifetime. From this, the total number of photons emitted before photobleaching can be calculated by multiplication of the photobleaching lifetime and the average count-rate.
We tested three frequently used fluorophores, namely ATTO647N, Alexa555 and Cy5, and compared them to their respective photostabilizer–dye conjugates (nitrophenyl alanine (NPA)-ATTO647N, NPA-Alexa555, NPA-Cy5, Cy5-COT Fig. 3 and Fig. S1–S20, ESI†). The performance of the dyes was evaluated under either deoxygenated conditions (glucose oxidase based oxygen scavenging, GOX), or deoxygenated conditions with additional photostabilizers: 2 mM aged TX13 (antioxidant) or 2 mM COT11 (triplet-state quencher). The GOX–buffer system in Fig. 2 and 3 contains 200 μM TCEP, which was added for enzyme stability.2
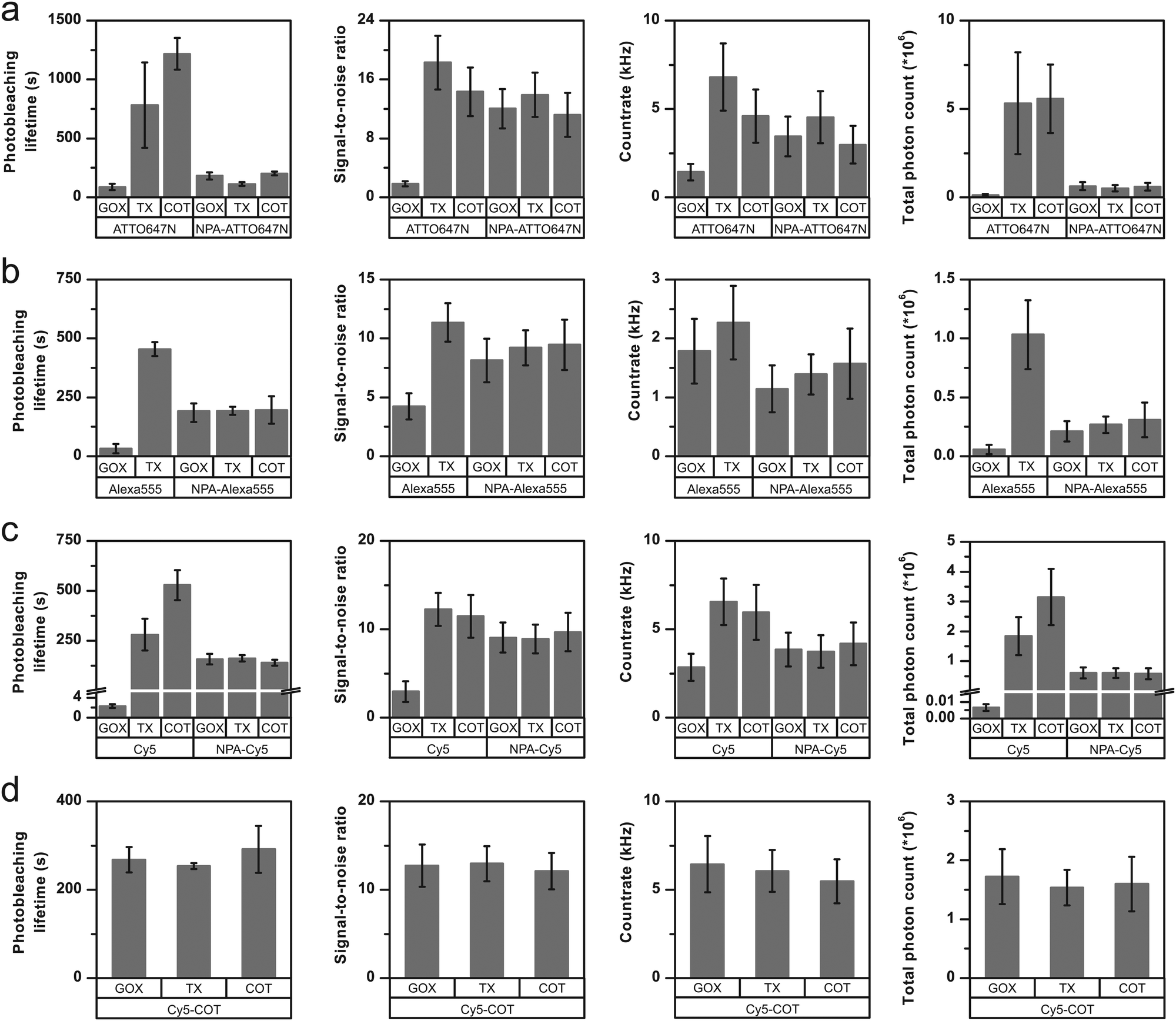 | ||
| Fig. 3 Photophysical parameters of (a) ATTO647N and NPA-ATTO647N (b) Alexa555 and NPA-Alexa555 (c) Cy5 and NPA-Cy5 (d) Cy5-COT under deoxygenated conditions without the addition of solution-based photostabilizers (GOX) and in the presence of 2 mM TX or 2 mM COT. The glucose-oxidase catalase enzyme cocktail contains 200 μM of TCEP. Photobleaching lifetime numbers were obtained by fitting several photobleaching decay curves and averaging the obtained time constants. Error bars are standard deviation of 3 independent measurements. Signal-to-noise ratio and count-rate were determined from individual fluorescent transients for ≥200 molecules. The total photon count is the product of the photobleaching lifetime and count-rate. The error bar shows the standard deviation of repeats on two different days. A detailed description is given in the “Material and methods” section. Individual data sets for each condition are shown in the ESI.† | ||
In agreement with previously published results,19,20,23 we found an improvement of photophysical parameters in the presence of solution additives or when comparing unmodified fluorophores with their NPA- or COT-conjugates as shown in Fig. 3. This included increased photobleaching lifetimes, signal-to-noise ratio, count-rates and total number of collected photons, and can be attributed to the depopulation of the triplet-state (Fig. 3a–c and Fig. S1–S20, ESI†).
Inter- and intramolecular quenching processes compete with each other
For all fluorophores and stabilizer combinations, we observed no improvement of photophysical properties upon combining inter- and intramolecular photostabilization. Instead, the photobleaching lifetimes did not increase beyond values observed for NPA- or COT-conjugates. For example, when comparing the photobleaching lifetimes of the carbopyronine fluorophore ATTO647N under different conditions (Fig. 3a), there was a clear increase between the parent dye in GOX buffer (oxygen removal only) and TX-based or COT-based buffers. A smaller increase was also observed for covalent linkage of NPA to the dye (NPA-ATTO647N, GOX) with respect to the parent compound (ATTO647N, GOX). However, the combination of both inter- and intramolecular stabilization, e.g., NPA-ATTO647N with either TX or COT as buffer additives, did not result in an increased photostability with respect to NPA-ATTO647N (Fig. 3a).Similar results were obtained for Alexa555 (Fig. 3b) and Cy5 (Fig. 3c) in combination with the covalent stabilizer NPA (Fig. 3a–c) or COT (Fig. 3d). It can thus be concluded that the photostabilizing effect observed for the two approaches individually is not additive. Instead, the photobleaching lifetimes were similar to that of the photostabilizer conjugates and were not affected further by stabilizers in solution. Besides these general trends, there were subtle differences in the photophysics of NPA-fluorophores in GOX buffer and TX/COT-containing buffers. For NPA-ATTO647N and NPA-Cy5, addition of TX removed faster signal fluctuations (Fig. S4 vs. Fig. S5 & Fig. S15 vs. Fig. S16, ESI†) whereas addition of COT induced short up-blinks of unknown origin (Fig. S4 vs. Fig. S6, ESI†). Also, the addition of TX/COT increased the overall count-rate for NPA-based dyes (Fig. S4 vs. Fig. S5 & Fig. S9 vs. Fig. S11, ESI†) but not for intramolecular-linked COT that fully shields the fluorophore from other photostabilizers in solution (Fig. S18–S20, ESI†). This could be rationalized as being due to the presence of a transient dark state in the NPA-based quenching pathway, which does not contribute to photobleaching, and which was removed by TX and COT in solution, thus improving the other photophysical parameters.
These findings suggest that in the competition for the triplet-state, the covalently bound stabilizer effectively outcompetes the solution based stabilizers, i.e., it quenches the triplet-state faster. This can be understood in view of the fact that in the intramolecular case, the photostabilizer is present in much higher local concentrations as compared to the solution additives. However, faster quenching of the triplet-state (which is difficult to observe directly in single-molecule experiments) did not result in higher levels of photostability. These conclusions are supported by our earlier findings that two covalently tethered stabilizers (e.g., a fluorophore with both TX and NPA attached) did not show improved performance beyond that of the individual stabilizers.20 In this case too, photostabilization was dominated by one of the two stabilizers – presumably the one which quenches the triplet faster.
In our previous work, we also investigated the importance of stabilizer-fluorophore arrangement and linker length for self-healing processes.22 Different linking chemistries or orientations of the photostabilizer resulted in different photostabilities. Our interpretation of the data in Fig. 3 is further corroborated by experiments in which covalently linked NPA was arranged differently in space with respect to Cy5 (Fig. S29, ESI†). The nitrophenyl group used as a photostabilizer was either linked directly to the fluorophore through an amino-acid scaffold on the 3′-end of a DNA strand (NPA-Cy5), or was attached on its own to the 5′-end of the complementary DNA strand (NPAA) such that upon annealing it was brought into proximity with the fluorophore directly attached to the other strand (Cy5-NPAA); Fig. S29 (ESI†). The photobleaching lifetimes of NPA-Cy5 and Cy5-NPAA were found to be different (207 ± 25 s and 647 ± 241 s, respectively). Significantly, annealing the DNA strand NPA-Cy5 with the NPAA strand to place Cy5 in proximity of two stabilizers moieties (Fig. S29, ESI†) did not lead to an additive effect on the photobleaching lifetime of Cy5. While there is a slight difference between the spatial arrangement of NPAA and Cy5 in the absence and presence of the amino-acid NPA-moiety, this data suggests again that the photophysics is dominated by the closest photostabilizer unit.
A simple combination of inter- and intramolecular approaches (or multiple intramolecular stabilizers) was found not to be beneficial for photostability.22 These findings allowed us to draw further mechanistic conclusions which we discuss in detail below.
Photoswitching competes with intramolecular triplet-state quenching and photobleaching
Our results have direct implications for the use of self-healing dyes in localization-based super-resolution microscopy, because these techniques require controlled on/off-switching of single fluorophores via solution additives.34 In STORM, compounds such as mercaptoehtylamine (2-aminoethane-1-thiol, MEA), β-mercaptoethanol (2-sulfanylethan-1-ol, β-ME) or TCEP are used to from a meta-stable dark state. Typically, this dark state can be recovered to the singlet ground state by UV-illumination, thus achieving reversible photoswitching. The effect of reducing compounds and particularly that of thiols has been extensively studied.4,34–39 A suitable concentration of photoswitching additive allows for the control of the on/off-duty cycle needed for optimal temporal separation of fluorophore emission in STORM.26,36,40,41To study the interplay of intramolecular photostabilizers with photoswitching compounds in solution and explore whether self-healing dyes could be useful for STORM imaging, we investigated the influence of the photoswitching agent TCEP and MEA on the photophysical properties of Cy5 and Cy5-COT. In STORM microscopy, the off-switching events are either (i) photo-induced or (ii) proceed via an equilibrium step. As an example, the reversible addition reaction between TCEP and Cy5 occurs with an equilibrium constant of 0.91 mM−1.34 With this information at hand, we first examined the impact of 200 μM of TCEP as a buffer additive on the photophysics of Cy5 and Cy5-COT. TCEP was typically present in our buffers at this concentration to maintain GOX enzyme stability (Fig. 3). According to the equilibrium constant, 16% of the Cy5 molecules should be quenched. Strikingly, however, the presence of 200 μM TCEP reduced the signal duration by 11-fold, which was a much larger effect than expected (Fig. 4a and c, Cy5 ± 200 μM TCEP).
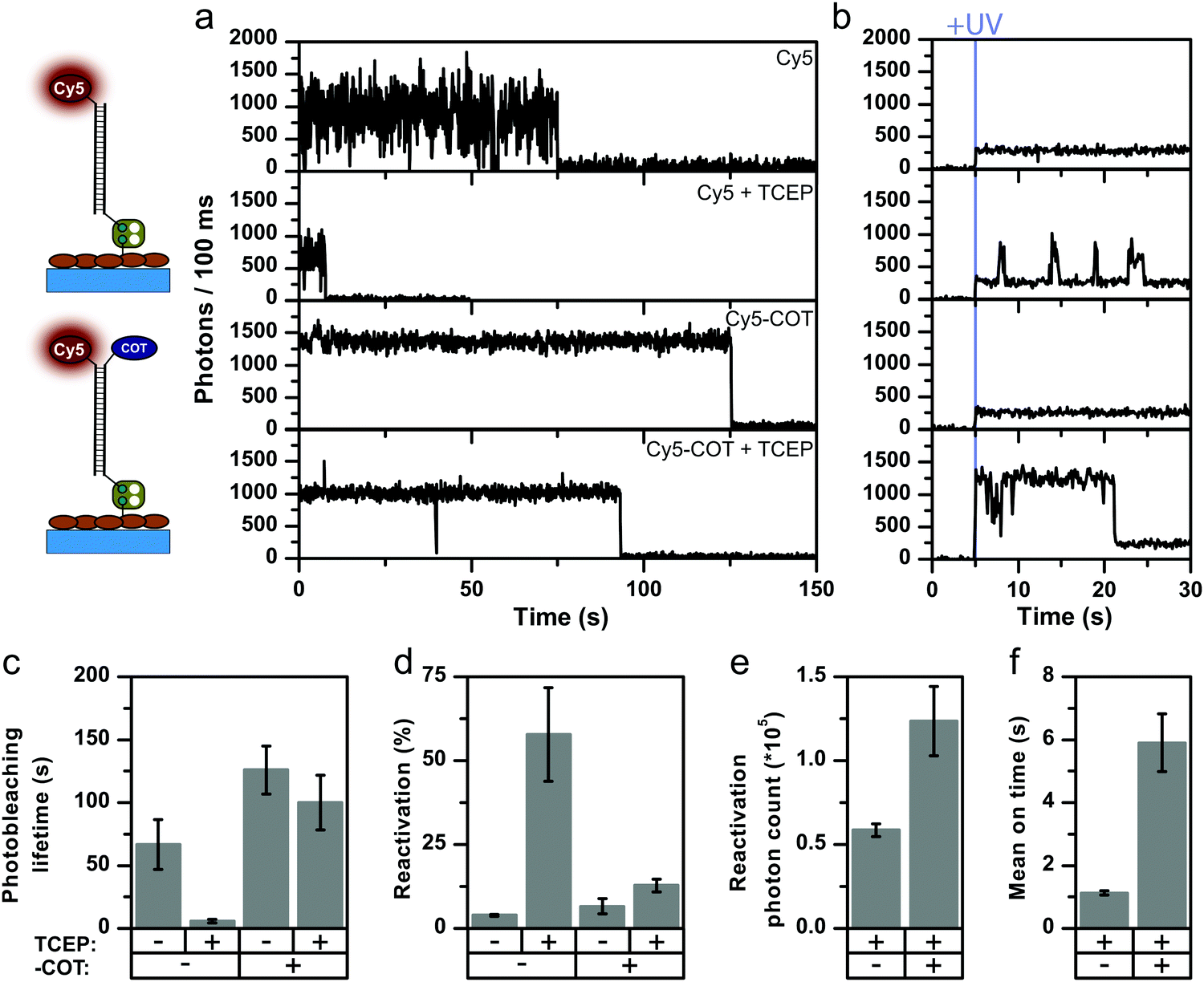 | ||
| Fig. 4 Comparison of Cy5 parent fluorophore and stabilized Cy5-COT in the absence and presence of 200 μM TCEP. Imaging performed under deoxygenated conditions (GOX) with excitation at 637 nm, 0.4 kW cm−2. (a) Representative fluorescence transients. (b) Representative fluorescence transient under UV illumination (375 nm, 0.1 kW cm−2). (c) Photobleaching lifetimes. (d) Reactivation yield during UV illumination. (e) Mean photon count per activated fluorophore and (e) mean on-time during activation. Error bars are either standard deviations (c and d) of repeats on 3 different days or SEM (e and f). A detailed description is given in the “Material and methods” section. Individual data sets of all conditions are found in the ESI.† | ||
This result cannot be explained solely by equilibrium quenching. We postulate that the faster signal loss observed in the presence of TCEP involves a photo-induced off-switching. Apparently, TCEP uses the Cy5 triplet-state for off-switching even at very low concentrations similar to the mechanism of thiols.35,39,42 Experiments with Cy5-COT support this notion of photo-induced off-switching via triplet-state quenching since the impact of TCEP was reduced for Cy5-COT (Fig. 4a). In Cy5-COT, both TCEP and COT apparently compete for the triplet-state and the intramolecular quencher successfully outcompetes TCEP due to its high local concentration.
This conclusion was further corroborated by photoactivation experiments using 375 nm UV light (Fig. 4a, second column). In our experiments, both irreversible photobleaching and off-switching result in signal loss, which we relate to “apparent photobleaching”. After apparent photobleaching, the UV laser was turned on to monitor potential fluorescence recovery and thus to disentangle the reversible off-switching contribution caused by TCEP from actual irreversible photobleaching. As expected, when TCEP is omitted, the fluorophore reactivation upon applying UV light is minimal and considered to be background signal.
This indicates that the observed signal loss in the absence of TCEP is indeed irreversible photobleaching. In contrast, a significant fraction, >50% of the fluorophores, can be reactivated in the presence of TCEP (Fig. 4d). When reactivating Cy5-COT, the activation yield in the presence of TCEP is much lower than with Cy5 (Fig. 4d), but always significantly higher than in the absence of TCEP (Fig. 4d). Consequently, the mean relative on-times and photon counts after photoactivation were higher for Cy5-COT. Longer on-times of Cy5-COT compared to Cy5 are attributed to prevention of TCEP-induced darkening but not improved photophysics of Cy5 in the on-state. The influence of TCEP on the reactivation yield relates to the fact that TCEP controls the ratio of photobleached to off-switched Cy5 molecules, but does not imply a direct influence of the UV-photoactivation step by TCEP. The blinking kinetics of the parent fluorophore Cy5 were also characterized by confocal microscopy (Fig. S30, ESI†) to determine the origin of fast blinking. By calculating the autocorrelation function of fluorescent transients, the cis–trans and triplet-related off times of Cy5 blinking were determined. We found that these values were not influenced by TCEP, suggesting that TCEP caused only the appearance of photoactivatable long off states, but did not induce short-timescale blinking.
Similar effects were observed for PET-based photostabilizer–dye conjugates NPA-Cy5 and TX-Cy5 conjugates in the presence of TCEP (Fig. S33, ESI†). Also the presence of these distinct photostabilizers protects the fluorophore against reversible off-switching, resulting in longer apparent photobleaching times and on-duration after UV-activation. Overall, however, lower reactivation yields are observed for both conjugates. In the presence of 5 mM MEA, (Fig. S34, ESI†) a reducer frequently used for STORM imaging, the reactivation yields are generally higher, suggesting under these conditions a dark state is able to efficiently form.
Since we and other groups in the field2,11,18–25 use reducing agents such as TCEP, MEA and β-ME as additives in imaging buffers when characterizing the photophysics of (self-healing) dyes on the single-molecule level, we determined the apparent photostability values of our dyes in the presence and absence of 200 μM TCEP for representative buffer conditions, i.e., comparable to those in Fig. 3. While there was only a small impact on Alexa555 and no impact on ATTO647N (Fig. S31, ESI†), the apparent bleaching lifetime of Cy5 was drastically shortened by TCEP (Fig. 4a and c). This suggests that the previously reported photostability increases of Cy5 fluorophores upon linkage to stabilizers represent an overestimation.2,11,18–25 Here, the dye-standard without the linked stabilizer showed artificially low photon numbers that likely represent the kinetics of reversible Cy5-off-switching by TCEP/MEA/β-ME, but not those of irreversible photobleaching. Fast signal loss via off-switching has been described earlier by Aitken et al.14 for TCEP, β-ME, DTT and other reducing agents when added at 10 mM concentrations. Lower concentrations of e.g., 200 μM TCEP had, however, not been characterized until now, but seem to demand similar attention. Our findings suggest that reducing compounds should not be used when the photophysics of fluorophores are to be characterized, and reveal that intramolecular photostabilizers protect fluorophores from reversible photoswitching reactions caused by reducing agents. The implications of our findings for design criteria and mechanistic understanding of self-healing dyes and their use in super-resolution microscopy will be discussed in detail below.
Self-healing dyes for STORM-type super-resolution microscopy
We previously demonstrated that self-healing dyes are useful for STED-microscopy because their superior photostability increases the number of available successive images.23 In STORM, imaging is a more complex interplay of photoswitching kinetics (to temporally separate the labels) and photostability. The available photon number N in the on-state relates to a lower bound of the localization error which can be approximated by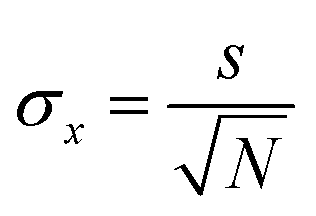 | (1) |
To improve the situation, we systematically tested variations of laser power and TCEP concentration in experiments shown in Fig. 5. We also varied the UV activation conditions, and as expected from previous work,34 increasing UV activation power was found to shorten the off-times (Fig. S32a, ESI†). In our experiments in Fig. 5, we used 375 nm activation light at 90 W cm−2. In practical applications these tuneable off-times can be used to optimize the on–off duty ratio according to the needs of the specific experiment. Fig. 5a and b show the details of how we extracted quantitative parameters to evaluate the suitability of Cy5-COT for STORM. Fig. 5a shows an initial field of view of individual Cy5 or Cy5-COT molecules that are continually irradiated with 637 nm excitation light to induce photobleaching or photoswitching. An algorithm counted the number of molecules in the initial field of view until only a few to none remain. Then the 375 nm excitation laser was switched on and all subsequently reactivated molecules were counted. The reactivation yield provided in Fig. 5d is the ratio between the number of reactivated molecules and the initial molecule count. Single activation events were found by thresholding the fluorescent transients, where short off blinks (<400 ms) were ignored. For every activation event, the on-time and the total number of photons was calculated; the mean values for the different conditions are shown in Fig. 5e and f, respectively. The localization error was calculated according to eqn (1) and the resulting values were multiplied by 2.35 to give the theoretically obtainable resolution.38 The mean values per condition are a shown in Fig. 5g.
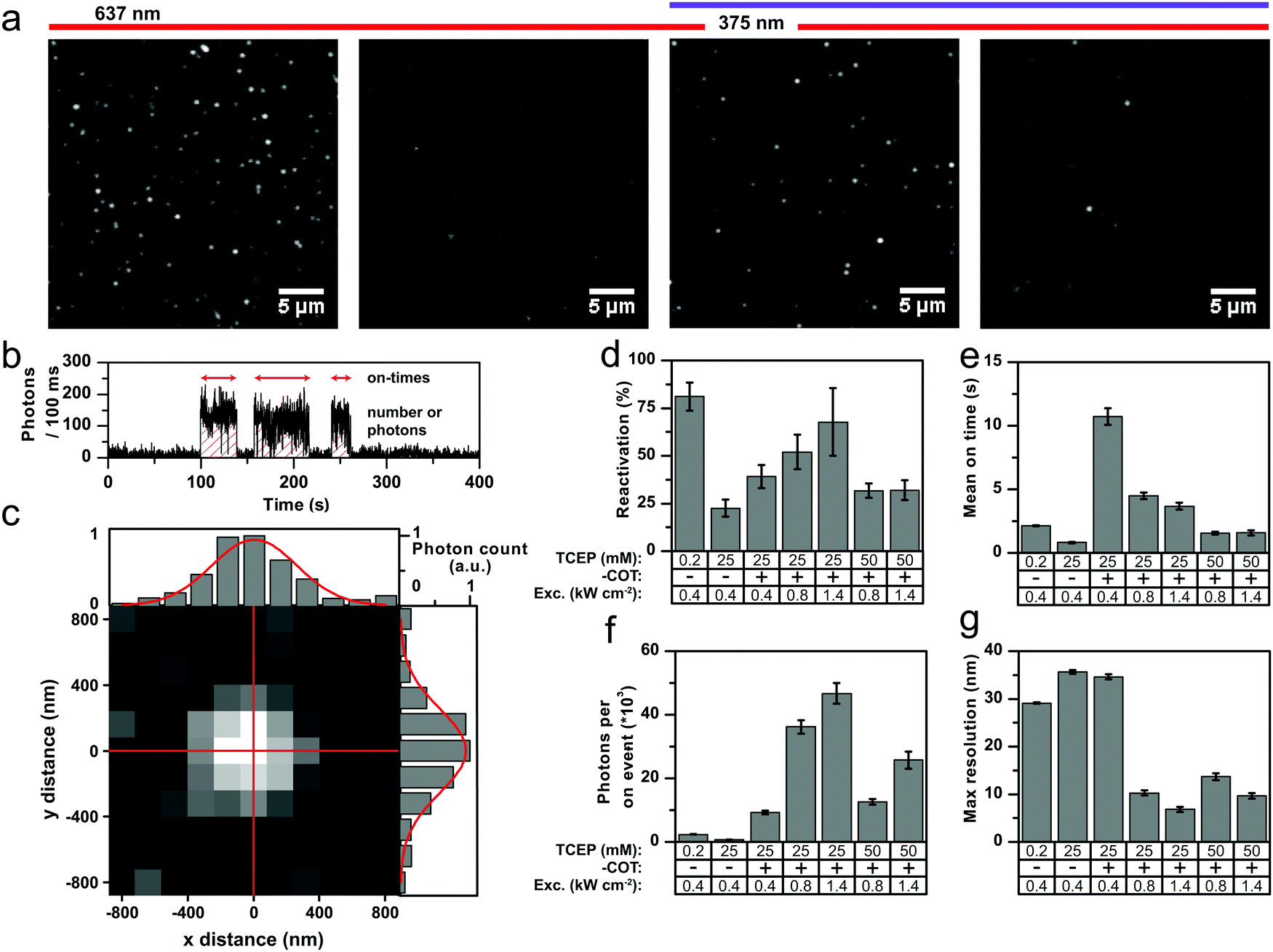 | ||
| Fig. 5 Characterization of Cy5 and Cy5-COT for application in STORM-type super-resolution microscopy. Imaging was done under deoxygenated conditions (GOX) with different TCEP concentrations. (a) TIRF images of (1) frames 1–10, (2) last frame of image acquisition, (3) frames 1–500 averaged, and (4) last frame of subsequent photoactivation acquisition. (b) A representative fluorescent transient showing the photoactivation of Cy5 in the presence of 200 μM TCEP. (c) A single photoactivated emitter along with x and y cross-section showing the result of 2D Gaussian fitting (red). The standard deviation of the Gaussian fit was found to be 201 nm. (d–g) Parameters describing the performance of the dyes for super-resolution. Error bars are standard deviation of 2–3 different days (d) or SEM for each different condition. See main text and materials and methods for further details. | ||
Our analysis shows that Cy5-COT can become a superior dye for STORM microscopy compared to Cy5; the optimal settings were found to be 1.4 kW cm−2 of laser excitation and 25 mM of TCEP. In the ideal case, the reactivation yield (Fig. 5d) of both Cy5 and Cy5-COT can be tuned to be >60%, where the parent Cy5 performs slightly better than the photostabilizer–dye conjugate (but only at low TCEP concentrations). Consistent with expectations, the on-duration decreased with higher laser power (Fig. 5e). Interestingly, the photons per on-period decreased for parent Cy5 but increased for Cy5-COT with higher laser power (Fig. 5f). This unique feature of Cy5-COT enables optimum photon-yields and with it maximum obtainable resolution. As mentioned before, the on–off duty ratio, which needs to be small to allow temporal fluorophore separation in STORM-imaging, can be adjusted further by decreasing UV excitation or increasing the TCEP concentration (Fig. 5e). In these experiments, Cy5-COT was clearly revealed to be a superior dye for STORM imaging. The number of reactivation events per fluorophore (Cy5 vs. Cy5-COT) suggests that the gain likely comes from a 6-fold lower number of activation events for Cy5-COT compared to Cy5 (Fig. S32b, ESI†), and from an overall higher total photon yield (Fig. S32c, ESI†).
When comparing the Cy5-COT conjugates (Fig. 4, 5 and Fig. S32, ESI†) to redox-based NPA-Cy5 and TX-Cy5 (Fig. S33, ESI†), TCEP reveals poor reactivation yields and on-times for the latter. In case of using MEA as photoswitching agent of Cy5, a better reactivation yield is achieved for NPA-Cy5 and TX-Cy5 compared to Cy5-COT. NPA-Cy5 in combination with MEA has also extremely short apparent photobleaching lifetimes and thus represents a good combination to achieve low on/off-duty ratios.
Proximal tryptophan residues can reduce the photostabilization effects of solution-based healers
In the previous sections, we have shown that covalently coupled triplet-state quenchers effectively outcompete solution-based additives for photostabilization and – switching, whenever the underlying quenching mechanism involves the triplet-state. These findings prompted us to investigate whether natural aromatic amino-acids might have a similar effect, since they are known to quench both singlet- and triplet-states of xanthene and oxazine dyes, via a PET quenching mechanism.48–50 Our hypothesis was that aromatic amino-acids in close proximity to the fluorophore might reduce photostabilization by intermolecular stabilizers in a similar manner to covalently-linked NPA or COT moieties. Such a mechanism could explain the large variations in photostabilization effects observed with identical buffer cocktails and dye pairs on different targets such as proteins and oligonucleotides.2,51,52To test our hypothesis, we prepared DNA-conjugates of Cy5 and ATTO647N with a single tryptophan residue (Fig. 6a). The photostability of the compounds, i.e., fluorophore and its corresponding tryptophan conjugate, were investigated in the presence and absence of Trolox. As a result of the conclusions drawn in the previous section, all experiments were now performed without TCEP in the GOX buffer, and photobleaching lifetimes for Cy5 and ATTO647N of ∼100 s and ∼50 s, respectively, were observed (Fig. 6b and c).
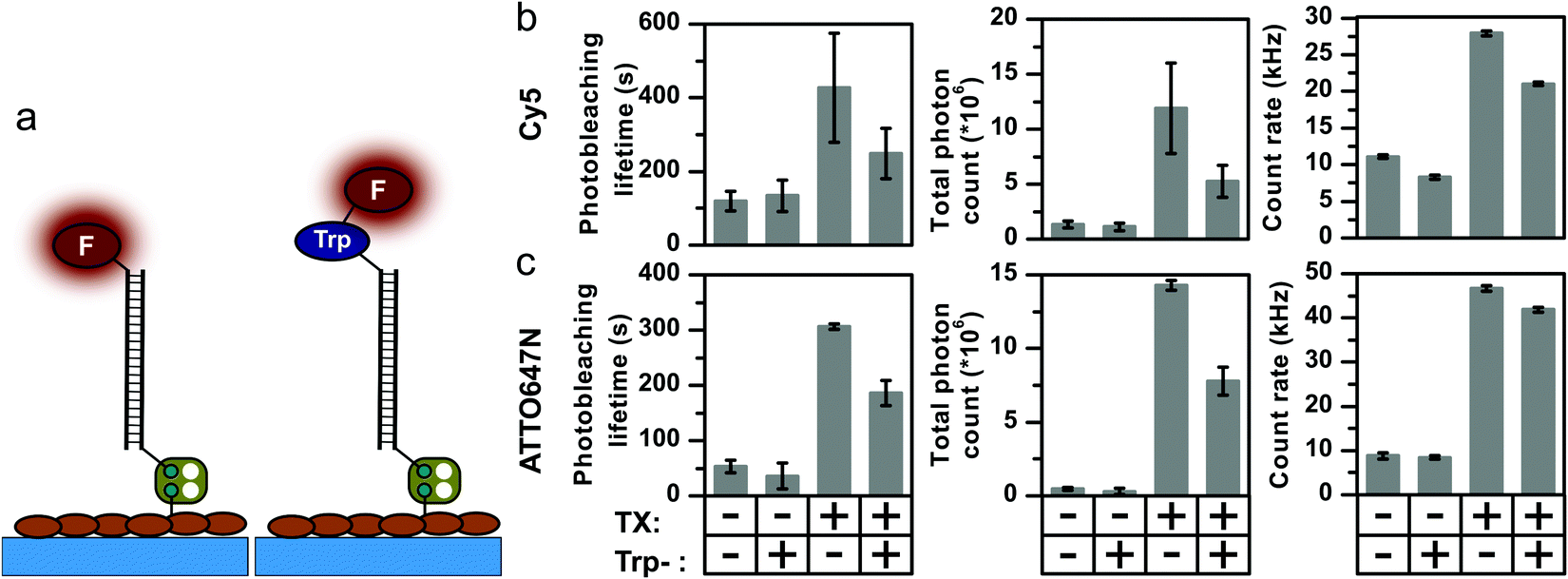 | ||
| Fig. 6 The influence of a covalently coupled tryptophan residue (a) on the photophysical properties of (b) Cy5 and (c) ATTO647N in the absence and presence of 2 mM TX. All measurements were done in the absence of oxygen with 637 nm laser power of 800 W cm−2. Error bars show standard deviation of repeated experiments as described in the “Material and methods” section. | ||
Linking a tryptophan residue covalently to the fluorophore did not drastically influence the photophysics of the two dyes (Fig. S21–S28, ESI†). A small decrease in Cy5 brightness was observed, in line with reported relative quantum yields in literature,53 likely due to singlet-state quenching. Since the total photon output is not increased upon tryptophan conjugation, it appears that it is not able to quench both the Cy5 or ATTO647N triplet states. However, the photostabilizing effect of TX in solution was significantly affected. Whereas the addition of 2 mM TX increased the photobleaching lifetime of Cy5 to ∼450 s and that of ATTO647N to ∼300 s, both tryptophan conjugates showed a ∼2-fold lower stability and thus also lower total photon counts (Fig. 6b). Identical experiments to those shown in Fig. 6 were also performed with phenylalanine and tyrosine; both had no notable influence on the observed photostability (Fig. S31, ESI†). These experiments are suggestive of a key role for tryptophan in the photostabilization of organic dyes (as also suggest previously by others53), and support the notion that the biochemical environment of a fluorophore has a strong influence on its photophysical properties.
Interestingly, tryptophan-dependent effects on photostability were not observed when examining Cy5 and Trp-Cy5 in combination with 2 mM COT in the buffer (data not shown), suggesting that COT might compete more efficiently with Trp for triplet-state quenching. An alternative explanation is that Trp interacts with fluorophore radical states which are only formed during the solution-based quenching pathway with TX but not in the case of COT. This might be the mechanistic reason for the observation that COT is often a more potent photostabilizer for proteins in single-molecule fluorescence studies.13 The notion that tryptophan interacts with fluorophore radical states might have implications for super-resolution microscopy (in particular STORM), which relies on the formation of a stable dark state to obtain the desired blinking kinetics using TCEP, MEA or β-ME.54 The presence of tryptophan in close proximity to the fluorophore label could explain observed differences in blinking kinetics when coupled to different biological targets or antibodies.
3. Discussion & outlook
The work presented in this study revealed the underlying mechanisms of competing inter- and intramolecular pathways for quenching of triplet-states in organic fluorophores (Fig. 7). We found that (i) inter- and intramolecular processes compete with each other limiting the photostability of the fluorophore (Fig. 7a–c). This is caused by a high local concentration and faster triplet-state quenching by intramolecular processes either via NPA (Fig. 7a, PET) or direct triplet-state quenching using COT. (ii) Inter- and intramolecular pathways have different photostabilization efficiencies; although the first step of the intramolecular pathway is fast, long-lasting and reactive subsequent intermediates prevent effective overall photostabilization. (iii) A verified function of intramolecular photostabilizers is the protection55 of fluorophores from reversible off-switching reactions caused by solution-additives (Fig. 7avs.Fig. 7c), which can be misinterpreted as irreversible photobleaching. (iv) Self-healing dyes can be used for STORM-type super-resolution microscopy and provide improved photon-yields and potentially higher spatial resolution. (v) Proximity of tryptophan residues significantly reduces the photostabilization efficiency of commonly used buffer cocktails. These findings have various implications for the design and characterization of self-healing dyes, as well as for their applications.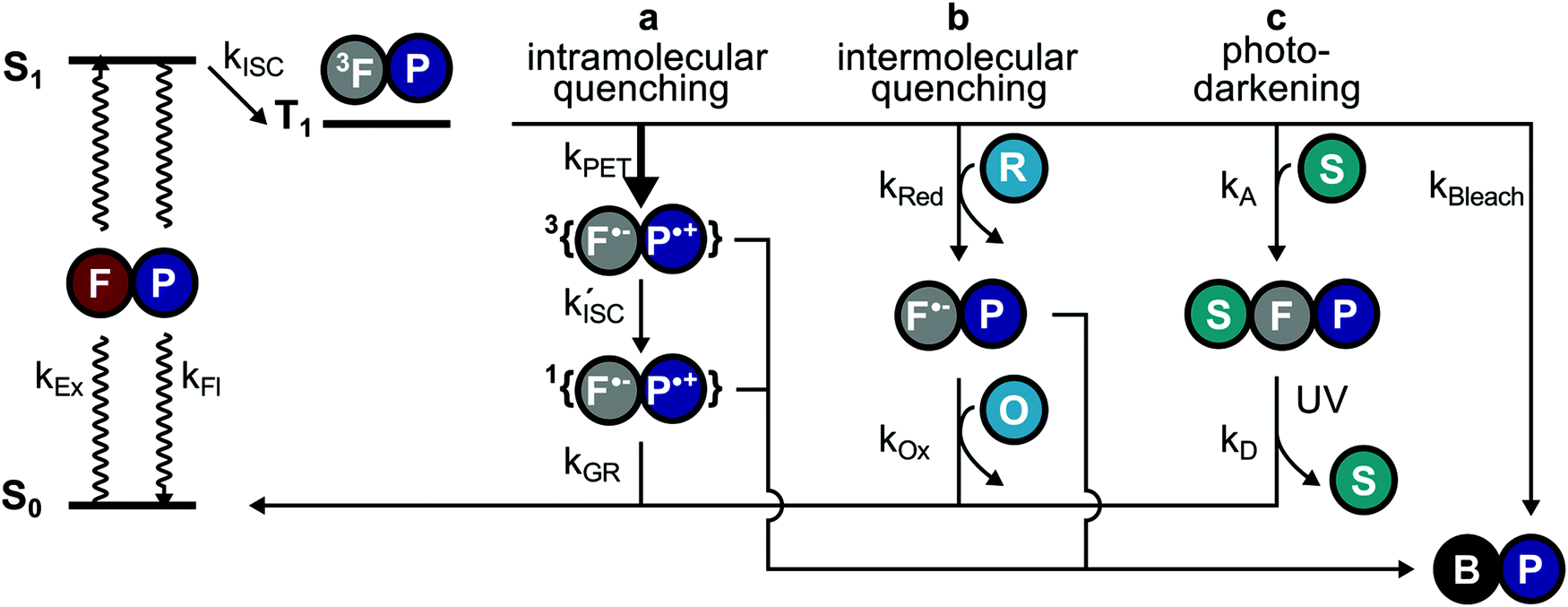 | ||
| Fig. 7 Schematic view of the main pathways through which photostabilization occurs by depletion of the triplet-state. (a) In self-healing dyes, the first step of Intramolecular quenching kPET outcompetes the other pathways due to a higher localized concentration of the photostabilizer P. However, bleaching from subsequent radical intermediates can still limit the photostability. (b) Intermolecular quenching (ROXS) takes places in two steps with reduction (kRed) followed by oxidation (kOx) to recover the fluorophore ground state. (c) Off-switching for STORM-type super resolution is facilitated by thiols or other reducing agents that form a dark state likely via the triplet-state route. This pathway can be misinterpreted as apparent photobleaching since the fluorophore can be recovered through illumination with UV light. | ||
A remaining unanswered question is whether buffer additives could successfully support the photostabilizer and with that positively influence fluorophore photophysics.33 Assuming that intramolecular healing is faster at triplet-state quenching than solution based methods, it would have been expected that intramolecular healing outperforms intermolecular healing. This prediction, however, is not supported in the present work by the observed photobleaching lifetimes and total photon counts. One possible explanation might be that the limiting factor is the stability of the photostabilizing group. We have previously reported photophysical events19,56 in which the fluorescent signal abruptly transitions from a stable signal into parent fluorophore photophysics with blinking. In reality, these events are rare and can thus not account for the observed discrepancy, unless photochemical destruction of the photostabilizer also results in immediate bleaching of the parent fluorophore. Another possible explanation is that additional photobleaching pathways exist and these are facilitated by the intramolecular quenching mechanism (Fig. 7a). The mechanism of self-healing might possibly include reactive states such as charge-separated states (e.g., for NPA) or triplet-states (e.g., for COT) which can contribute to photobleaching and thereby limit the maximal photobleaching lifetime. This is supported by recent work from the Cosa lab,57 where it was shown that efficient charge recombination in self-healing dyes requires intersystem crossing (ISC) of the biradical (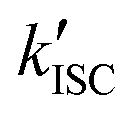 , Fig. 7a). This slow ISC, and therefore long-lasting biradical, might be the cause for differences in photostability between different fluorophore-stabilizer geometries (Fig. S29, ESI†), because a longer linker may allow faster ISC and therefore faster recovery of the fluorophore ground state.
, Fig. 7a). This slow ISC, and therefore long-lasting biradical, might be the cause for differences in photostability between different fluorophore-stabilizer geometries (Fig. S29, ESI†), because a longer linker may allow faster ISC and therefore faster recovery of the fluorophore ground state.
Our findings also have various implications for the rational design of self-healing dyes with photoswitching kinetics tailored for super-resolution imaging and might explain why the same dye has completely different photoswitching in different biochemical surroundings, e.g., antibody vs. DNA or protein. They should also be taken into account when quantitative determinations of photostabilization effects are being made, since TCEP, MEA or β-ME are often found in buffers to ensure sample stability. The thiol compounds MEA and β-ME are known to reversible switch-off dyes through a similar mechanism as proposed here for TCEP.35,39 We and others have shown that some dyes, particularly cyanine-based ones, have a strong tendency for fast but reversible signal loss, which can be totally unrelated to photobleaching. In the presence of TCEP (and also for MEA or β-ME), it was impossible with our methods to distinguish between irreversible photobleaching and reversible photoswitching. Consequently, these compounds have to be omitted for quantitative determination of photobleaching lifetimes. We found these effects to be most pronounced for Cy5 dyes but almost negligible for Alexa555 and ATTO647N. These observations suggest that conclusions drawn in studies reporting enhancement factors for self-healing cyanine dyes should be revisited.2,11,18–25 In these earlier investigations, competition between reversible off-switching and triplet-state quenching of the photostabilizer most likely would have varied, hindering a quantitative determination of the degree of photostabilization, and impeding a true understanding of the mechanism of self-healing. And finally, self-healing dyes can be used for STORM-microscopy as demonstrated for Cy5-COT, and probably also in a more straightforward fashion for techniques that do not rely on photoswitching, e.g., PAINT or DNA PAINT.58,59
In conclusion, we suggest an improved protocol for determining photostabilization of self-healing dyes, where triplet-reactive solution additives are omitted. When using self-healing dyes, photostabilizing buffer additives have little or no effect, and the presence of photoswitching reagents can lead to the misinterpretation of reversible signal loss (off-switching) as apparent photobleaching. We also show that the biochemical surroundings of “normal” fluorophores have to be carefully chosen to ensure no harmful residues such as tryptophan might impact stabilization or switching efficiencies. Taken together our results serve as a guide on how to rationally optimize the parameters needed to use the higher photon output of dyes in general and that of self-healing ones for STORM-type super-resolution microscopy. Finally, our study suggests that intramolecular triplet-state quenchers act as a protector of fluorophores from reversible photoswitching reactions. The overall role of oxygen and its competition with intramolecular healers, however, still has to be clarified and was not taken into account in our work here.
4. Materials and methods
Synthesis
All chemical compounds were purchased from commercial suppliers and used without further purification. A Varian 400 MHz was used to record 1H-NMR and 13C-NMR spectra.The dsDNA-fluorophore samples with the labels ATTO647N, NPA-ATTO647, Alexa555, NPA-Alexa555, Cy5, and NPA-Cy5, were synthesized and prepared as previously described.19,23 Cy5-COT was prepared by labelling a biotinylated ssDNA-NH2 strand named P2 (biotin-5′-CGT CCA GAG GAA TCG AAT ATT A-3′-NH2) with NHS-COT; see ESI† for synthetic details using a modification of a procedure from the literature.18 The resulting oligonucleotide was characterized by MALDI-TOF mass spectrometry (Fig. S46, ESI†) using a ABI Voyager DE-PRO MALDI-TOF (delayed extraction reflector) Biospectrometry Workstation mass spectrometer. Hybridizing this strand to ssDNA-Cy5 (Cy5-5′-TAA TAT TCG ATT CCT CTG GAC G-3′) gave the Cy5-COT sample. The ssDNA-Trp-Cy5/ATTO647N constructs were synthesized by subsequently coupling tryptophan (Fmoc-Trp-NHS) and the different dyes to a ssDNA-NH2 named P1 (NH2-5′-TAA TAT TCG ATT CCT CTG GAC G-3′), according to previously established procedures.23 The resulting oligonucleotide was characterized by ESI-MS (Fig. S47 and S48, ESI†). The final ssDNA product was hybridized to a biotin-modified complementary DNA strand (unmodified P2) to allow immobilization.
Widefield TIRF imaging was performed on an inverted microscope (Olympus IX-71, UPlanSApo ×100 NA 1.49 Objective, Olympus, Germany) in a similar manner as described before.19 Images were collected with a back-illuminated electron multiplying charge-coupled device camera (512 × 512 pixel, C9100-13, Hammamatsu, Japan) with matching filters and optics. They were stored using either MetaMorph or MicroManager.60 Whereas all the data shown in Fig. S1–S21 (ESI†) were obtained on an Olympus IX-71 setup, that for Fig. S22–S28 (ESI†) and the related main text Fig. 5 were obtained on an Olympus IX-83 setup. The photon numbers and count-rates on the latter setup were slightly different which we attribute to differences in effective excitation power, collection efficiency, detection efficiency and count-to-photons calibration.
Confocal microscopy was performed on a home-built setup as described previously.19
Fluorescent transients were extracted (3 × 3 pixel area as above) and background corrected for every activated molecule. The transients were thresholded by Otsu's method63,64 to find activation events. Short off-blinks (<400 ms) were excluded from analysis. For every activation event, the on-time and number of photons was determined. A single emitter was fitted to a 2D Gaussian to find its standard deviation, σ. Then, using this value of σ and eqn (1), the localization error can be calculated for every on-event. Finally, these values were multiplied by 2.35 to obtain the theoretical maximum resolution and then averaged to give a single maximum resolution for the different imaging conditions.
Autocorrelation analysis was performed on fluorescent transients obtained from confocal microscopy (Fig. S30, ESI†). The transients were fitted to bi-exponential decays to obtain on and off times.65
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financed by an ERC Starting Grant (ERC-STG 638536 – SM-IMPORT to T. C.) and an ERC Advanced Grant (ERC-ADG 694610 – SUPRABIOTICS to A. H.). J. H. M. v. d. V. acknowledges a PhD stipend from the Ubbo-Emmius funds (University of Groningen). T. C. was supported by the Center of Nanoscience Munich (CeNS), Deutsche Forschungsgemeinschaft within GRK2062, LMUexcellent and the Center for integrated protein science Munich (CiPSM). We thank S. C. Blanchard, D. Griffith, V. Glembockyte and P. Tinnefeld for thoroughly reading the manuscript and for their critical and useful comments.References
- I. Rasnik, S. A. McKinney and T. Ha, Nonblinking and Long-Lasting Single-Molecule Fluorescence Imaging, Nat. Methods, 2006, 3(11), 891–893 CrossRef CAS PubMed.
- J. Vogelsang, R. Kasper, C. Steinhauer, B. Person, M. Heilemann, M. Sauer and P. Tinnefeld, A Reducing and Oxidizing System Minimizes Photobleaching and Blinking of Fluorescent Dyes, Angew. Chem., Int. Ed., 2008, 47(29), 5465–5469 CrossRef CAS PubMed.
- J. Widengren, A. Chmyrov, C. Eggeling, P.-Å. Löfdahl and C. A. M. Seidel, Strategies to Improve Photostabilities in Ultrasensitive Fluorescence Spectroscopy, J. Phys. Chem. A, 2007, 111(3), 429–440 CrossRef CAS PubMed.
- M. Heilemann, S. van de Linde, M. Schüttpelz, R. Kasper, B. Seefeldt, A. Mukherjee, P. Tinnefeld and M. Sauer, Subdiffraction-Resolution Fluorescence Imaging with Conventional Fluorescent Probes, Angew. Chem., Int. Ed., 2008, 47(33), 6172–6176 CrossRef CAS PubMed.
- S. van de Linde, A. Löschberger, T. Klein, M. Heidbreder, S. Wolter, M. Heilemann and M. Sauer, Direct Stochastic Optical Reconstruction Microscopy with Standard Fluorescent Probes, Nat. Protoc., 2011, 6(7), 991–1009 CrossRef CAS PubMed.
- A. P. Gorka and M. J. Schnermann, Harnessing Cyanine Photooxidation: From Slowing Photobleaching to near-IR Uncaging, Curr. Opin. Chem. Biol., 2016, 33, 117–125 CrossRef CAS PubMed.
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, The Contribution of Reactive Oxygen Species to the Photobleaching of Organic Fluorophores, Photochem. Photobiol., 2014, 90(2), 448–454 CrossRef CAS PubMed.
- F. Wilkinson, D. J. McGarvey and A. F. Olea, Excited Triplet State Interactions with Molecular Oxygen: Influence of Charge Transfer on the Bimolecular Quenching Rate Constants and the Yields of Singlet Oxygen [O* 2 (1. Delta. g)] for Substituted Naphthalenes in Various Solvents, J. Phys. Chem., 1994, 98(14), 3762–3769 CrossRef CAS.
- M. Heupel, I. Gregor, S. Becker and E. Thiel, Photophysical and Photochemical Properties of Electronically Excited Fluorescent Dyes: A New Type of Time-Resolved Laser-Scanning Spectroscopy, Int. J. Photoenergy, 1999, 1(3), 165–172 CrossRef CAS.
- P. Targowski, B. Ziętek and A. Bączyński, Luminescence Quenching of Rhodamines by Cyclooctatetraene, Z. Naturforsch., A: Phys. Sci., 1987, 42(9), 1009–1013 CAS.
- R. Dave, D. S. Terry, J. B. Munro and S. C. Blanchard, Mitigating Unwanted Photophysical Processes for Improved Single-Molecule Fluorescence Imaging, Biophys. J., 2009, 96(6), 2371–2381 CrossRef CAS.
- D. Pfiffi, B. A. Bier, C. M. Marian, K. Schaper and C. A. M. Seidel, Diphenylhexatrienes as Photoprotective Agents for Ultrasensitive Fluorescence Detection, J. Phys. Chem. A, 2010, 114(12), 4099–4108 CrossRef CAS PubMed.
- T. Cordes, J. Vogelsang and P. Tinnefeld, On the Mechanism of Trolox as Antiblinking and Antibleaching Reagent, J. Am. Chem. Soc., 2009, 131(14), 5018–5019 CrossRef CAS PubMed.
- C. E. Aitken, R. A. Marshall and J. D. Puglisi, An Oxygen Scavenging System for Improvement of Dye Stability in Single-Molecule Fluorescence Experiments, Biophys. J., 2008, 94(5), 1826–1835 CrossRef CAS PubMed.
- C. Eggeling, J. Widengren, R. Rigler and C. A. M. Seidel, Photobleaching of Fluorescent Dyes under Conditions Used for Single-Molecule Detection:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Evidence of Two-Step Photolysis, Anal. Chem., 1998, 70(13), 2651–2659 CrossRef CAS PubMed.
Evidence of Two-Step Photolysis, Anal. Chem., 1998, 70(13), 2651–2659 CrossRef CAS PubMed. - E. M. S. Stennett, M. A. Ciuba and M. Levitus, Photophysical Processes in Single Molecule Organic Fluorescent Probes, Chem. Soc. Rev., 2014, 43(4), 1057–1075 RSC.
- G. W. Byers, S. Gross and P. M. Henrichs, Direct and Sensitized Photooxidation of Cyanine Dyes, Photochem. Photobiol., 1976, 23(1), 37–43 CrossRef CAS PubMed.
- R. B. Altman, D. S. Terry, Z. Zhou, Q. Zheng, P. Geggier, R. A. Kolster, Y. Zhao, J. A. Javitch, J. D. Warren and S. C. Blanchard, Cyanine Fluorophore Derivatives with Enhanced Photostability, Nat. Methods, 2012, 9(1), 68–71 CrossRef CAS PubMed.
- J. H. M. van der Velde, E. Ploetz, M. Hiermaier, J. Oelerich, J. W. de Vries, G. Roelfes and T. Cordes, Mechanism of Intramolecular Photostabilization in Self-Healing Cyanine Fluorophores, ChemPhysChem, 2013, 14(18), 4084–4093 CrossRef CAS.
- J. H. M. van der Velde, J. Oelerich, J. Huang, J. H. Smit, M. Hiermaier, E. Ploetz, A. Herrmann, G. Roelfes and T. Cordes, The Power of Two: Covalent Coupling of Photostabilizers for Fluorescence Applications, J. Phys. Chem. Lett., 2014, 3792–3798 CrossRef CAS.
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Ultra-Stable Organic Fluorophores for Single-Molecule Research, Chem. Soc. Rev., 2014, 43(4), 1044–1056 RSC.
- J. H. M. van der Velde, J. J. Uusitalo, L.-J. Ugen, E. M. Warszawik, A. Herrmann, S. J. Marrink and T. Cordes, Intramolecular Photostabilization via Triplet-State Quenching: Design Principles to Make Organic Fluorophores “Self-Healing, Faraday Discuss., 2015, 184, 221–235 RSC.
- J. H. M. van der Velde, J. Oelerich, J. Huang, J. H. Smit, A. Aminian Jazi, S. Galiani, K. Kolmakov, G. Guoridis, C. Eggeling and A. Herrmann, et al., A Simple and Versatile Design Concept for Fluorophore Derivatives with Intramolecular Photostabilization, Nat. Commun., 2016, 7, 10144 CrossRef CAS.
- Q. Zheng, S. Jockusch, Z. Zhou, R. Altman, H. Zhao, W. Asher, M. Holsey, S. Mathiasen, P. Geggier and J. A. Javitch, et al., Electronic Tuning of Self-Healing Fluorophores for Live-Cell and Single-Molecule Imaging, Chem. Sci., 2017, 8, 755–762 RSC.
- R. B. Altman, Q. Zheng, Z. Zhou, D. S. Terry, J. D. Warren and S. C. Blanchard, Enhanced Photostability of Cyanine Fluorophores across the Visible Spectrum, Nat. Methods, 2012, 9(5), 428–429 CrossRef CAS.
- T. Ha and P. Tinnefeld, Photophysics of Fluorescent Probes for Single-Molecule Biophysics and Super-Resolution Imaging, Annu. Rev. Phys. Chem., 2012, 63(1), 595–617 CrossRef CAS.
- V. Glembockyte, R. Lincoln and G. Cosa, Cy3 Photoprotection Mediated by Ni2+ for Extended Single-Molecule Imaging: Old Tricks for New Techniques, J. Am. Chem. Soc., 2015, 137(3), 1116–1122 CrossRef CAS.
- V. Glembockyte, J. Lin and G. Cosa, Improving the Photostability of Red- and Green-Emissive Single-Molecule Fluorophores via Ni2+ Mediated Excited Triplet-State Quenching, J. Phys. Chem. B, 2016, 120(46), 11923–11929 CrossRef CAS.
- R. Kasper, B. Harke, C. Forthmann, P. Tinnefeld, S. W. Hell and M. Sauer, Single-Molecule STED Microscopy with Photostable Organic Fluorophores, Small, 2010, 6(13), 1379–1384 CrossRef CAS PubMed.
- M. Heilemann, S. van de Linde, A. Mukherjee and M. Sauer, Super-Resolution Imaging with Small Organic Fluorophores, Angew. Chem., Int. Ed., 2009, 48(37), 6903–6908 CrossRef CAS PubMed.
- B. Liphardt, B. Liphardt and W. Lüttke, Laser Dyes with Intramolecular Triplet Quenching, Opt. Commun., 1981, 38(3), 207–210 CrossRef CAS.
- B. Liphardt, B. Liphardt and W. Lüttke, Laser Dyes III: Concepts to Increase the Photostability of Laser Dyes, Opt. Commun., 1983, 48(2), 129–133 CrossRef CAS.
- P. Tinnefeld and T. Cordes, “Self-Healing” Dyes: Intramolecular Stabilization of Organic Fluorophores, Nat. Methods, 2012, 9(5), 426–427 CrossRef CAS.
- J. C. Vaughan, G. T. Dempsey, E. Sun and X. Zhuang, Phosphine Quenching of Cyanine Dyes as a Versatile Tool for Fluorescence Microscopy, J. Am. Chem. Soc., 2013, 135(4), 1197–1200 CrossRef CAS PubMed.
- G. T. Dempsey, M. Bates, W. E. Kowtoniuk, D. R. Liu, R. Y. Tsien and X. Zhuang, Photoswitching Mechanism of Cyanine Dyes, J. Am. Chem. Soc., 2009, 131(51), 18192–18193 CrossRef CAS PubMed.
- L. Nahidiazar, A. V. Agronskaia, J. Broertjes, B. Broek and K. van den; Jalink, Optimizing Imaging Conditions for Demanding Multi-Color Super Resolution Localization Microscopy, PLoS One, 2016, 11(7), e0158884 CrossRef PubMed.
- M. Heilemann, E. Margeat, R. Kasper, M. Sauer and P. Tinnefeld, Carbocyanine Dyes as Efficient Reversible Single-Molecule Optical Switch, J. Am. Chem. Soc., 2005, 127(11), 3801–3806 CrossRef CAS.
- J. C. Vaughan, S. Jia and X. Zhuang, Ultrabright Photoactivatable Fluorophores Created by Reductive Caging, Nat. Methods, 2012, 9(12), 1181–1184 CrossRef CAS.
- N. R. Conley, J. S. Biteen and W. E. Moerner, Cy3–Cy5 Covalent Heterodimers for Single-Molecule Photoswitching, J. Phys. Chem. B, 2008, 112(38), 11878–11880 CrossRef CAS.
- G. T. Dempsey, J. C. Vaughan, K. H. Chen, M. Bates and X. Zhuang, Evaluation of Fluorophores for Optimal Performance in Localization-Based Super-Resolution Imaging, Nat. Methods, 2011, 8(12), 1027–1036 CrossRef CAS.
- M. Minoshima and K. Kikuchi, Photostable and Photoswitching Fluorescent Dyes for Super-Resolution Imaging, JBIC, J. Biol. Inorg. Chem., 2017, 1–14 Search PubMed.
- P. Holzmeister, A. Gietl and P. Tinnefeld, Geminate Recombination as a Photoprotection Mechanism for Fluorescent Dyes, Angew. Chem., Int. Ed., 2014, 53(22), 5685–5688 CrossRef CAS.
- R. E. Thompson, D. R. Larson and W. W. Webb, Precise Nanometer Localization Analysis for Individual Fluorescent Probes, Biophys. J., 2002, 82(5), 2775–2783 CrossRef CAS.
- R. J. Ober, S. Ram and E. S. Ward, Localization Accuracy in Single-Molecule Microscopy, Biophys. J., 2004, 86(2), 1185–1200 CrossRef CAS.
- C. E. Shannon, Communication in the Presence of Noise, Proc. IRE, 1949, 37(1), 10–21 Search PubMed.
- H. Shroff, C. G. Galbraith, J. A. Galbraith and E. Betzig, Live-Cell Photoactivated Localization Microscopy of Nanoscale Adhesion Dynamics, Nat. Methods, 2008, 5(5), 417–423 CrossRef CAS.
- T. Cordes, M. Strackharn, S. W. Stahl, W. Summerer, C. Steinhauer, C. Forthmann, E. M. Puchner, J. Vogelsang, H. E. Gaub and P. Tinnefeld, Resolving Single-Molecule Assembled Patterns with Superresolution Blink-Microscopy, Nano Lett., 2010, 10(2), 645–651 CrossRef CAS PubMed.
- I. Kraljić and L. Lindqvist, Laser photolysis study of triplet eosin and thionine reactions in photosensitized oxidations, Photochem. Photobiol., 1974, 20(4), 351–355 CrossRef.
- G. I. Jones, C. W. Farahat and C. Oh, Photoinduced Electron Transfer Involving Eosin-Tryptophan Conjugates. Long-Lived Radical Pair States for Systems Incorporating Aromatic Amino Acid Side Chains, J. Phys. Chem., 1994, 98(28), 6906–6909 CrossRef CAS.
- N. Periasamy, S. Bicknese and A. S. Verkman, Reversible Photobleaching of Fluorescein Conjugates in Air-Saturated Viscous Solutions: Singlet and Triplet State Quenching by Tryptophan, Photochem. Photobiol., 1996, 63(3), 265–271 CrossRef CAS.
- M. Mickler, M. Hessling, C. Ratzke, J. Buchner and T. Hugel, The Large Conformational Changes of Hsp90 Are Only Weakly Coupled to ATP Hydrolysis, Nat. Struct. Mol. Biol., 2009, 16(3), 281–286 CrossRef CAS.
- C. Ratzke, B. Hellenkamp and T. Hugel, Four-Colour FRET Reveals Directionality in the Hsp90 Multicomponent Machinery, Nat. Commun., 2014, 5, 4192 CrossRef CAS.
- N. Marmé, J.-P. Knemeyer, M. Sauer and J. Wolfrum, Inter- and Intramolecular Fluorescence Quenching of Organic Dyes by Tryptophan, Bioconjugate Chem., 2003, 14(6), 1133–1139 CrossRef.
- S. van de Linde, I. Krstić, T. Prisner, S. Doose, M. Heilemann and M. Sauer, Photoinduced Formation of Reversible Dye Radicals and Their Impact on Super-Resolution Imaging, Photochem. Photobiol. Sci., 2011, 10(4), 499–506 RSC.
- S. C. Blanchard, Reply to “‘Self-Healing’ Dyes: Intramolecular Stabilization of Organic Fluorophores, Nat. Methods, 2012, 9(5), 427–428 CrossRef CAS.
- S. W. Hell, S. J. Sahl, M. Bates, X. Zhuang, R. Heintzmann, M. J. Booth, J. Bewersdorf, G. Shtengel, H. Hess and P. Tinnefeld, et al., The 2015 Super-Resolution Microscopy Roadmap, J. Appl. Phys., 2015, 48(44), 443001 Search PubMed.
- V. Glembockyte and G. Cosa, Redox-Based Photostabilizing Agents in Fluorescence Imaging: The Hidden Role of Intersystem Crossing in Geminate Radical Ion Pairs, J. Am. Chem. Soc., 2017, 139(37), 13227–13233 CrossRef CAS.
- A. Sharonov and R. M. Hochstrasser, Wide-Field Subdiffraction Imaging by Accumulated Binding of Diffusing Probes, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(50), 18911–18916 CrossRef CAS.
- R. Jungmann, C. Steinhauer, M. Scheible, A. Kuzyk, P. Tinnefeld and F. C. Simmel, Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami, Nano Lett., 2010, 10(11), 4756–4761 CrossRef CAS.
- A. D. Edelstein, M. A. Tsuchida, N. Amodaj, H. Pinkard, R. D. Vale and N. Stuurman, Advanced Methods of Microscope Control Using MManager Software, J. Biol. Methods, 2014, 1(2), e10 CrossRef PubMed.
- H. Yang, Change-Point Localization and Wavelet Spectral Analysis of Single-Molecule Time Series, in Single-Molecule Biophysics, ed. T. Komatsuzaki, S. Kawakami, S. Takahashi, H. Yang and R. J. Silbey, John Wiley & Sons, Inc., 2011, pp. 217–243 Search PubMed.
- F. R. Hill, A. M. van Oijen and K. E. Duderstadt, Detection of Kinetic Change Points in Piece-Wise Linear Single Molecule Motion, J. Chem. Phys., 2018, 148(12), 123317 CrossRef PubMed.
- N. Otsu, A Threshold Selection Method from Gray-Level Histograms, IEEE Trans. Syst. Man Cybern., 1979, 9(1), 62–66 Search PubMed.
- S. Walt, J. L. van der Schönberger, J. Nunez-Iglesias, F. Boulogne, J. D. Warner, N. Yager, E. Gouillart and T. Yu, Scikit-Image: Image Processing in Python, PeerJ, 2014, 2, e453 CrossRef.
- J. Yu, R. Lammi, A. J. Gesquiere and P. F. Barbara, Singlet−Triplet and Triplet−Triplet Interactions in Conjugated Polymer Single Molecules, J. Phys. Chem. B, 2005, 109(20), 10025–10034 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cp05063e |
| ‡ Present address: Physical Chemistry, Department of Chemistry, Ludwig-Maximilians-Universität München, Butenandtstr. 11, 81377 Munich, Germany. |
| § Present address: School of Chemistry, University of Wollongong, Northfields Avenue Wollongong NSW 2522, Australia. |
| This journal is © the Owner Societies 2019 |