
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of an ionic liquid and processing conditions on the β-polymorph crystal formation in poly(vinylidene fluoride)†
Tom
Pickford
a,
Xu
Gu
a,
Ellen L.
Heeley
 *b and
Chaoying
Wan
*a
*b and
Chaoying
Wan
*a
aInternational Institute for Nanocomposites Manufacturing (IINM), WMG, University of Warwick, CV4 7AL, UK. E-mail: chaoying.wan@warwick.ac.uk
bFaculty of Science, Technology, Engineering and Mathematics, Open University, Walton Hall, Milton Keynes, MK7 6AA, UK. E-mail: Ellen.Heeley@open.ac.uk
First published on 8th August 2019
Abstract
The piezoelectric properties of poly(vinylidene fluoride) (PVDF) are determined by the prevalence and orientation of its polar β-crystal phase, which can be tuned by processing conditions, additives, mechanical stretching or post-treatment. Here, the effects of three types of processing conditions on the crystalline structure evolution of PVDF were investigated; electrospinning, solution-casting and melt-compression. An ionic liquid (1-allyl-3-methylimidazolium chloride (AMIM)) used as an additive in aiding the electrospinning process, also affected the crystalline structure of the electrospun PVDF nanofibers. The total crystallinity, crystalline phase content and dielectric properties of the PVDF samples prepared by the different conditions were evaluated. The FTIR and DSC analyses show that the melt-compressed PVDF contains a high proportion of the paraelectric α-phase with a low total crystallinity, whereas the solution-cast PVDF contains a high proportion of polar γ-phase and a higher total crystallinity. In the case of the electrospun PVDF nanofibres, the addition of AMIM improved the morphology, uniformity and promoted the formation of the polar β and γ crystalline phases. X-Ray scattering analysis refined the crystal phase contents derived from FTIR, and also determined the ordered lamellar macromorphology formed by the three processing techniques. Furthermore, AMIM enhanced the a.c. conductivity and relative permittivity of the electrospun PVDF nanofibres by an order of magnitude, showing the effectiveness of using the ionic liquid AMIM, to improve the morphology and properties of electrospun PVDF nanofibers.
1. Introduction
Polyvinylidene fluoride (PVDF) is a semi-crystalline piezoelectric polymer with good mechanical strength, flexibility and chemical resistance. PVDF has several crystalline polymorphs, the most common being denoted as the α, β and γ-phases.1 The α phase has a trans–gauche conformation and is the most thermodynamically stable form, whereas the β-phase conformation is all trans and the γ-phase is trans–trans–trans–gauche.2,3 The phase that predominates in the polymer depends upon the processing conditions. The α-phase is generally formed above the Curie temperature of PVDF (170 °C), or at rapid cooling rates.4,5 The α-phase is transformed into the β-phase under mechanical deformation, high pressure and temperatures and applying high electric fields during processing.3,6 The γ-phase can be obtained from the α-phase but requires high annealing temperatures and again mechanical deformation.6,7 In previous studies, the relationships between processing parameters and crystalline transitions in PVDF have been investigated both theoretically and experimentally. It is often speculated that the formation of the α-phase and subsequent relaxation to the β-phase is a primary mechanism for β-phase formation.8 Alternatively, in solution, the α and β-phases may relax to the γ-phase when using highly polar solvents or high casting temperatures.9–11 Furthermore, the addition of polar additives to melts or solutions of PVDF, or application of electric fields are seen to amplify this relaxation process.12PVDF has attracted great interest in the field of energy harvesting as a flexible piezoelectric material13 due to its piezoelectric properties, which originate from the polar β-crystal conformation of its crystalline structure. A high proportion of the β-phase in PVDF, can increase the relative permittivity by over 10 fold.14 Thus, various methods have been employed to enhance formation of the β-phase while suppressing the paraelectric α-phase in PVDF;15via tailoring the polymer chain structure,16–18 optimising processing parameters (temperature, pressure, cooling rate and by applying shearing forces),19–22 or by use of post-treatment techniques.6,23 Furthermore, the addition of nanoparticles such as carbon nanotubes or ferrite particles can also significantly enhance the relative permittivity of PVDF and its copolymers.16,17,24,25
Solution-casting and melt-compression are two typical polymer processing techniques for PVDF. Solution-casting using highly polar, low boiling point solutions or melt-compression with high pressures, high temperatures and rapid cooling rates can all promote β-phase formation in the polymer.6,26–28 However, the relationship between processing parameters and optimal crystallisation conditions is still not fully understood. Moreover, electrospinning can also promote the formation of the β-phase in PVDF nanofibers. As such, electrospun PVDF has been used in the fields of medicine, environmental engineering and energy.29–31 The high shearing and electrostatic forces exerted on the polymer jet by the electrostatic field is effective at promoting β-phase crystallisation within the PVDF nanofibres32 whereby the negatively charged CF2 groups align themselves within the solution stream as it travels toward the collector, enhancing the trans β-phase formation. This eliminates the need for post-processing shearing and annealing, often used to increase the piezoelectric coefficient in PVDF films.33–35
Electrospun fibre membranes often exhibit very high piezoelectric coefficients due to the electrical poling of the fibres during spinning, the electrical poling induced by the electrostatic field used to form the fibres causes alignment of the PVDF molecules into a dipolar configuration as the solvent dries, eliminating the need for poling post-processing.36 Also, rapid evaporation of the volatile solvents used during electrospinning causes rapid formation of the β-phase, similar to solution-casting.9,20,26 Finally, the high mechanical stress exerted on the nanofibres during electrospinning along with the poling effect greatly induces dipole formation, leading to a high proportion of electroactive β-phase content.11,15 Thus, electrospinning is advantageous in utilizing a combination of polar solvents, electrical poling and mechanical stretching to maximise the electroactive phase content in one single processing step.
The key challenge with electrospinning PVDF is in optimising the processing parameters to maximise the β-phase content, which largely involves achieving stable spinning at high voltages to form thin nanofibres. Spinning thin fibres causes the formation of nanoscale ordered crystalline domains within the nanofibres oriented along the fibre axis.37,38 Similarly, the solution properties must be fine-tuned to achieve high quality fibres, as balancing the viscosity and conductivity of the solution in turn determines the flow rate-voltage balance.
This work is to investigate the effects of three processing methods (electrospinning, solution-casting and melt-compression) on the crystal polymorph transitions of PVDF. An ionic liquid 1-allyl-3-methylimidazolium chloride (AMIM) was used to improve the electrospinning process, its effect on the fibre morphology and crystalline structure are discussed. The morphology of the electrospun fibres was characterised with scanning electron microscopy (SEM), while the crystalline structure was characterised using Fourier transform infrared spectroscopy (FTIR), small- and wide-angle X-ray scattering (SAXS/WAXS) and differential scanning calorimetry (DSC). Finally, the dielectric properties of the PVDF nanofibres were measured using electrical impedance spectroscopy (EIS).
2. Experimental
2.1 Materials
Commercial poly(vinylidene fluoride) pellets (PVDF, Kynar 740, Tg = −40 °C, Tm = 168 °C) used for fabrication of all samples was purchased from Arkema. Dimethylformamide, DMF (>99%) was purchased from Fisher Scientific UK. Acetone was purchased from Sigma-Aldrich, UK and 1-allyl-3-methylimidazolium chloride (AMIM, 98%) was purchased from Alfa Aesar. Carbon black grease, used for electrical impedance spectroscopy, was purchased from MG Chemicals, UK.2.2 Sample preparation
PVDF samples were prepared by three different methods: solution-casting, melt-compression and electrospinning. For solution-casting, PVDF and PVDF/AMIM (1 wt%) solutions were directly cast onto glass dishes and dried under vacuum at 60 °C for 24 hours. The resulting film thickness was approximately 120 ± 50 μm. Dried PVDF and PVDF/AMIM films were further melt-compressed at 190 °C, under 8 MPa for 5 minutes. The thickness of resultant samples was approximately 120 ± 50 μm.Electrospun PVDF nanofibres were prepared with the Spraybase CAT000001 electrospinning instrument. Firstly, PVDF pellets were dissolved in DMF at 60 °C for at least 6 hours under magnetic stirring to form a homogeneous solution. After cooling down to room temperature, acetone was added to form a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 volumetric ratio to DMF, and the solution was stirred for further 12 hours. To study the effects of additives, AMIM was added at 1 wt% and 3 wt% to the solutions during the stirring process. PVDF fibre membranes were electrospun with a spinneret–collector distance of 15 cm with a range of voltage and spinning rates in order to determine the optimal spinning conditions for stable fibres.
:7 volumetric ratio to DMF, and the solution was stirred for further 12 hours. To study the effects of additives, AMIM was added at 1 wt% and 3 wt% to the solutions during the stirring process. PVDF fibre membranes were electrospun with a spinneret–collector distance of 15 cm with a range of voltage and spinning rates in order to determine the optimal spinning conditions for stable fibres.
Ionic liquids (ILs), can significantly ease electrospinning and improve nanofibre morphologies even at these low loadings, while also increasing conductivity of the nanofibre membranes due to their ionic nature.10,39,40 ILs are also easily dispersed in comparison to many solid nanofillers, which tend to agglomerate or stack in the electrospinning solution, and also alter the morphology of the nanofibres. AMIM was selected over other ILs after it was found that it produced better enhancements to nanofibre morphology at low concentrations.
2.3 Characterisation
Fourier transform infrared spectroscopy (FTIR) was performed on all samples using a Bruker Tensor 27 in a wavenumber range of 500–4000 cm−1. Samples were cut and removed from substrates, and an ambient background reading was subtracted from each spectrum. Phase content may be estimated with the method detailed by Cai et al.41 Firstly, a total ‘electroactive’ (EA) phase content – the sum of β and γ-phase contents, F(EA) – is firstly found by comparing the strong (β + γ) peak at 840 cm−1 to the α-phase peak at 763 cm−1. This can be determined according to the Beer–Lambert law as shown in eqn (1): | (1) |
2D small-angle X-ray scattering and 2D wide-angle X-ray scattering (2D SAXS/WAXS) data was collected using a Xenocs Xeuss 2.0 X-ray instrument operating with a Cu Kα source (λ = 1.54 Å). The 2D SAXS/WAXS data were collected on the Pilatus 300 K and Pilatus 100 K detector systems, respectively. Both detectors were calibrated with silver behenate. An evacuated chamber of 1.2 m was situated between the sample and SAXS detector to reduce air scattering and absorption. The WAXS detector was positioned in the evacuated sample chamber at a distance of 162 mm. The PVDF and composite samples were positioned vertically in the evacuated sample chamber and static 2D SAXS/WAXS data was taken with a collection time of 120 s. All SAXS/WAXS data were normalized for sample thickness, transmission and background scattering. X-ray data reduction and analysis was performed using the Xeuss 2.0 instrument data processing and analysis software. The 2D SAXS/WAXS data were reduced to 1D scattering profiles of intensity (I) versus scattering vector (q) for SAXS and 2θ for WAXS (where q = (4π/λ) sin(θ), 2θ is the scattering angle and λ is the X-ray wavelength), by sector averaging around the beam stop by a fixed angle and radius, q. To determine information on the long-range ordering in the samples, 1D correlation functions were computed from the 1D SAXS profiles using the Corfunc software incorporated into the SasView SAXS analysis package.42,43 The 1D correlation function, γ(R) is expressed as:
 | (2) |
 | (3) |
The 1D correlation functions were computed by the extrapolation of the 1D SAXS profile (q → ∞) according to Porod's law and a Guinier model back extrapolation (q → 0).44,45 The correlation function analysis assumes an ideal two-phase lamellar morphology of the PVDF polymer and various parameters including long period Lp, crystalline layer thickness Hb, amorous layer thickness Sb, and estimated bulk percent crystallinity Xc, can be extracted.42,44,46
Scanning electron microscopy (SEM) imaging was performed using a Carl Zeiss Sigma field SEM at voltages of 5.00–10.00 kV. Samples were sputter coated using an Au/Pd target to a thickness of ∼5 nm.
Differential scanning calorimetry (DSC) was performed using a Mettler Toledo STARe1 instrument on samples weighing ∼10 mg under a nitrogen atmosphere at a flow rate of 20 cm3 min−1. Samples were subjected to a heating and cooling cycle between −50 to 230 °C at a rate of 10 °C min−1.
Electrical impedance spectroscopy (EIS) measurements were carried out using a Princeton Applied Research Parstat MC with a PMC-2000 card and a two-point probe. Samples were coated with silver paint in order to fix the conductive area. Measurements were taken between 100 to 105 Hz.
3. Results and discussion
3.1 Effects of an ionic liquid on the morphology of electrospun PVDF nanofibres
The selection of an appropriate solvent is necessary for electrospinning homogeneous nanofibres. Here, DMF was used due to its moderate boiling point and evaporation rate which promotes the production of homogenous fibres. Acetone was initially added to increase the solvent volatility and assist in fibre formation.47–49 This may also serve to promote β-phase formation due to rapid solvent evaporation.6,22 However, the presence of acetone in the PVDF/DMF solution of 20 wt% made the electrospinning process unstable and generally resulted in the formation of beads rather than fibres, as observed by SEM in Fig. S1 in the ESI.† In the absence of acetone, the PVDF/DMF (20 wt%) solution can be electrospun into more homogeneous nanofibres with relatively few beads, with average diameter of 50–200 nm (Fig. 1(a)). The optimisation of the electrospinning process is further detailed in the ESI.† | ||
| Fig. 1 SEM micrographs of electrospun PVDF fibres containing (a) no additives, spun at a flow rate 3 ml h−1 and a voltage of 12.7 kV; (b) 1 wt% AMIM and (c) 3 wt% AMIM spun at a flow rate of 1.5 ml h−1 with a voltage of 7.4–8 kV. | ||
The ionic liquid 1-allyl-3-methylimidazolium chloride (AMIM) was incorporated into the PVDF/DMF solution to improve the conductivity of the solution and assist the electrospinning process. The morphologies of the electrospun PVDF fibres are shown in Fig. 1. The resultant fibre diameters were ∼100 nm in all samples, although the range distribution of diameters became more uniform upon the addition of AMIM, 100 ± 30 nm (Fig. 1b and c), compared to the neat PVDF fibres which are in the range of 50 nm up to 200 nm (Fig. 1a). AMIM clearly improves the nanofibre morphology, with fewer beads compared with the neat PVDF nanofibre. Moreover, the PVDF/AMIM nanofibres possess a more uniform diameter distribution of 50–150 nm with a lower mean diameter of ∼90 nm. It was found that 1 wt% AMIM addition was sufficient to form (almost) beadless fibres (Fig. 1(b)). Increasing the amount of AMIM to 3 wt% (Fig. 1(c)) adversely affects the fibre quality, with more beads and lower uniformity seen in the membrane. This may be due to the limited compatibility of AMIM with PVDF, leading to inhomogeneity in the solution conductivity and the fibre formation process at higher AMIM content.37
3.2 Crystallinity of PVDF samples
 | (4) |
| Sample | T m/°C | X dsc/% | T c/°C |
|---|---|---|---|
| a Very broad melting peak seen due to presence of multiple phases. Value given is at the absolute peak. | |||
| PVDF melt-compressed | 167.3 | 39 | 136.5 |
| PVDF solution-cast | 170.2 | 50 | 137.2 |
| PVDF electrospun | 166.5 | 47 | 136.3 |
| PVDF/AMIM melt-compressed | 173.1 | 44 | 139.5 |
| PVDF/AMIM solution-cast | 173.0 | 54 | 136.8 |
| PVDF/AMIM electrospun | 169a | 52 | 134.5 |
 | ||
| Fig. 2 DSC heating thermograms of (a) neat PVDF and (b) PVDF/AMIM; and cooling thermograms of (c) neat PVDF and (d) PVDF/AMIM fabricated by various processing methods (note that the absolute value of the heat flow is arbitrary). | ||
Comparing the values of Tm, the PVDF/AMIM melt-compressed and solution-cast samples peaked at ∼173 °C, while the corresponding neat PVDF solution-cast and melt-compressed samples peaked at ∼170 °C and ∼167 °C, respectively. A higher Tm is often referred to as being indicative of the β-phase.51,52 However, the value of Tm is dependent on the crystallisation history of the material, and so Tm alone does not provide a complete qualitative description of the phase content.3,6
In Fig. 2(a), the melting peak of all the pure PVDF samples are relatively broad indicating a range of crystallize sizes, crystal perfection and phases are present. In Fig. 2(b), (and Table 1), AMIM is also seen to increase Tm by ∼3 °C in all of the samples, and thus AMIM is seen to unilaterally increase Tm compared to the neat PVDF samples. However, it should be noted that the electrospun PVDF/AMIM melting peak (Fig. 2(b)) is quite broad (consisting of several shoulders) compared with the neat electrospun PVDF sample, implying a more diverse crystallite structure. Anousheh et al. suggest that in pure crystals α-phase PVDF displays a higher temperature melting peak, but that regioisomeric defects in the material will influence the melting peak of the α-phase more so than the β-phase.53 Hence, greater disorder may shift the α-phase melting peak down towards that of the β-phase, causing the illusion of a high β-phase content. We expect that in the PVDF/AMIM electrospun sample, the defect proportion will be lower than that of the other samples, leading to an initial β-phase melting peak at 169 °C, and an α-phase peak at 173 °C, matching the melt-compressed and solution-cast PVDF/AMIM melting peaks (Fig. 2(b)).
From the data in Table 1 and melting enthalpies of the DSC thermograms, it was found that the addition of AMIM to the electrospun PVDF enhances Xdsc by 5%. The improved fibre morphology consisting of thinner fibres and a low number of beads likely induces a higher degree of molecular ordering in the fibres correlating with a higher Xdsc. The β-phase content will also be higher in the more uniform nanofibres as the large electrostatic forces exerted on the polymer during fibre formation induces the formation of the polar β-phase. This is seen as a shoulder in the melting thermogram of electrospun PVDF/AMIM (Fig. 2(b)).
Xing et al. observed a broadening in their melting peaks as the concentration of the ionic liquid in their melt blended PVDF increases, although this is only observed in our electrospun sample.39 In fact, the inverse is seen in our solution-cast and melt-compressed samples; the peaks are made thinner with the addition of AMIM, and the melting enthalpy gives a higher Xdsc. The low Xdsc and spread of crystalline phases in the neat PVDF melt-compressed sample will contribute to the broader peak. Furthermore, the melt-compressed samples (with and without AMIM), showed noticeably lower values of Xdsc than samples made by other processing methods. This may be due to the high temperature used in the process, which keeps the PVDF in a less viscous solution during crystallisation, promoting the formation of the high bonding energy and kinetically favourable α-phase.8,53
The solution-cast samples show similar or higher crystallinity than those which were electrospun, although the sharp peaks in both samples indicates a low variation in crystallite sizes and greater crystalline perfection. This is supported by the Xdsc values being the greatest from this method within the neat PVDF and PVDF/AMIM sample sets. The solution-cast samples show a sharpening and an upward shift of the melting peak by 3 °C with the addition of AMIM. The broader form and small hump on the neat solution-cast melting peak is likely due to the lower crystallinity measured in this sample.6 Surprisingly however, there is no indication of the γ-phase in the melting peak, as this generally appears between 180–190 °C. This may indicate a transition of the γ-phase to the α-phase during the heating or melting process. Generally, the high temperature and sharp form of the solution-cast melting peak compared to the other methods with and without AMIM, may also imply a lack of the β-phase compared to the other samples.
The crystallisation peaks (Fig. 2(c and d)) offers some insight into the effects of AMIM on the nucleation and crystal phase formation in the samples. The electrospun samples show only a slight difference in Tc on the addition of AMIM, although the peak broadens considerably, implying that AMIM slows and diversifies the nucleation processes. As for the solution-cast samples, the addition of AMIM has a less significant effect on Tc and has little effect on the form of the peak, thus AMIM seems to make a minimal difference to nucleation and recrystallisation process. Conversely, the melt-compressed samples show a large increase in Tc of 3.0 °C and a sharper peak with AMIM addition. This implies that the AMIM has acted as a nucleating agent and has induced a crystallisation processes not seen in the other samples to any extent.
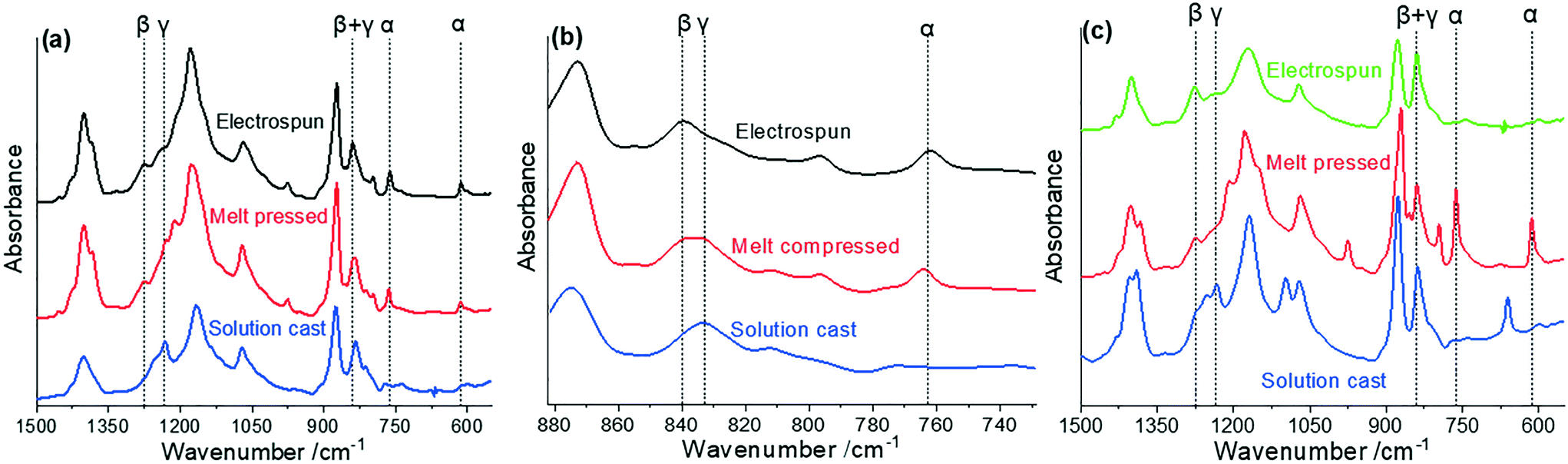 | ||
| Fig. 3 (a) FTIR of neat PVDF made with various processing methods; (b) expanded FTIR region between 740 and 880 cm−1 (c) equivalent FTIR for PVDF/AMIM samples. | ||
| Sample | α (%) | β (%) | γ (%) | β + γ (%) |
|---|---|---|---|---|
| PVDF electrospun | 41 | ≲59 | — | 59 |
| PVDF/AMIM electrospun | 26.5 | 68 | 5.5 | 73.5 |
| PVDF melt-compressed | 41 | 41 | 18 | 57 |
| PVDF/AMIM melt-compressed | 55 | ≲45 | — | 45 |
| PVDF solution-cast | 26 | — | ≲74 | 74 |
| PVDF/AMIM solution-cast | 27 | — | ≲73 | 73 |
For the neat PVDF samples (Fig. 3(a and b)), comparing the melt-compressed and electrospun samples, while they show similar α-phase and β + γ-phase fractions, the electrospun sample lacks signatures of the γ-phase. It can be inferred that the β to γ-phase relaxation is occurring in the melt-compressed samples before a significant proportion of the solvent has boiled off, but not enough to have removed all of the β-phase content. Conversely, this relaxation is being inhibited in the electrospun samples in the early stages of fibre formation, preventing the formation of any significant amount of the γ-phase. This can be concluded from the presence of the peak at 833 cm−1 shown in Fig. 3(c) – ascribed to γ-phase crystallites formed by this relaxation – which is seen in the melt-compressed but not in the electrospun sample.41 The electric field poling during electrospinning may prevent this relaxation, while the faster solvent evaporation rate in melt-compression likely assists in stopping γ-phase formation as solution viscosity increases too quickly for the β to γ-phase relaxation to occur. As a result, the β-phase content by melt-compression is ∼18% lower than neat PVDF obtained by electrospinning. However, the high pressure forces exerted on the PVDF during melt-compression facilitates the confirmation from α-phase to β-phase, meaning the β-phase content is still significant.15,27
When comparing the neat PVDF solution-cast sample to both the melt-compressed and electrospun PVDF (Fig. 3(a and b)), the α-phase signatures at 763 and 614 cm−1 are both diminished, while the 833 cm−1 and 1234 cm−1 γ-phase peaks dominate the 840 cm−1 and 1275 cm−1 β-phase peaks respectively. Due to the slow evaporation rate during solution-casting, the β to γ-phase relaxation occurs considerably, resulting in a high γ-phase content and the strong 833 cm−1 β to γ-phase relaxation peak.
In the PVDF/AMIM samples, shown in Fig. 3(c), the 833 cm−1 β to γ relaxation peak is not seen in either the melt-compressed or electrospun sample. In fact, in the melt-compressed samples, the electroactive phase content was decreased as a result of AMIM addition. The solution-cast samples, both with and without AMIM, instead show strong peaks in the γ-phase. It is likely α or β to γ-phase transitions are prominent during the crystallisation process and that the solvent conditions did not encourage β-phase formation.9 This is confirmed by the strong 833 cm−1 γ peak seen in the neat solution-cast sample. Interestingly, the phase content shows no change when AMIM is added to the solution, and the 833 cm−1 peak is far less prominent. A low rate of solvent evaporation is likely to have occurred in these samples, providing an environment for γ-phase formation. Since there are no other processing conditions during solution-casting which promote the formation of other phases (such as the lack of mechanical stress), the γ-phase seems to completely dominate these samples. However, the lack of detectability of the β-phase from this FTIR likely means that its content is underestimated.
As seen in Table 2, the addition of AMIM to the electrospinning solution clearly increases the electroactive phase composition, with β + γ-phase contents of over 70%. As expected, the highest α-phase content is in the neat PVDF electrospun sample, whereas the lowest is seen in the PVDF/AMIM samples.
 | ||
| Fig. 4 2D SAXS/WAXS data of pure PVDF and PVDF/AMIM composites from the three different processing techniques (casting, melt-compression and electrospinning). Major Bragg reflections are indexed on the 2D WAXS patterns for the α, β and γ crystal phases of PVDF. | ||
The corresponding isotropic 2D WAXS scattering for the pure PVDF and PVDF/AMIM, have some of the major Bragg reflections indexed on the patterns for the α, β and γ crystal phases of the polymer.54 Again, the different processing methods affect the major crystalline phase occurring in the polymer. The solution-cast samples (with and without AMIM) shows mainly the β and γ crystal phases predominate in the PVDF. However, both the pure PVDF melt-compression and electrospun samples are predominantly composed of the α and γ crystal phases. On addition of AMIM, the crystal phase shifts largely to the β and γ crystal phases. To gain further insight in to both the macro and micromorphology of the PVDF and composites 1D SAXS/WAXS profiles were obtained from the 2D patterns in Fig. 4.
 | ||
| Fig. 5 1D SAXS profiles of (a) pure PVDF and (b) PVDF–AMIM composites prepared by the three different processing conditions; (c) corresponding 1D correlation function of solution cast pure PVDF. | ||
To obtain details about the crystalline macrostructure of the PVDF, 1D correlation functions were computed from the 1D SAXS profiles. Fig. 5(c) shows an example 1D correlation function for pure solution-cast PVDF. This allows the extraction of parameters (as labelled in Fig. 5(c)) such as a more accurate determination of the long period, Lp, the crystalline and amorphous layer thicknesses, Hb and Sb respectively, and estimated bulk percent crystallinity Xc,42 to be obtained. The bulk crystallinity Xc, is determined from the correlation function by way of eqn (5):
| χc = γmin/(γmin + γ*) | (5) |
Table 3 collates the correlation function analysis data from the 1D SAXS profiles. However, some of the data did not give reliable fits to the model and so the lamellar parameters could not be extracted in those cases from the correlation function. Comparing the results for the pure PVDF melt-compressed and solution-cast samples, the Lp is clearly reduced in the solution-cast sample and there is a reduction in the amorphous layer thickness but no real change in the crystalline lamellae layer. This results in an increased bulk crystallinity, Xc. Hence, the solution-casting processing technique tends to induce more crystallinity in the PVDF sample compared with the in the melt-compressed processing technique.3 However, with the addition of AMIM to the melt-compressed sample, we observe a decrease in Lp, which is due to an increase in crystalline layer thickness (and reduction of amorphous layer thickness), which increases Xc slightly. Similarly, Xing et al. showed that the addition of ILs in to PVDF, increased the scattering intensity of SAXS and shifted the Lp to lower q values and concluded that the ILs reside in the amorphous fraction of the macrostructure.39 Here, the addition of AMIM influences the total crystallinity (as seen in DSC, Fig. 2) and crystallite size, potentially acting as a nucleating agent as well as inducing β-phase formation.
| Sample | L p/Å | H b/Å | S b/Å | X c (±2)/% |
|---|---|---|---|---|
| PVDF melt-compressed | 99 | 24 | 75 | 49 |
| PVDF solution-cast | 62 | 24 | 38 | 55 |
| PVDF/AMIM melt-compressed | 82 | 29 | 53 | 51 |
1D WAXS is used to confirm the prominence of the different crystal phases, which have been identified with FTIR, for the three processing methods and addition of AMIN. Fig. 6, shows the 1D WAXS for all samples highlighting the major peaks for the α, β and γ-phrases. A full breakdown of the crystallographic planes54 identified from the 1D WAXS profiles for all samples is provided in Table S1 of the ESI.† From the data in Table S1,† the three different processing methods of neat PVDF influence the crystal phase content, although it is seen that all samples have mixtures of the three major crystalline phases. Typically, the α-phase tends to dominate in the electrospun and melt-compressed neat PVDF samples,3,6,24 whereas the β-phase is dominant in the electrospun PVDF/AMIN samples.39 The solution-cast neat PVDF and PVDF/AMIN show that the β-and γ-phases are prevalent in these samples due to the fast solvent evaporation at low temperatures.6 The 1D WAXS profiles for the electrospun samples are shown in Fig. 6(a and b). The α-phase signature in both samples is present but appears to be more dominant in the neat PVDF sample. The α-phase (120)/(012) peak at 26.6° in particular is absent in the PVDF/AMIM sample, but strong in the neat PVDF. Conversely, the γ phase peaks in the region of 38.8° to 42° are enhanced the PVDF/AMIM sample. Fig. 6(b) displays the form of the peaks around 20°. The β-phase signature is identified by the (110) peak at 20.6°, which is partially obscured by peaks at 20.0° and 20.3°, representing the (110) α-phase and γ-phase respectively. The PVDF/AMIM sample clearly shows a broader form to this peak, implying significant amounts of all three crystalline phases are present but the β-phase is mainly dominant, which agrees with the FTIR analysis.
 | ||
| Fig. 6 (a and b) Electrospun; (c and d) solution-cast and (e and f) melt-compressed 1D-WAXS spectra (note that the absolute value of the absorbance is arbitrary). | ||
The 1D-WAXS profiles of the solution-cast samples (Fig. 6(c and d)) show a high proportion of the γ-phase with little to no α-phase signature, again supported by the FTIR data. The position of the (110) peak at 20–21°, shown in Fig. 6(d), highlights the γ-phase is dominant in in both samples. The data also shows the (310) β-phase peaks in both samples, confirming at least a partial split in the electroactive phase content undetectable by FTIR. Therefore, the addition of AMIM to the casting solution seems to have little effect on the phase content of PVDF, again this agrees with the FTIR data.
The 1D-WAXS profiles of both melt-compressed samples in Fig. 6(e and f), predominantly show α-phase signatures. These are more prominent in the neat PVDF data. On the addition of AMIM the proportion of the β-phase content is increased, where the combined α–β peaks around 35.9–36.3° are shifted toward higher 2θ, implying a high proportion of the β-phase is present. Similarly, the combined peaks around 20.0–20.6° (shown in Fig. 6(f)) are shifted toward higher 2θ, also confirming a β and/or γ-phase enhancement. Again, the addition of AMIM was seen to increase β-phase content in our FTIR data. Finally, the peaks around the 32–34° region in the neat melt-compressed sample correspond with the (121) and (130) crystallographic planes of the δ-phase; a more elusive phase similar to the α conformation, but with alternate molecules rotated by 180° in the unit cell.6 Similarly, the neat PVDF solution-cast sample shows (041) and (221) δ-phase signatures around 42–43°.
The 1D WAXS data (Fig. 6 and Table S1†) confirms that the AMIM is effective at inducing the α to β crystal phase relaxation of PVDF in both the electrospun and melt-compressed samples but does not alter the unit cells associated with these forms, (monoclinic to orthorhombic respectively). Therefore, it is reasonable to assume that the AMIM is not incorporated into the PVDF unit cell, but will be part of the amorphous region in the lamellar macrostructure.41
From the FTIR and SAXS/WAXS data here, it can be concluded that while the solution-cast samples have an electroactive phase content just as high as the best electrospun samples, electrospinning with an ionic substance remains the most reliable method for producing a high β-phase fraction.
3.3 Electrical properties of PVDF nanofibre membranes
The effect of AMIM on the electrical properties of the PVDF nanofibres was investigated, shown in Fig. 7. Both the conductivity and relative permittivity of the PVDF nanofibres increase by almost an order of magnitude when spun with AMIM. The more homogenous fibre structure of the PVDF/AMIM samples forms due to stable electrospinning in which the electric field exerts a force on the polymer jet constant in time and position. This should increase the degree of crystalline order within the fibres, likely contributing to the higher conductivity, as demonstrated by the larger crystallinity seen in Table 1. Similarly, the electroactive phase content of the AMIM incorporated fibres (Table 2) will be a key component of this enhancement. | ||
| Fig. 7 (a) a.c. conductivity and (b) relative permittivity of the PVDF nanofibre membranes. | ||
The incorporation of an ionic substance into the fibres leads to a higher charge mobility, as their presence in the polymer matrix should impede the accumulation of static charges on the polymer surface.55,56 Xing et al. report an order of magnitude decrease to the volume resistivity of their PVDF/ionic liquid melt mixed blends when increasing their ionic liquid concentration from 2 wt% to 4 wt%, and further order of magnitude drops when increased to 10 wt% and 20 wt% in turn, while they were unable to probe the conductivity in their neat PVDF, which they attribute to electrostatic charge build-up.39 They later report that similar conductivity enhancements were seen in their PVDF/ionic liquid electrospun fibres at the same ionic liquid contents due to a decrease in surface charge accumulation. This leads to a conductive, hydrophobic, porous and piezoelectric polymer membrane, highlighting the multifunctional potential of these materials.10 Hence, increasing the proportion of AMIM in our PVDF samples to similar concentrations (e.g. 10 wt%) could lead to further increases in conductivity. However, it should be noted that the conductivity of the polymer solution needs to be well balanced in the case of electrospinning. A higher conductivity of the solution may cause the solution jet to fall back on to itself between the collector and the spinneret due to charge movement within the jet, inhibiting the formation of beadless, thin and well dispersed fibres.
4. Conclusions
The advantages of electrospinning have been demonstrated by producing neat PVDF nanofibres which exhibit higher β-phase content compared to melt-compressed and solution-cast samples. The melt-compressed samples are dominant in the paraelectric α-phase and possess low total crystallinity, rendering them impractical for piezoelectric applications. Conversely, the solution-cast PVDF films show high γ-phase content but similarly lacked a significant β-phase crystalline fraction. Since the solution-cast sample has a slightly higher total crystallinity than the electrospun sample, the technique may be useful if highly crystalline γ-phase PVDF films are desired.Furthermore, the ionic liquid AMIM has been shown to assist the electrospinning of PVDF by producing more homogeneous nanofibres with a higher proportion of the electroactive β-phase. As seen in DSC, AMIM has also had a positive impact on total crystallinity of samples made via solution-casting and melt-compression as well as electrospinning. Interestingly, the solution-cast samples show the highest crystallinity, and the PVDF/AMIM electrospun samples also show a high total crystallinity. Combined with the high β-phase contents of the samples, electrospinning of PVDF with AMIM is found to be the optimal method of producing crystalline, piezoelectric phase-dominant PVDF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Tom Pickford thanks the PhD studentship and research facility support from the International Institute for Nanocomposites Manufacturing (IINM), WMG at the University of Warwick, UK. The authors acknowledge the support from Dr. Steve Huband for SAXS/WAXS data collection and processing.References
- A. J. Lovinger, Science, 1983, 220, 1115 CrossRef CAS PubMed.
- L. Ruan, X. Yao, Y. Chang, L. Zhou, G. Qin and X. Zhang, Polymer, 2018, 10, 228 Search PubMed.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS.
- J. S. Green, B. L. Farmer and J. F. Rabolt, J. Appl. Phys., 1986, 60, 2690–2693 CrossRef CAS.
- G. Teyssedre, A. Bernes and C. Lacabanne, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 879–890 CrossRef CAS.
- Z. L. Cui, N. T. Hassankiadeh, Y. B. Zhuang, E. Drioli and Y. M. Lee, Prog. Polym. Sci., 2015, 51, 94–126 CrossRef CAS.
- M. Li, H. J. Wondergem, M.-J. Spijkman, K. Asadi, I. Katsouras, P. W. M. Blom and D. M. de Leeuw, Nat. Mater., 2013, 12, 433 CrossRef CAS PubMed.
- W. J. Kim, M. H. Han, Y. H. Shin, H. Kim and E. K. Lee, J. Phys. Chem. B, 2016, 120, 3240–3249 CrossRef CAS PubMed.
- R. Gregorio and D. S. Borges, Polymer, 2008, 49, 4009–4016 CrossRef CAS.
- C. Y. Xing, J. P. Guan, Y. J. Li and J. Y. Li, ACS Appl. Mater. Interfaces, 2014, 6, 4447–4457 CrossRef CAS PubMed.
- G. J. Zhong, L. F. Zhang, R. Su, K. Wang, H. Fong and L. Zhu, Polymer, 2011, 52, 2228–2237 CrossRef CAS.
- Y. Li, S. D. Tang, M. W. Pan, L. Zhu, G. J. Zhong and Z. M. Li, Macromolecules, 2015, 48, 8565–8573 CrossRef CAS.
- K. S. Ramadan, D. Sameoto and S. Evoy, Smart Mater. Struct., 2014, 23, 033001 CrossRef.
- P. Martins, J. S. Nunes, G. Hungerford, D. Miranda, A. Ferreira, V. Sencadas and S. Lanceros-Mendez, Phys. Lett. A, 2009, 373, 177–180 CrossRef CAS.
- C. Y. Wan and C. R. Bowen, J. Mater. Chem. A, 2017, 5, 3091–3128 RSC.
- M. Aldas, G. Boiteux, G. Seytre, Z. Ghallabi and Ieee, in Proceedings of the 2010 Ieee International Conference on Solid Dielectrics, 2010 Search PubMed.
- M. Lallart, P. J. Cottinet, L. Lebrun, B. Guiffard and D. Guyomar, J. Appl. Phys., 2010, 108, 034901 CrossRef.
- H. S. Xu, J. Appl. Polym. Sci., 2001, 80, 2259–2266 CrossRef CAS.
- B. Khomami and C. A. Langton, Polym. Eng. Sci., 1991, 31, 803–811 CrossRef CAS.
- W. Z. Ma, J. Zhang, S. J. Chen and X. L. Wang, J. Macromol. Sci., Part B: Phys., 2008, 47, 434–449 CrossRef CAS.
- B. Mohammadi, A. A. Yousefi and S. M. Bellah, Polym. Test., 2007, 26, 42–50 CrossRef CAS.
- W. A. Yee, M. Kotaki, Y. Liu and X. H. Lu, Polymer, 2007, 48, 512–521 CrossRef CAS.
- J. E. Lee and S. N. Leung, CrystEngComm, 2018, 20, 4080–4089 RSC.
- P. Martins, C. M. Costa, M. Benelmekki, G. Botelho and S. Lanceros-Mendez, CrystEngComm, 2012, 14, 2807–2811 RSC.
- S. S. Yu, W. T. Zheng, W. X. Yu, Y. J. Zhang, Q. Jiang and Z. D. Zhao, Macromolecules, 2009, 42, 8870–8874 CrossRef CAS.
- D. L. Chinaglia, R. Gregorio, J. C. Stefanelo, R. A. P. Altafim, W. Wirges, F. P. Wang and R. Gerhard, J. Appl. Polym. Sci., 2010, 116, 785–791 CAS.
- D. D. Song, D. C. Yang and Z. L. Feng, J. Mater. Sci., 1990, 25, 57–64 CrossRef CAS.
- M. M. Tao, F. Liu, B. R. Ma and L. X. Xue, Desalination, 2013, 316, 137–145 CrossRef CAS.
- M. A. Qasaimeh, S. Sokhanvar, J. Dargahi and M. Kahrizi, J. Microelectromech. Syst., 2009, 18, 195–207 CAS.
- J. R. Kim, S. W. Choi, S. M. Jo, W. S. Lee and B. C. Kim, Electrochim. Acta, 2004, 50, 69–75 CrossRef CAS.
- R. Balamurugan, S. Sundarrajan and S. Ramakrishna, Membranes, 2011, 1, 232–248 CrossRef PubMed.
- Y. M. Shin, M. M. Hohman, M. P. Brenner and G. C. Rutledge, Polymer, 2001, 42, 9955–9967 CrossRef CAS.
- J. Nunes-Pereira, V. Sencadas, V. Correia, J. G. Rocha and S. Lanceros-Mendez, Sens. Actuators, A, 2013, 196, 55–62 CrossRef CAS.
- M. Sharma, V. Srinivas, G. Madras and S. Bose, RSC Adv., 2016, 6, 6251–6258 RSC.
- H. Guo, Y. Zhang, F. Xue, Z. Cai, Y. Shang, J. Li, Y. Chen, Z. Wu and S. Jiang, CrystEngComm, 2013, 15, 1597–1606 RSC.
- V. Sencadas, C. Ribeiro, I. K. Bdikin, A. L. Kholkin and S. Lanceros-Mendez, Phys. Status Solidi A, 2012, 209, 2605–2609 CrossRef CAS.
- L. Fan, Y. T. Xu, X. Zhou, F. Chen and Q. Fu, Polymer, 2018, 153, 61–69 CrossRef CAS.
- X. Q. Ma, J. L. Liu, C. Y. Ni, D. C. Martin, D. B. Chase and J. F. Rabolt, ACS Macro Lett., 2012, 1, 428–431 CrossRef CAS.
- C. Y. Xing, M. M. Zhao, L. P. Zhao, J. C. You, X. J. Cao and Y. J. Li, Polym. Chem., 2013, 4, 5726–5734 RSC.
- L. He, J. Sun, X. Wang, C. Wang, R. Song and Y. Hao, Polym. Int., 2013, 62, 638–646 CrossRef CAS.
- X. M. Cai, T. P. Lei, D. H. Sun and L. W. Lin, RSC Adv., 2017, 7, 15382–15389 RSC.
- A. Ryan, Fibre Diffraction Review, 1994, 3, 25–29 Search PubMed.
- SasView for Small Angle Scattering Analysis, http://www.sasview.org/, (accessed 02/04/2019).
- F. J. Baltá-Calleja and C. G. Vonk, X-ray scattering of synthetic polymers, Elsevier, 1989 Search PubMed.
- G. Porod, Colloid Polym. Sci., 1951, 124, 22 CrossRef.
- G. R. Strobl and M. Schneider, J. Polym. Sci., Part B: Polym. Phys., 1980, 18, 1343–1359 CAS.
- E. S. Cozza, O. Monticelli, E. Marsano and P. Cebe, Polym. Int., 2013, 62, 41–48 CrossRef CAS.
- Y. R. Wang, J. M. Zheng, G. Y. Ren, P. H. Zhang and C. Xu, Smart Mater. Struct., 2011, 20, 045009 CrossRef.
- J. F. Zheng, A. H. He, J. X. Li and C. C. Han, Macromol. Rapid Commun., 2007, 28, 2159–2162 CrossRef CAS.
- J. Gomes, J. S. Nunes, V. Sencadas and S. Lanceros-Mendez, Smart Mater. Struct., 2010, 19, 065010 CrossRef.
- J. S. Andrew and D. R. Clarke, Langmuir, 2008, 24, 670–672 CrossRef CAS PubMed.
- L. H. He, J. Sun, X. X. Wang, C. D. Wang, R. Song and Y. M. Hao, Polym. Int., 2013, 62, 638–646 CrossRef CAS.
- N. Anousheh and A. Soldera, Polymer, 2017, 125, 154–160 CrossRef CAS.
- K. Jurczuk, A. Galeski, M. Mackey, A. Hiltner and E. Baer, Colloid Polym. Sci., 2015, 293, 1289–1297 CrossRef CAS PubMed.
- A. X. Lu, M. McEntee, M. A. Browe, M. G. Hall, J. B. DeCoste and G. W. Peterson, ACS Appl. Mater. Interfaces, 2017, 9, 13632–13636 CrossRef CAS PubMed.
- J. Pernak, A. Czepukowicz and R. Pozniak, Ind. Eng. Chem. Res., 2001, 40, 2379–2383 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce01051c |
| This journal is © The Royal Society of Chemistry 2019 |