
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular π–hole interactions with nitro aromatics†
Antonio
Franconetti
 a,
Antonio
Frontera
a and
Tiddo J.
Mooibroek
*b
a,
Antonio
Frontera
a and
Tiddo J.
Mooibroek
*b
aDepartment of Chemistry, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma, Baleares, Spain
bvan't Hoff Institute for Molecular Sciences, Universiteit van Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: t.j.mooibroek@uva.nl
First published on 7th August 2019
Abstract
A thorough evaluation of the CSD and DFT computations were conducted to assess if intramolecular π–hole interactions can stabilize a conformer of nitro aromatics. It was found that this can only be the case when the nitro N-atom and an interacting electron-rich atom are separated by at least four bonds. Data from the solid state correspond well to the gas phase calculations and stabilizing energies were estimated to be as large as about 2–3 kcal mol−1, which is in the order of weak hydrogen bonding interactions.
Introduction
Intramolecular interactions such as hydrogen bonding can direct the formation of the three-dimensional structure of large molecules such as synthetic polymers,1 artificial foldamers2 and proteins.3 The structure and function of proteins can also be affected by other intramolecular forces such as n → π* interactions4 and halogen bonding.5 Conformation-directing effects of intramolecular hydrogen bonds are also prevalent in small molecules,6 which raises the question what other types of intramolecular non-covalent forces can stabilize conformers.It is known that the N-atom in nitro (–NO2) compounds bears a positive potential, which has been classified as a so-called π–hole (see Fig. 1a).7 Such π–holes can establish favorable intermolecular interactions with lone-pair electrons, for example in nitrate esters.8 This is even the case for the ubiquitous nitrate anion when its negative charge is sufficiently spread out by hydrogen bonding9 or coordination to a metal.10 The same holds for nitro aromatics,11 which are synthetically very accessible and thermally robust. In fact, small nitro aromatic molecules are widely studied for their pharmacological properties‡ and many are approved by the U.S. Food and Drug Administration and regularly administered.12 For example, nitro aromatic dihydropyridine derivatives such as nifedipine (Adalat)13 and nisoldipine (Sular)14 are calcium channel blockers used to treat hypertension and angina.15
 | ||
| Fig. 1 Molecular electrostatic potential map of nitrobenzene indicating the location of the π–hole (a, DFT/MP2/6-311+G**, from −22 (red) tot +24 kcal mol−1 (blue)) and the general queries used to retrieve data from the CSD for nitro aromatics where a possibly interacting atom (X or ElR) is three, four, or ≥ five bonds removed from the nitro's N-atom (i.e. five bonds for n = 1, six bonds for n = 2 and so forth). X can be any atom except H; ElR = N, P, As, O, S, Se, Te, F, Cl, Br, I or At. α = the angle between the NO2 plane and the plane of the aryl ring. The blue dashed lines indicate that all types of bonds were allowed and the red dashed lines indicate the intramolecular NNO2⋯ElR distance. The O-atoms of the nitro moiety were constrained to those bound only to N (thus excluding structures that were coordinated to a metal). | ||
As it is known that conformation-directing effects can be relevant for medicinal chemistry,16 we wondered if there is any evidence for conformer stabilization of intramolecular π–hole interactions within nitro aromatics. To this end, we scrutinized the geometric data of small molecules that are present in the Cambridge Structure Database (CSD) and conducted computational studies on model systems.
Materials and methods
The CSD version 5.39 including two updates (until February 2018) was inspected using ConQuest version 1.21 (build 168![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 220) and all searches were limited to single crystal X-ray structures that contained 3D coordination and had an R-factor ≤ 0.1. As depicted in Fig. 1, several separate analyses were performed on datasets where the number of bonds between the nitro's N (NNO2) atom and the possibly interacting electron-rich atom (ElR, i.e., N, P, As, O, S, Se, Te, F, Cl, Br, I or At) was confined to three, four or ≥ five bonds (depending on n).
220) and all searches were limited to single crystal X-ray structures that contained 3D coordination and had an R-factor ≤ 0.1. As depicted in Fig. 1, several separate analyses were performed on datasets where the number of bonds between the nitro's N (NNO2) atom and the possibly interacting electron-rich atom (ElR, i.e., N, P, As, O, S, Se, Te, F, Cl, Br, I or At) was confined to three, four or ≥ five bonds (depending on n).
DFT calculations were performed either with the Turbomole 7.0 program (energy profiles at the PBE017-D318/def2-TZVP19 level of theory) or with the Amsterdam Density Functional (ADF)20 modelling suite at the B3LYP21-D318/TZ2P19 level of theory (no frozen cores, also used for ‘atoms in molecules’22 analyses). The def2-TZVP/TZ2P basis sets give an accurate energy at reasonable computational cost and a very low basis set superposition error (BSSE).19
Results and discussion
Three bonds of separation
The dataset involving entries with three bonds between NNO2 and ElR actually comprises substituents directly ortho to the nitro moiety. Because the distance between NNO2 and ElR is thus relatively fixed, the overlap of van der Waals shells cannot be taken as an indicator for stabilizing interactions. To evaluate these data, the angle between the aryl ring plane and the NO2 plane was measured (α). The search was done for specific substituents X, namely, o-H, o-Me, o-OCR3, o-SCR3, o-F, o-Cl, o-Br, and o-I (R = any atom). The number of CIFs and hits within these CIFs are collected in Table 1, together with the angle range that characterized most of the hits within a certain dataset (see Fig. S1† for distributions). Also shown in the table are the angle α of the energetic minimum and the barrier of α rotation for model nitro aromatics (R = H) computed with 10 degree intervals at the DFT/PBE0-D3/def2-TZVP level of theory (see Fig. S2† for full energy profiles).| X | N CIFs | N Hits | α in CSDa (DFT)b | Barrierc |
|---|---|---|---|---|
| a The CSD data were plotted at 10 degree intervals of α as shown in Fig. S1; listed in the table are the intervals with most data. b Conformational isomers were computed with 10 degree intervals of α at the DFT/PBE0-D3/def2-TZVP level of theory, as shown in Fig. S2. c Energies in kcal mol−1. All the energetic maxima were characterized by α = 90°. d In the CSD analysis, R can be any atom and in the DFT calculations R = H. These datasets were also used to generate Fig. 2. | ||||
| o-H | 16058 |
24959 |
0–10 (0) | 6.0 |
| o-CH3 | 165 | 323 | 30–50 (30) | 2.8 |
| o-OCR3 | 239 | 328 | 30–40 (40)d | 1.5 |
| o-SCR3 | 67 | 72 | 0–10 (0)d | 5.2 |
| o-F | 34 | 47 | 10–20 (20) | 2.5 |
| o-Cl | 113 | 167 | 40–50 (40) | 1.2 |
| o-Br | 29 | 136 | 80–90 (40) | 1.0 |
| o-I | 20 | 31 | 30–40 (40) | 1.2 |
For X = H, the CSD data are tightly grouped near α = 0° and DFT predicts the energetic minimum at α = 0° with a rotational barrier of 6.0 kcal mol−1. These data are congruent with the absence of any steric constraints for a co-planar arrangement of the aryl ring and the nitro group, and perhaps some stabilization of NO2⋯HC hydrogen bonding interactions. Indeed, for the more sterically demanding X = CH3, most CSD data are found near α = 40° and the minimum is computed at α = 30° with a smaller barrier of rotation (3.8 kcal mol−1). Interestingly, the distribution is similar for o-methoxy nitro arenes with the calculated minimum at α = 40°. The energetic penalty for adopting the α = 90° position is merely 1.5 kcal mol−1, half that for the sterically similar o-CH3. Unexpectedly, the data for o-SR3 are skewed to a geometry with α = 0° and DFT indeed predicts a coplanar geometry with the second highest rotational barrier of 5.2 kcal mol−1. In accordance with DFT, the o-F data are nearly co-planar (α = 10–20°) while the CSD data of the other halogens are not (α = 40–90°). The rotational barrier for all halogens is rather low (∼1 kcal mol−1), which might explain the wide distribution of CSD data (spanning nearly the whole region α = 0–90°).
The rather large difference in (α) between o-SCR3 (α = 0°) and both its O-analogue and the larger halides (α = 40°) prompted us to scrutinize the o-SCR data further. To this end, comparative heat plots were generated for α versus the C–C–O/S–CR3 torsion angle (β) for o-OCR3 and o-SCR3. From these plots shown in Fig. 2, it is evident that the data for o-SCH3 are much more tightly grouped near α = 0° and β = 180°. An energy minimization at the DFT/B3LYP-D3-TZ2P level of theory for both nitro arenes is shown at the top of Fig. 2 and corroborates the observed difference in α.
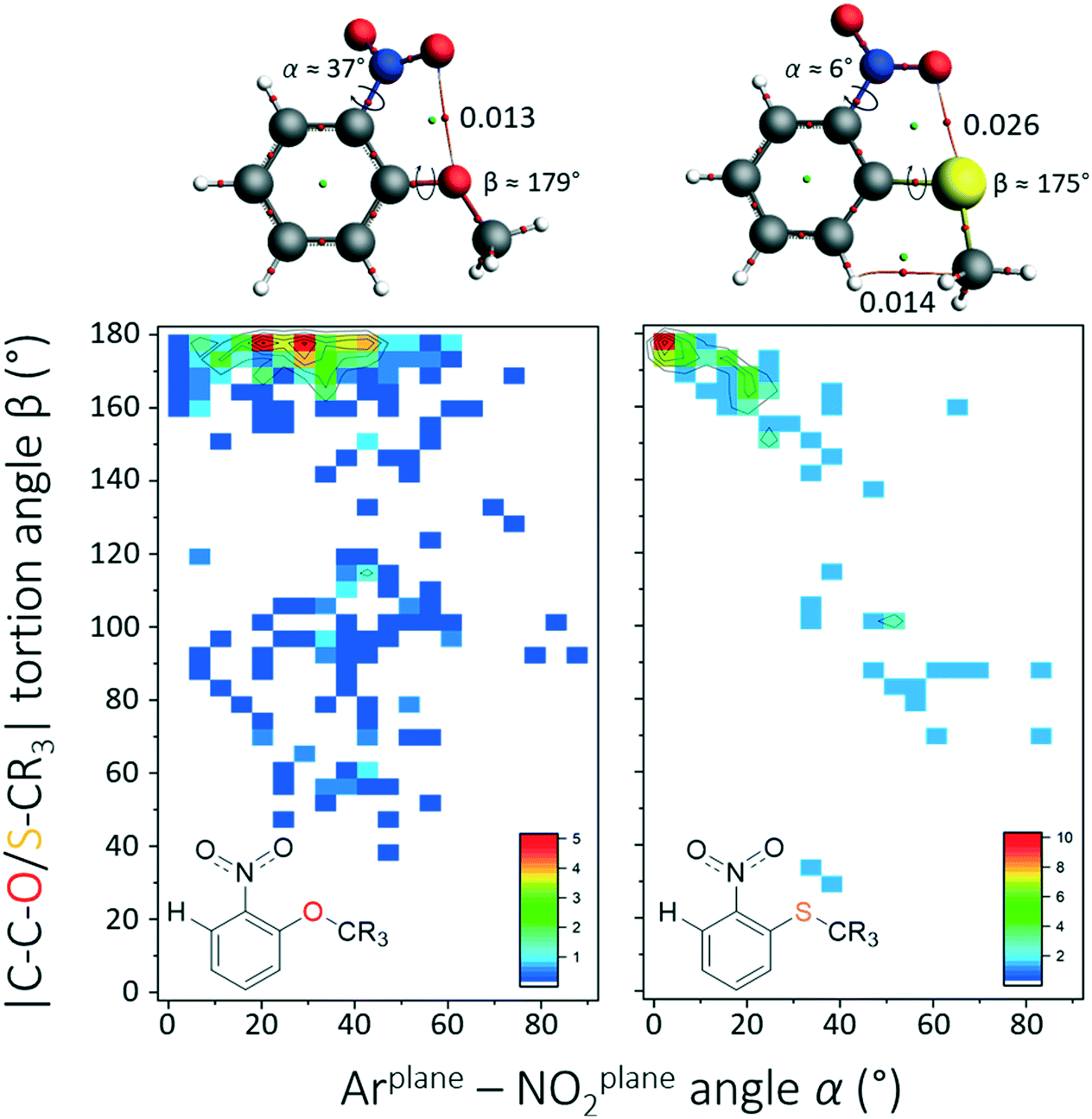 | ||
| Fig. 2 β(α) heat plots of the CSD data involving derivatives of o-methoxynitrobenzene (left, N = 328) and o-methylsulphanylnitrobenzene (right, N = 72). The colour code from blue to red is in percentages as indicated in the inset figures. The molecules displayed at the top are the energy minima at the DFT/B3LYP-D3/def2-TZ2P level of theory with accompanying atoms in molecules analysis. Indicated bond critical points are in arbitrary units. | ||
An ‘atoms in molecules’ (AIM) analysis reveals a bond critical point (bcp) between an NO2–O atom and O or S with bond densities of 0.013 and 0.026, respectively, which is indicative of a σ–hole interaction (O/S–C). The larger rotational barrier and bcp density for o-SCH3 suggest that this interaction is significant with sulphur and similar to weak hydrogen bonding interactions (similar barrier of α rotation as in nitrobenzene). This is in accordance with literature data that the larger and more polarizable S-atoms readily engage in σ–hole interactions.7b,23 Based on the above results, it is concluded that there are no intramolecular π–hole interactions within nitroarenes and electron-rich atoms that are separated by three bonds. Surprisingly, it was found that weak ONO2 → S σ–hole interactions are likely with o-methylsulphanylnitrobenzene derivatives.
Four bonds of separation
In total, 5780 hits were found within 2582 CIFs where the nitro N-atom and an electron-rich atom were four bonds removed from each other (see middle query in Fig. 1). These data were analyzed using heat plots of the van der Waals corrected NNO2⋯ElR distance d′ (i.e.: d – the van der Waals radius of N and ElR) as a function of α. Structures where X = NH (730 hits) were omitted because NO2⋯H–N hydrogen bonding interactions skewed the data to a feature at d′ ≈ 1.2 Å and α ≈ 5°(see Fig. S3†). The resulting dataset contained 5031 hits and the d′(α) heat plot is shown in Fig. 3.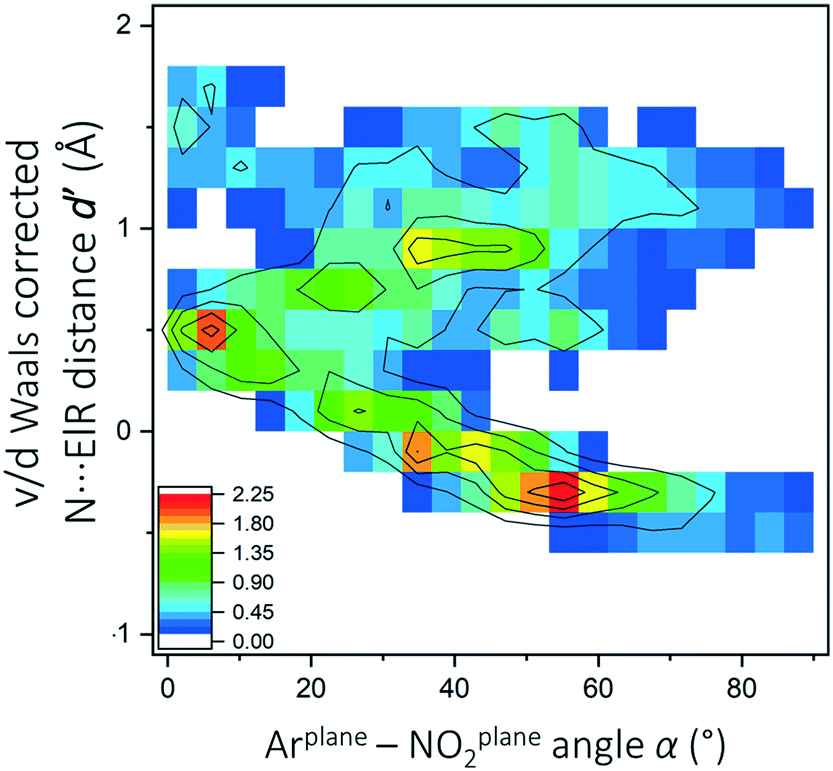 | ||
| Fig. 3 d′ (α) heat plot for data (N = 5301) obtained with query b in Fig. 1 where X ≠ NH (see also Fig. S3†). The color code represents percentages from low (blue) to high (red) as indicated in the inset figure. Contour lines were added as a guide to the eye. | ||
The data are clearly not randomly distributed and there is a correlation between α and d′ in the region 0° ≥ α ≤ 70° and −0.6 ≥ d ≤ 0.6 with a significant amount of data involved in the van der Waals overlap, particularly near α ≈ 55°. A systematic evaluation of X and ElR is detailed in Fig. S4† and revealed mostly datasets that are too small for further analysis. Shown in Fig. 4 are the d′(α) heat plots for datasets that contained a sufficient amount of data: imides (a), diazenes (b) O/N methylenes (c), amides (d), acids and esters (e) and sulfonyls (f). For model compounds, geometry optimizations and subsequent AIM analysis were performed for a conformer where ElR is pointing towards (ElR → NNO2) or away from the nitro moiety (top in each figure in Fig. 4). Energy profiles as a function of the C–C–X–ElR torsion angle were also obtained (see Fig. S5†).
 | ||
| Fig. 4 d′ (α) heat plots for: a) C = NR (N = 121 within 121 CIFs), b) N = NR (N = 93 within 51 CIFs), c) CR2OR and CR2NR2 (N = 302 within 239 CIFs), d) C(O)NR2 (N = 122 within 91 CIFs), e) CO2 (N = 847 within 538 CIFs) and f) SO2 (N = 533 within 521 CIFs). For both CO2 and SO2, only the shortest N⋯O distance was used in the plot. The color code represents percentages from low (blue) to high (red) as indicated in the inset figures. The inset molecules are simple geometry optimized models (DFT/B3LYP-D3/def2-TZ2P) starting from a conformer where ElR is pointing towards (left) or away from (right) the nitro group. Energies are in kcal mol−1 and the angles α are given in each case. The bond critical points (small red dots) are <0.018 a.u. in each case except the H-bonding interactions with the amide in (d) (0.027) and the carboxylic acid in (e) (0.042). | ||
For imide-like structures (Fig. 4a), there is a tight grouping around [1.2;30] and virtually no NNO2⋯N van der Waals overlap is present. Computations of the model imides reveal that the N → NNO2 conformer is 3.6 kcal mol−1 higher in energy than the conformer where ElR is pointing away from the nitro moiety. The observed energy minimum might well be due to weak H-bonding with the imide CH and/or a ONO2 → C = NH π–hole interaction. Similarly, most data in the dataset for diazenes (Fig. 4b) have ElR (N) pointing away from the nitro group (d′ ≈ 1 Å) and this conformer of a model diazene is indeed the most stable (1.3 kcal mol−1). The feature around [−0.25;35] is indicative of compounds with an N → NNO2 interaction geometry. That such a feature is observed for diazenes and not for imides is ascribed to the much smaller energetic difference between the two conformers. It is also consistent with the energy profiles of a rotating C–C–X–ElR torsion angle, which revealed one clear minimum for the imide but another (smaller) minimum for the diazene (see Fig. S5†).
The dataset involving O/N methylenes (Fig. 4c) again has the majority of data in a conformer that cannot have a N → NNO2 interaction at [1.4;25]. Interestingly, a small feature is present at [−0.25;55], which – on manual inspection – turned out to be mostly amines. Indeed, model computations of methylamines show that the geometry congruent with a π–hole interaction is 1.3 kcal mol−1 more stable. The reverse (also 1.3 kcal mol−1) is the case for the methoxy analogue (not shown). This is in line with the energy profiles of C–C–C–ElR rotation (Fig. S5†) which shows one true minimum for ElR = O and two minima of the same magnitude for ElR = N.
The datasets involving amides (Fig. 4d) and –CO2 moieties (Fig. 4e) are similar and display a clear d′(α) correlation with a large portion of van der Waals overlap (31% and 45%, respectively). In both cases, the most stable conformer has an O-atom pointing towards the nitro moiety. The energy difference between the conformers considered is negligible for the methylamide (0.21 kcal mol−1) and significant for the carboxylic acid (2.1 kcal mol−1). This is in line with the energy profiles of C–C–C–O rotation (Fig. S5†) revealing two minima of similar magnitude for the amide and one clear minimum for the carboxylic acid. It is noteworthy that in both cases the conformer with an intramolecular H-bonding interaction is the least stable. This is likely due to the stabilising effect of complementary N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO π–hole interactions.
O⋯CO π–hole interactions.
Nearly all the data involving sulfonyls (Fig. 4f, 78%) are involved in the van der Waals overlap and grouped around [−0.25;55]. In agreement with this, calculations of a methylsulfonyl revealed only one clear minimum (see also Fig. S5†). Starting from a geometry where the methyl group is pointing toward the nitro moiety led to the same minimum. The small feature at [0.75;55] is caused by the N-atoms of sulfonamides, which are likely pointing away from NO2 due to steric hindrance of the R-groups in –SO2NR2.
In summary, the AIM analysis never yielded a clear NNO2⋯N bcp. However, geometries congruent with intramolecular π–hole interactions involving the nitroarene are most stable when X-ElR is methylamine (−1.3 kcal mol−1), methylamide (−0.2 kcal mol−1), carboxylic acid (−2.1 kcal mol−1) or a methylsulfonyl (only minimum). These stabilizations are rather small, yet are reflected in the CSD as represented by the d′(α) plots in Fig. 4.
Five bonds of separation
Shown in the left-hand side of Fig. 5 is the d′(α) plot obtained for query c in Fig. 1 (with n = 1) where structures containing metals were omitted; metals skewed the data by acting as a bridge (X) between ElR and a nitro aromatic compound (see Fig. S6† for details). The data are clearly distributed non-randomly and the feature at [2,10] was found to be due to structures with an o-NH substituent (detailed in Fig. S6†).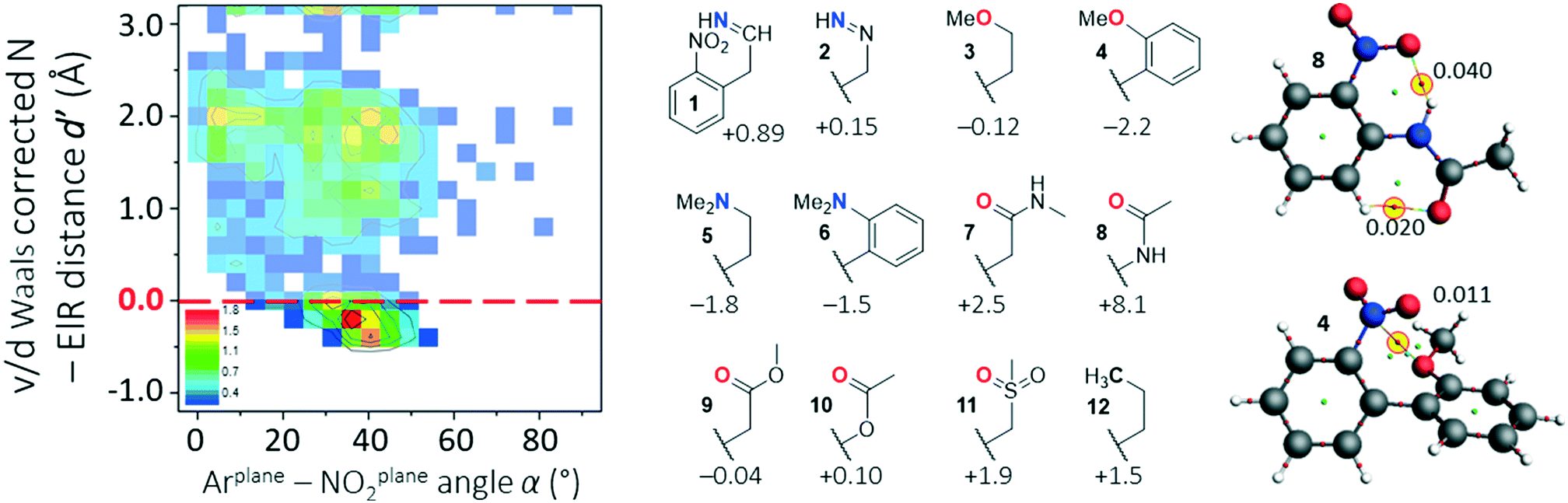 | ||
| Fig. 5 Overview of data for nitro aromatics potentially interacting with an ElR atom five bonds away from the nitro functionality. Left: d′(α) heat plot generated using query c in Fig. 1 (with n = 1, excluding structures with X = a metal; N = 2736). Middle: Several nitro aromatics that were subjected to computational analysis by geometry optimization of a conformer where ElR is pointing as far away from NNO2 as possible and one in which ElR is pointing towards NNO2. Relative energies of ElR → NNO2 geometries are given in kcal mol−1 (see also Fig. S6†). Right: AIM analysis of geometry optimized structures of 4 and 8. Calculations were done at the DFT/B3LYP-D3/def2-TZ2P level of theory. | ||
The feature around [1.8,40] cannot involve structures with ElR → NNO2 interactions while the feature at [−0.25,35] clearly can. Manual inspection of all 321 CIFs with structures where −0.25 Å ≤ d′ ≥ −0.25 Å and 20° ≤ d′ ≥ 60° revealed a very broad range of structures, none of which was numerous enough for an individualized d′(α) plot. The structures around [−0.25,35] included most of the nitro aromatics 1–12 shown in the middle of Fig. 5. Structures 1–12 have similar groups to the structures that were evaluated for data with four bonds of separation (see Fig. 4). For each of these structures, two geometry optimizations were done: one for a conformer where ElR is pointing as far away from NNO2 as possible and one in which ElR is pointing towards NNO2 (such as schematically shown in the figure, see Fig. S7† for details). The relative energies of these ElR → NNO2 interaction geometries are shown in the figure (in kcal mol−1).
Apparently, structures 1, 2, 7, 8 and 10–12 are most stable when the potentially interacting atom is pointing away from NNO2. This is most likely due to the minimization of steric hindrance (12 in particular) and/or the formation of intramolecular hydrogen bonding interactions (see also Fig. S7†). The large energy difference of 8.1 kcal mol−1 for amide 8 can be rationalized by the formation of two intramolecular hydrogen bonding interactions that form six-membered rings. This is illustrated in the right-hand side of Fig. 5 (top) by means of an AIM analysis. This analysis shows two clear bcps; one between NO2⋯HN (ρ = 0.040 a.u.) and one between CO⋯HC (ρ = 0.020 a.u.).
For nitro aromatics 3–6 and 9, the geometry congruent with an O/N → NNO2 interaction is the most stable. That 4 is the most stable (−2.2 kcal mol−1) can be rationalized by an intramolecular N⋯O π–hole interaction; indeed, an AIM analysis of 4, shown in the bottom right of Fig. 5, reveals a clear bcp between NNO2⋯O (ρ = 0.010 a.u.).
It is worth pointing out that the strain of bending the propyl chain in 12 from linearity towards the nitro moiety has an energy penalty of about 1.5 kcal mol−1. Such a penalty is likely of similar magnitude in many of the other structures considered.24 This in turn entails that, for example, the negligible stabilization computed for ether 3 (−0.12 kcal mol−1) is actually about 1.5 kcal mol−1; there is indeed a NNO2⋯O bcp found for 3 (ρ = 0.008 a.u.). Applying such a rationale to amine 5, which is already stabilized by −1.8 kcal mol−1 and has a N⋯N bcp (ρ = 0.010 a.u.), suggests that the total amount of stabilization is about −3.3 kcal mol−1. This is slightly smaller than that of the water dimer (−4.7 kcal mol−1)25 and the intermolecular adducts of nitrobenzene with dimethyl ether (−4.4 kcal mol−1) and trimethylamine (−6.2 kcal mol−1).11a Such intermolecular adducts have to cope with an intrinsic translational entropic penalty that is often ignored in computations. Despite the entropy penalty, the N → NO2 π–hole adduct between trimethylamine and nitro ethane (computed enthalpy of −5.7 kcal mol−1) has been observed with rotational spectroscopy.26 The counteracting effect of a positive translational entropy does not apply to the intramolecular π–hole interactions discussed here. Moreover, while the intramolecular π–hole interactions with nitro aromatics are weak, it is well known that weak interactions can be relevant, also in biological systems. For example: weak H–π interactions can be directional,27 such as by directing the crystal packing of indolcarbazoles;28 weak H-bonding interactions are present between water and the α-H of amino acid side chains;29 weak S⋯π+ interactions can be relevant for bio-catalysis;30 carbonyl–carbonyl π–hole interactions31 can direct protein folding;4b weak C–H⋯π interactions with carbohydrates in protein pockets32 or artificial receptors.33
In summary, there is ample evidence that π–hole interactions can stabilize conformers when the interacting atom is five bonds away for the N-atom of a nitro aromatic compound. The stabilization energy can be as large as −2.2 kcal mol−1 calculated for 4. This might actually be higher when considering that bending towards the nitro moiety involves an energy penalty of about 1.5 kcal mol−1 as calculated for 12.
Six bonds of separation
A search in the CSD for structure with six bonds between a nitro moiety and ElR (c in Fig. 1 with n = 2) resulted in merely 139 CIFs with structures too diverse to generate meaningful statistical plots. Manual inspection of these entries led to several structures with short NNO2⋯ElR distances. For two of these structures, QEPZAO and UVACAW, the experimental coordinates were truncated and a single point computation and subsequent AIM analysis were conducted as displayed in Fig. 6. In both instances, a NNO2⋯O bcp is present with the ester group. Interestingly, both optimized geometries are −1.1 kcal mol−1 more stable than their conformational isomers where the nitro aromatic compound has been rotated by 180° (thus pointing ‘down’ in Fig. 6, not shown).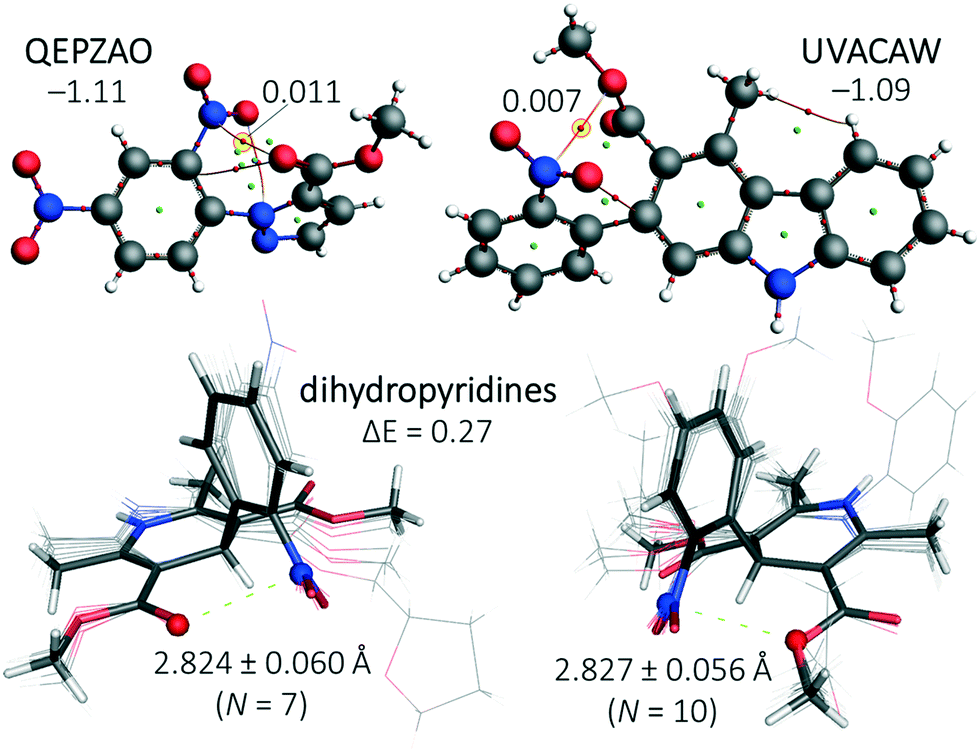 | ||
| Fig. 6 Top: Truncated structures of QEPZAO (left) and UVACAW (right) and an accompanying AIM analysis. The energies shown indicate the stabilization of the structures shown versus a conformation where the nitro aromatic compound has been flipped along the xz plane (i.e., the nitro pointing ‘downwards’, not shown). Bottom: Structural overlays of nitro aromatic dihydropyridines found within the CSD that have a close contact between the nitro N-atom and the ester carbonyl O-atom (left, involving structures ASATOD, BELHIJ, BICCIZ, JENMOH, KIXQIU, QUPRUP and YARGON01) or the sp3 O-atom of an ester (right, involving structures BICCIZ01, BICCIZ02, BICCIZ03, FULPAD, JENMUN, JENMUN01, MALFEN, PAZCUP, SUZGEA and XITMAP). Both isomers differ by merely 0.3 kcal mol−1 and an AIM analysis of their energy minima reveals a NNO2⋯O bcp with ρ = 0.01 a.u. (see Fig. S8† for details). All calculations were done at the DFT/B3LYP-D3/def2-TZ2P level of theory and relative energies are given in kcal mol−1. | ||
In addition, the CSD contained many dihydropyridine derivatives, including nifedipine (Adalat)13 and nisoldipine (Sular).14 Both are calcium channel blockers and approved by the U.S. Food and Drug Administration (FDA) to treat hypertension and angina.15 Shown in the bottom of Fig. 6 are structural overlays of two conformational isomers of nifedipine and very similar molecules where a nitro aromatic moiety is flanked by two esters.§ The nitro moiety is always in close proximity to one of the two O-atoms of an ester with short NNO2⋯O distances of (on average) 2.824 Å (with the carbonyl-O, bottom left in Fig. 6) or 2.827 Å (with an sp3 hybridized O, bottom right in Fig. 6). The energy difference between these two isomers is negligible (0.3 kcal mol−1, see Fig. S8† for details). The conformations where both carbonyl O-atoms point towards or away from the nitro aromatic are, respectively, 3.0 kcal mol−1 less and more stable than the structures shown in the bottom of Fig. 6 (see Fig. S8† for details). The most stable conformer is also found in the nifedipine ligand present within structure 6jp5,34 an Oryctolagus cuniculus calcium channel membrane protein. The observed NNO2⋯O distance of 2.58 Å is 0.49 Å within the sum of the van der Waals radii of N (1.55) + O (1.52) = 3.07 Å.35 Structures where an ElR atom and a nitro group were 7 bonds or more from one another were not scrutinized (using query c in Fig. 1 with n = 3, 4, 5 and so forth).
In summary, the CSD contains too few structures with short NNO2⋯ElR distances in structures with six or more bonds between NNO2 and ElR to perform a meaningful statistical evaluation. However, in several highlighted structures such as the drugs nifedipine and nisoldipine a stabilizing effect of an intramolecular π–hole interaction was encountered.
Summary and conclusion
No evidence was found for the stabilizing effect of NNO2⋯ElR interactions in nitro aromatic molecules when NNO2 and ElR are separated by three bonds. The d′(α) plots for such structures separated by four or five bonds clearly reflect a stabilizing effect, which was estimated by DFT to be as large as about 2–3 kcal mol−1. Several structures were highlighted where NNO2⋯ElR interactions stabilize a conformer of structures where NNO2 and ElR are six bonds apart, including the drugs nifedipine and nisoldipine. It is thus concluded that intramolecular π–hole interactions can stabilize conformers by as much as 3 kcal mol−1 when NNO2 and ElR are at least four bonds apart from one another. These conformer-directing interactions might find application in the fine-tuning of the energy minimum 3D structure of drugs or organo(metallic) catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. F. and A. F. thank the MINECO of Spain (project CTQ2017-85821-R, FEDER funds) for funding. We also thank the “Centre de Tecnologies de la Informació” (CTI) at the UIB for computational facilities. T. J. M. conducted the work with funds from the research program ‘VIDI’ with project number 723.015.006, which is financed by the Netherlands Organization for Scientific Research (NWO).References
- R. Nomura, J. Tabei and T. Masuda, J. Am. Chem. Soc., 2001, 123, 8430 CrossRef CAS PubMed.
- N. Chandramouli, Y. Ferrand, G. Lautrette, B. Kauffmann, C. D. Mackereth, M. Laguerre, D. Dubreuil and I. Huc, Nat. Chem., 2015, 7, 334 CrossRef CAS PubMed.
- (a) G. D. Rose and R. Wolfenden, Annu. Rev. Biophys. Biomol. Struct., 1993, 22, 381 CrossRef CAS PubMed; (b) D. W. Bolen and G. D. Rose, Annu. Rev. Biochem., 2008, 77, 339 CrossRef CAS PubMed.
- (a) A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244 CrossRef CAS PubMed; (b) G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615 CrossRef CAS PubMed; (c) R. W. Newberry, G. J. Bartlett, B. VanVeller, D. N. Woolfson and R. T. Raines, Protein Sci., 2014, 23, 284 CrossRef CAS PubMed.
- (a) A. R. Voth, F. A. Hays and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6188 CrossRef CAS PubMed; (b) E. Danelius, H. Andersson, P. Jarvoll, K. Lood, J. Grafenstein and M. Erdelyi, Biochemistry, 2017, 56, 3265 CrossRef CAS PubMed; (c) M. R. Scholfield, M. C. Ford, A. C. C. Carlsson, H. Butta, R. A. Mehl and P. S. Ho, Biochemistry, 2017, 56, 2794 CrossRef CAS PubMed.
- (a) S. H. Gellman, G. P. Dado, G. B. Liang and B. R. Adams, J. Am. Chem. Soc., 1991, 113, 1164 CrossRef CAS; (b) G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 1054 CrossRef CAS; (c) P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef CAS.
- (a) J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541 CrossRef CAS PubMed; (b) A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef CAS PubMed.
- A. Bauza, A. Frontera and T. J. Mooibroek, Cryst. Growth Des., 2016, 16, 5520 CrossRef CAS.
- A. Bauza, A. Frontera and T. J. Mooibroek, Nat. Commun., 2017, 8, 14522 CrossRef PubMed.
- T. J. Mooibroek, CrystEngComm, 2017, 19, 4485 RSC.
- (a) A. Bauza, T. J. Mooibroek and A. Frontera, Chem. Commun., 2015, 51, 1491 RSC; (b) A. Bauza, A. V. Sharko, G. A. Senchyk, E. B. Rusanov, A. Frontera and K. V. Domasevitch, CrystEngComm, 2017, 19, 1933 RSC.
- D. Olender, J. Zwawiak and L. Zaprutko, Pharmaceuticals, 2018, 11, 29 CrossRef PubMed.
- P. A. Poole-Wilson, J. Lubsen, B. A. Kirwan, F. J. van Dalen, G. Wagener, N. Danchin, H. Just, K. A. A. Fox, S. J. Pocock, T. C. Clayton, M. Motro, J. D. Parker, M. G. Bourassa, A. M. Dart, P. Hildebrandt, A. Hjalmarson, J. A. Kragten, G. P. Molhoek, J. E. Otterstad, R. Seabra-Gomes, J. Soler-Soler, S. Weber and A. Investigators, Lancet, 2004, 364, 849 CrossRef CAS.
- (a) R. S. Kass, J. Pharmacol. Exp. Ther., 1982, 223, 446 CAS; (b) R. O. Estacio, B. W. Jeffers, W. R. Hiatt, S. L. Biggerstaff, N. Gifford and R. W. Schrier, N. Engl. J. Med., 1998, 338, 645 CrossRef CAS PubMed.
- W. J. Elliott and C. V. S. Ram, J. Clin. Hypertens., 2011, 13, 687 CrossRef CAS PubMed.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 19 CrossRef PubMed.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931 CrossRef CAS.
- (a) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- (a) J. Skolnick and E. Helfand, J. Chem. Phys., 1980, 72, 5489 CrossRef CAS; (b) A. Bauza, T. J. Mooibroek and A. Frontera, Phys. Chem. Chem. Phys., 2014, 16, 19192 RSC; (c) K. B. Wiberg and M. A. Murcko, J. Am. Chem. Soc., 1988, 110, 8029 CrossRef CAS; (d) G. D. Smith and R. L. Jaffe, J. Phys. Chem., 1996, 100, 18718 CrossRef CAS.
- (a) P. Reinhardt and J. P. Piquemal, Int. J. Quantum Chem., 2009, 109, 3259–3267 CrossRef CAS; (b) J. M. Andric, G. V. Janjic, D. B. Ninkovic and S. D. Zaric, Phys. Chem. Chem. Phys., 2012, 14, 10896–10898 RSC.
- W. Li, L. Spada, N. Tasinato, S. Rampino, L. Evangelisti, A. Gualandi, P. G. Cozzi, S. Melandri, V. Barone and C. Puzzarini, Angew. Chem., Int. Ed., 2018, 57, 13853 CrossRef CAS PubMed.
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 8462 RSC.
- H. Jiang, P. Hu, J. Ye, A. Chaturvedi, K. K. Zhang, Y. X. Li, Y. Long, D. Fichou, C. Kloc and W. P. Hu, Angew. Chem., Int. Edit., 2018, 57, 8875–8880 CrossRef CAS PubMed.
- S. Scheiner, T. Kar and Y. L. Gu, J. Biol. Chem., 2001, 276, 9832 CrossRef CAS PubMed.
- K. E. Ranaghan, J. E. Hung, G. J. Bartlett, T. J. Mooibroek, J. N. Harvey, D. N. Woolfson, W. A. van der Donk and A. J. Mulholland, Chem. Sci., 2014, 5, 2191 RSC.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- (a) N. K. Vyas, M. N. Vyas and F. A. Quiocho, Science, 1988, 242, 1290 CrossRef CAS PubMed; (b) K. L. Hudson, G. J. Bartlett, R. C. Diehl, J. Agirre, T. Gallagher, L. L. Kiessling and D. N. Woolfson, J. Am. Chem. Soc., 2015, 137, 15152 CrossRef CAS PubMed.
- (a) T. J. Mooibroek, M. P. Crump and A. P. Davis, Org. Biomol. Chem., 2016, 14, 1930 RSC; (b) P. Rios, T. J. Mooibroek, T. S. Carter, C. Williams, M. R. Wilson, M. P. Crump and A. P. Davis, Chem. Sci., 2017, 8, 4056 RSC; (c) T. S. Carter, T. J. Mooibroek, P. F. N. Stewart, M. P. Crump, M. C. Galan and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 9311 CrossRef CAS PubMed; (d) T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS PubMed; (e) P. Rios, T. S. Carter, T. J. Mooibroek, M. P. Crump, M. Lisbjerg, M. Pittelkow, N. T. Supekar, G. J. Boons and A. P. Davis, Angew. Chem., Int. Ed., 2016, 55, 3387–3392 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce01015g |
| ‡ The Reaxys® database lists 156,991 nitro aromatic structures with available pharmacological data (April 4th 2019). |
| § Besides the structures stacked in Fig. 6, there were several simple and good quality structures of dihydropyridines where one of the esters was replaced by an H-atom (CIZQAE), an aldehyde (ECUYIL, COSCOF, XAWCAB), or a nitrile (FIJFAF), or where both esters were mutated to amides (SEMKUQ), a ketone and an amine (LOHPIH), a ketone (TELNUT) or a nitrile (DAKBAS and RIJVAH). In half of these structures, a contact distance was found. In addition to dihydropyridine derivatives, there were 12 analogues with a pyran ring (BEQYIF, IROZIY, LERZEP, MEZXOE, MIYJUA, NODXEK, QIMPEK, SAKXIO, UXUSOX, VAQHOL, VAQHUR and ZAKWIU), one with a cyclohexadiene ring (IZOMUF) and one structure where the dihydropyridine was coordinated to Cu (BEFVIS, and thus formally negatively charged). In half of these structures, a contact distance was found. |
| This journal is © The Royal Society of Chemistry 2019 |