
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheobromine cocrystals with monohydroxybenzoic acids – synthesis, X-ray structural analysis, solubility and thermal properties†
Mateusz
Gołdyn
 *,
Daria
Larowska
,
Weronika
Nowak
and
Elżbieta
Bartoszak-Adamska
*,
Daria
Larowska
,
Weronika
Nowak
and
Elżbieta
Bartoszak-Adamska
Faculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: mateusz.goldyn@amu.edu.pl
First published on 9th August 2019
Abstract
Theobromine, an organic compound from the purine alkaloid group, is much less soluble in polar solvents than its analogues, i.e. caffeine and theophylline, that is why it has been used as an active pharmaceutical ingredient (API) model in cocrystal preparation. A series of theobromine (TBR) cocrystallization processes from solutions with such coformers as 2-hydroxybenzoic acid (2HBA), 3-hydroxybenzoic acid (3HBA) and 4-hydroxybenzoic acid (4HBA) were carried out. In addition, neat grinding and liquid-assisted grinding were performed. The obtained cocrystals TBR·2HBA and TBR·3HBA as well as TBR·2(4HBA)·H2O cocrystal monohydrate have been characterized by single crystal X-ray diffraction (SXRD), PXRD, UV-vis and STA (TGA/DSC) analyses. In all cases no proton transfer from the acid molecule to the imidazole nitrogen atom in theobromine was observed. TBR–acid heterosynthons are sustained by N⋯H–O interactions, where proton donors in TBR·2HBA and TBR·3HBA are carboxylic groups, and in TBR·2(4HBA)·H2O the proton donor is the hydroxyl group of the acid molecule. In each cocrystal, TBR–TBR homosynthon R22(8) formation by N–H⋯O hydrogen bonds was observed. Acid–acid dimers are created only in the crystal lattice of TBR·2(4HBA)·H2O. In the obtained cocrystals, similar supramolecular synthons were observed, such as in theophylline and caffeine cocrystals with the same coformers. C–H⋯O and π⋯π forces present in the described structures are responsible for 2D and 3D structure stabilization.
1. Introduction
The term “cocrystal” was used for the first time by Friedrich Wöhler in 1844 for quinhydrone, which consists of quinone and hydroquinone.1 Cocrystals can be defined as homogeneous solids, composed of two or more substances (molecular and/or ionic) with a well-defined stoichiometric ratio, except simple salts and solvates. Their components have to be solids under ambient conditions.2 In 2004, a subgroup of cocrystals was defined, where one of the components is an active pharmaceutical ingredient (API).3 A new drug has to undergo a series of clinical trials, before it is placed on the market. Unfortunately, a lot of medicines (60–70%), which have good pharmacological properties, are characterized by poor aqueous solubility, which results in poor bioavailability.4–6 In this case, improvement of the physicochemical properties such as solubility, stability, permeability or tabletability is crucial from the perspective of drug companies. The long times needed for introducing drugs on the market are usually connected with higher costs for pharmaceutical companies. A lot of methods like particle size reduction,7 nanoparticle formation,8 encapsulation9 self-emulsifying drug delivery system (SEDDS),10 salt formation,11 complexation,12etc. can be used to improve the solubility of APIs in water. Moreover, cocrystallization of APIs with selected coformers is also used to improve the medicine's properties.13 Cafcit (caffeine citrate),14,15 Steglatro (ertugliflozin L-pyroglutamic acid)16,17 and Lexapro (escitalopram oxalate)18,19 are examples of medicine cocrystals. When it comes to the advantages of this method, APIs can exist in a stable crystalline form and their pharmacological activity is maintained while improving the physicochemical properties.Theobromine is an organic compound, which belongs to the purine alkaloid group. It is present in cacao, yerba mate, kola nut, the guarana berry and the tea plant.20 It is one of the metabolites formed in the human liver as a result of caffeine demethylation.21 It affects the nervous system (cAMP deactivation). It is a vasodilator and heart stimulant and it has diuretic properties.22 For these reasons, it can be classified as an API but currently it is rarely used in the pharmaceutical industry. Although the theobromine molecule is structurally similar to paraxanthine, theophylline and caffeine, it is less soluble in water than them (0.33 g L−1 for theobromine, 1 g L−1 for paraxanthine, 7.4 g L−1 for theophylline, and 21.6 g L−1 for caffeine).23 That is why theobromine was chosen as an API model for cocrystallization and as a result, its solubility in water can be improved. Monohydroxybenzoic acids were used as coformers, because they have proton-donor groups. Our choice is also related to an earlier use of these coformers for theophylline and caffeine cocrystallization (Fig. 1).24,25
 | ||
| Fig. 1 Theobromine and monohydroxybenzoic acids. | ||
In this paper, two cocrystals of theobromine (TBR) with 2-hydroxybenzoic acid (2HBA) and 3-hydroxybenzoic acid (3HBA), and a cocrystal hydrate with 4-hydroxybenzoic acid (4HBA) were reported.
All of these solids were obtained by slow evaporation from different solutions and they were analyzed by a single-crystal X-ray diffraction method. Steady-state UV-vis spectroscopy was used to determine the cocrystal solubility. Additionally, simultaneous thermal analysis (STA) measurement was carried out.
2. Experimental section
2.1. Materials
TBR (99%) was purchased from Swiss Herbal Institute. 2HBA (≥99%), 3HBA (99%) and 4HBA (≥99%) were obtained from Sigma-Aldrich. Hydroxybenzoic acids were used for cocrystallization without purification. Methanol was purchased from Chempur and ethanol from Stanlab. In all absorption experiments, Millipore distilled water (18 MΩ cm) was used.2.2. Crystallographic database CSD search
To date, 31 different structures containing theobromine have been reported and they are available in the Cambridge Structural Database.26 10 of them contain carboxylic acid, such as 5-chlorosalicylic acid (CSATBR), oxalic acid (GORGUR), trifluoroacetic acid (HIJYAB), malonic acid (HIJYEF), gallic acid (MUPPET), acetic acid (NURYUV), salicylic acid (RUTHEV), anthranilic acid (ZIZRUX and ZIZRUX01), and 4-hydroxy-3-methoxybenzoic acid (ZOYBOG). Only five of them are composed of theobromine and a benzoic acid derivative: CSATBR, MUPPET, RUTHEV, ZIZRUX, and ZOYBOG. The ConQuest program was used for searching deposited structures containing theobromine.26,272.3. Cocrystal preparation
2.4. Single-crystal X-ray diffraction (SXRD)
X-ray diffraction data were collected on an Oxford Diffraction SuperNova diffractometer equipped with a CuKα radiation source (λ = 1.54178 Å) and with a Cryojet cooling system. CrysAlisPro28 and CrysAlisRed29 were used for data collection and data reduction, respectively. Multi-scan absorption correction was applied to the diffraction data.30 Olex2 software was used as an interface to facilitate the solution, refinement and structural analysis.31 The structures were solved using intrinsic phasing with SHELXT-2015 and were refined with SHELXL-2015 software.32 For non-hydrogen atoms, refinements were carried out with anisotropic atomic displacement parameters. All hydrogen atoms were derived from a difference Fourier map and they were refined isotropically. In TBR·2(4HBA)·H2O, hydrogen atoms of two carboxyl groups are disordered over two positions and they were refined with restraints (occupancies 0.5 for H1C and H2C atoms, 0.25 for H1D and 0.75 for H2D atoms). The extinction coefficient was applied in the refinement of TBR·2HBA and TBR·3HBA structures. The crystallographic data and refinement details are presented in Tables 1 and 2.| Detection wavelength (λdet) | |
|---|---|
| TBR·2HBA | 320 |
| TBR·3HBA | 310 |
| TBR·2(4HBA)·H 2 O | 300 |
| 1 | 2 | 3 | |
|---|---|---|---|
| TBR·2HBA | TBR·3HBA | TBR·2(4HBA)·H 2 O | |
| Deposition number | 1934751 | 1934758 | 1934759 |
| Molecular formula | C7H8N4O2·C7H6O3 | C7H8N4O2·C7H6O3 | C7H8N4O2·2(C7H6O3)·H2O |
| Formula weight, g mol−1 | 318.29 | 318.29 | 474.43 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
P |
| a, Å | 6.8096(2) | 6.4812(2) | 7.0166(2) |
| b, Å | 7.9530(3) | 23.7503(7) | 12.4761(4) |
| c, Å | 14.0826(4) | 9.3288(3) | 24.6450(8) |
| α, ° | 94.042(2) | 90 | 99.639(3) |
| β, ° | 103.561(2) | 101.841(3) | 91.740(3) |
| γ, ° | 103.726(3) | 90 | 99.464(3) |
| V, Å3 | 713.78(4) | 1405.44(8) | 2094.29(12) |
| Z, Z' | 2, 1 | 4, 1 | 4, 2 |
| F(000) | 332 | 664 | 992 |
| D x , g cm−1 | 1.481 | 1.504 | 1.505 |
| Radiation, Å | 1.54184 | 1.54184 | 1.54184 |
| μ, mm−1 | 0.975 | 0.990 | 1.02 |
| T, K | 150.0(1) | 150.0(1) | 130.0(1) |
| Crystal size, mm3 | 0.57 × 0.20 × 0.18 | 0.26 × 0.11 × 0.09 | 0.21 × 0.11 × 0.05 |
| 2θ range for data collection, ° | 6.514 to 152.442 | 7.444 to 152.012 | 7.290 to 153.214 |
| Index ranges (h, k, l) | −8 ≤ h ≤ 8 | −6 ≤ h ≤ 8 | −8 ≤ h ≤ 8 |
| −10 ≤ k ≤ 9 | −29 ≤ k ≤ 29 | −15 ≤ k ≤ 14 | |
| −17 ≤ l ≤ 15 | −11 ≤ l ≤ 10 | −30 ≤ l ≤ 30 | |
| Collected reflections | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 874 874 |
8760 | 16429 |
| Independent reflections | 2986 (Rint = 0.0287, Rsigma = 0.0184) | 2911 (Rint = 0.0216, Rsigma = 0.0214) | 8559 (Rint = 0.0302, Rsigma = 0.0369) |
| Reflections with I > 2σ(I) | 2795 | 2627 | 7299 |
| Data/restraints/parameters | 2986/0/265 | 2911/0/265 | 8559/0/797 |
| Final R indices with I > 2σ(I) | R 1 = 0.0352wR2 = 0.0982 | R 1 = 0.0384wR2 = 0.1052 | R 1 = 0.0452wR2 = 0.1325 |
| Final R indices with all data | R 1 = 0.0372wR2 = 0.1003 | R 1 = 0.0437wR2 = 0.1086 | R 1 = 0.0525wR2 = 0.1388 |
| GOF | 1.046 | 1.162 | 1.067 |
| Extinction coefficient | 0.0061(13) | 0.0013(3) | None |
| Δρmin., Δρmax, e Å−3 | −0.19, 0.28 | −0.20, 0.21 | −0.33, 0.33 |
2.5. Powder X-ray diffraction (PXRD)
Powder samples of components and cocrystals synthesized by grinding and by slow evaporation from solution were measured on an Oxford Diffraction Xcalibur diffractometer with a MoKα radiation source (λ = 0.71073 Å) at room temperature. The main goal of using the grinding method was to test this technique as an alternative way for this cocrystal synthesis. Experimental conditions: scanning intervals, 5–50° (2θ); step size, 0.01°; and time per step, 0.5 s. CrysAlisPro28 was used for data collection. The experimental and calculated powder patterns from the crystal structures were analyzed using Kdif software.332.6. Solubility studies of cocrystals by steady-state absorption spectroscopy
Steady-state UV-vis spectroscopy was used to determine the cocrystal solubility in distilled water. The powdered samples of cocrystals obtained by cocrystallization from solution were used for measurements. UV-vis absorption spectra were recorded using a two-beam spectrometer Cary 100 UV-vis scanning from 200 to 800 nm with 1 nm increments. Quartz cells with an optical length of 10 mm were used. Calibration curves of every cocrystal were prepared (Fig. S1†). Substance concentrations versus absorbance of the substance at detection wavelength (Table 1) were plotted. A linear relationship was obtained and the slope was calculated from the graph.To determine the solubility of the cocrystals, saturated aqueous solutions of each were prepared. The absorbance at detection wavelength (λdet) was measured and the concentration of the substance was calculated by applying the following relationship:
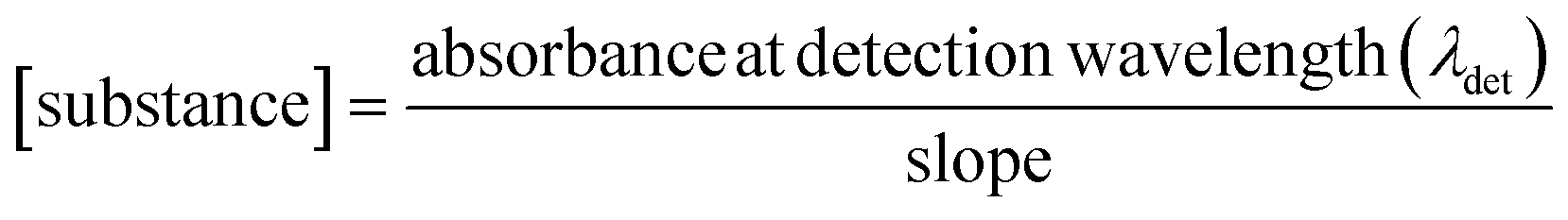 | (1) |
2.7. Simultaneous thermal analysis (STA)
The thermal properties of the samples were characterized using a STA analyser (Perkin-Elmer STA6000). The sample measurements were carried out under a nitrogen atmosphere from room temperature to 400 °C at 10 °C min−1.3. Results and discussion
In this paper, three solids containing theobromine and monohydroxybenzoic acids are presented. This dimethylxanthine cocrystallizes with 2HBA and 3HBA (1:1) as co-crystals. A hydrate was formed by combination of TBR and 4HBA (1:2). The bond lengths of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O indicate the carboxylic group geometry. The difference Fourier map clearly shows the position of the acidic hydrogen atom near the oxygen atom of the carboxyl group in 2HBA and 3HBA. There is no proton transfer from hydroxyl groups of the 4HBA molecules to the imidazole nitrogen atom of TBR (Table 3).
O indicate the carboxylic group geometry. The difference Fourier map clearly shows the position of the acidic hydrogen atom near the oxygen atom of the carboxyl group in 2HBA and 3HBA. There is no proton transfer from hydroxyl groups of the 4HBA molecules to the imidazole nitrogen atom of TBR (Table 3).
The ΔpKa parameter, described by the following equation:
| ΔpKa = pKa(base) − pKa(acid) | (2) |
allows prediction of salt or cocrystal formation.34 The determined values of ΔpKa are less than zero. So, there is a high probability that the combination of theobromine with monohydroxybenzoic acids would result in cocrystals.
3.1. Crystal structure of the investigated cocrystals
TBR·2HBA crystallizes in the triclinic space group P with one TBR and one 2HBA molecule in the asymmetric unit (Fig. 2a). Typical C–O and CO bond lengths of the carboxylic group were observed (1.310(1) Å for C7–O2 and 1.233(2) Å for C7O1), which confirmed the crystallochemical nature of this cocrystal. Each TBR molecule is hydrogen bonded to 2HBAvia O2–H2⋯N4 interaction (synthon III, Fig. 3 and Table 4). The ortho-hydroxyl group in 2HBA participates in intramolecular hydrogen bonding O3–H3⋯O1 and six-membered ring S11(6) is formed. In the crystal lattice, a finite centrosymmetric four-component system was identified (Fig. 2b). In this system two molecules of TBR interact via N1–H1⋯O4ii hydrogen bonds (synthon II).
 | ||
| Fig. 2 a) ORTEP representation of the asymmetric unit of the TBR·2HBA cocrystal (thermal ellipsoids were drawn with the 50% probability level); b) molecular layer composed of four-component centrosymmetric systems of TBR·2HBA connected by C–H⋯O hydrogen bonds; structural motifs S11(6), R22(8), R44(22) and R66(22) are marked; c) neighboring TBR·2HBA sheets represented by green and blue colors interlinked via π⋯π interactions; d) representation of stacking interaction in the TBR·2HBA cocrystal (symmetry codes: (v) 1 − x, 1 − y, 1 − z, (vi) 2 − x, 1 − y, 1 − z). | ||
 | ||
| Fig. 3 Theobromine synthons observed in the described cocrystal structures. | ||
| Cocrystal | D–H⋯A | D–H [Å] | H⋯A [Å] | D⋯A [Å] | D–H⋯A [Å] |
|---|---|---|---|---|---|
| TBR·2HBA | C6–H6⋯O5i | 0.99(2) | 2.42(2) | 3.146(2) | 130(1) |
| N1–H1⋯O4ii | 0.87(2) | 1.96(2) | 2.835(1) | 175(2) | |
| C14–H14B⋯O4iii | 0.99(2) | 2.55(2) | 3.387(2) | 142(2) | |
| C14–H14C⋯O3iv | 0.99(2) | 2.72(2) | 3.646(2) | 157(2) | |
| O3–H3⋯O1 | 0.92(2) | 1.75(2) | 2.587(1) | 149(2) | |
| O2–H2⋯N4 | 1.05(2) | 1.60(3) | 2.635(1) | 168(2) | |
| Symmetry codes: (i) −x + 1, −y, −z + 1; (ii) −x + 1, −y, −z; (iii) −x + 1, −y + 1, −z; (iv) −x + 2, −y + 2, −z + 1 | |||||
| TBR·3HBA | C5–H5⋯O3i | 0.98(2) | 2.85(2) | 3.716(2) | 148(2) |
| N1–H1⋯O4ii | 0.93(2) | 1.84(2) | 2.760(2) | 173(2) | |
| O3–H3⋯O5iii | 0.89(3) | 1.93(3) | 2.806(2) | 169(3) | |
| O2–H2⋯N4 | 0.98(3) | 1.75(3) | 2.722(2) | 177(3) | |
| Symmetry codes: (i) x, −y + 1/2, z + 1/2; (ii) −x + 3, −y + 1, −z + 1; (iii) −x + 1, −y + 1, −z | |||||
| TBR·2(4HBA)·H 2 O | O1–H1E⋯O4Ai | 0.87(3) | 1.93(3) | 2.798(2) | 173(3) |
| O1–H1F⋯O5Aii | 0.85(4) | 2.13(4) | 2.926(2) | 156(3) | |
| O2A–H2A⋯O1B | 0.89(4) | 1.68(4) | 2.564(2) | 172(4) | |
| O3A–H3A⋯O1 | 0.87(3) | 1.82(3) | 2.683(2) | 174(3) | |
| O2B–H2B⋯O1A | 0.90(4) | 1.78(4) | 2.683(2) | 176(4) | |
| O3B–H3B⋯N4A | 0.93(3) | 1.83(3) | 2.742(2) | 167(3) | |
| C5B–H5B⋯O2Aii | 0.94(3) | 2.43(3) | 3.308(2) | 155(2) | |
| C6B–H6B⋯O1Bii | 0.94(3) | 2.60(3) | 3.395(2) | 143(2) | |
| C5D–H5D⋯O2Civ | 0.96(3) | 2.40(3) | 3.253(2) | 147(2) | |
| C6D–H6D⋯O1Div | 0.98(2) | 2.53(2) | 3.344(2) | 141(2) | |
| O2C–H2C⋯O1D | 0.84(6) | 1.74(6) | 2.580(2) | 177(4) | |
| O3C–H3C⋯O2 | 0.91(4) | 1.73(3) | 2.635(2) | 173(3) | |
| O2D–H2D⋯O1C | 0.72(5) | 1.95(5) | 2.660(2) | 170(5) | |
| O3D–H3D⋯N4B | 0.87(3) | 1.95(3) | 2.804(2) | 169(3) | |
| N1B–H1B⋯O5Biii | 0.89(3) | 1.98(3) | 2.864(2) | 175(2) | |
| N1A–H1A⋯O5Avi | 0.85(3) | 2.01(3) | 2.861(2) | 178(2) | |
| O2–H2E⋯O5Biv | 0.84(4) | 2.08(4) | 2.884(2) | 163(3) | |
| O2–H2F⋯O4Bi | 0.85(3) | 1.93(3) | 2.781(2) | 173(3) | |
| C13A–H13F⋯O3Div | 0.99(3) | 2.76(3) | 3.703(3) | 158(2) | |
| C13B–H13B⋯O3Biv | 0.96(2) | 2.60(2) | 3.535(2) | 164(2) | |
| C14A–H14D⋯O4Avii | 1.00(3) | 2.56(2) | 3.252(2) | 127(2) | |
| C14B–H14A⋯O4Bv | 1.02(3) | 2.63(3) | 3.347(2) | 128(2) | |
| Symmetry codes: (i) x + 1, y, z + 1; (ii) −x + 2, −y + 1, −z + 2; (iii) −x − 1, −y + 1, −z; (iv) −x, −y + 1, −z + 1; (v) −x − 1, −y, −z; (vi) −x + 1, −y + 1, −z + 1; (vii) −x + 1, −y, −z + 1 | |||||
TBR-2HBA tetramers are connected by C–H⋯O forces creating a 2D layer parallel to the crystallographic plane (20). The oxygen atom of the endo-carbonyl group of TBR participates in C6–H6⋯O5i hydrogen bonding (Table 4), which together with the COOH⋯Nimidazole heterosynthon forms the R44(22) motif. Carbon atom C14 of the methyl group at the imidazole ring acts as a donor in C14–H14C⋯O3iv interaction, which takes part in cyclic array R66(22) formation (Fig. 2b). Two TBR molecules in neighboring sheets are connected through C14–H14B⋯O4iv hydrogen bonding. Layers are arranged in an offset manner and form stacks (Fig. 2c), which are held together by π(TBR)⋯π(2HBA) forces (Fig. 2d and Table 5).
| Cocrystal | ArM | ArN | ArM⋯ArNa [Å] | Dihedral angleb [°] | Interplanar distancec [Å] | Offsetd [Å] |
|---|---|---|---|---|---|---|
| a The distance between the ring centroids. b The angle between aromatic ring planes. c The distance between the ArN plane to the ArM centroid. d The distance between ArM and ArN projected onto the ring plane M. | ||||||
| TBR·K2HB | Ar1 | Ar3v | 3.454(1) | 2.35(1) | 3.337(1) | 0.89(1) |
| Ar2 | Ar3v | 3.578(1) | 1.96(1) | 3.348(1) | 1.26(1) | |
| Ar1 | Ar3vi | 3.633(1) | 2.35(1) | 3.332(1) | 1.45(1) | |
| Ar2 | Ar3vi | 3.714(1) | 1.96(1) | 3.322(1) | 1.66(1) | |
| Symmetry codes: (v) 1 − x, 1 − y, 1 − z, (vi) 2 − x, 1 − y, 1 – z | ||||||
| TBR·K3HB | Ar1 | Ar1iv | 3.318(1) | 0 | 3.226(1) | 0.78(1) |
| Ar2 | Ar1iv | 3.664(1) | 2.12(1) | 3.198(1) | 1.79(1) | |
| Ar2 | Ar3v | 3.756(1) | 5.52(1) | 3.224(1) | 1.93(1) | |
| Symmetry codes: (iv) 2 − x, 1 − y, 1 − z, (v) 1 + x, y, z | ||||||
| TBR·2(K4HB)·H 2 O | Ar2A | Ar3C | 3.925(1) | 10.35(1) | 3.088(1) | 2.42(1) |
| Ar2B | Ar3Aviii | 3.708(1) | 4.25(1) | 3.353(1) | 1.58(1) | |
| Symmetry codes: (viii) −2 + x, y, −1 + z | ||||||
O (1.217(2) Å) confirmed that a cocrystal was obtained. In the crystal lattice, four-component centrosymmetric motif R44(26) composed of two TBR and two 3HBA molecules is observed (Fig. 4b). This alkaloid is hydrogen bonded to two 3HBA molecules by O2–H2⋯N4 (synthon III) and O3–H3⋯O5iii (synthon V) hydrogen bonds (Fig. 3 and Table 4). TBR–TBR dimers R22(8) are held by N1–H1⋯O4ii (synthon II) interaction, where the exo-carbonyl of TBR is involved in this formation.
 | ||
| Fig. 4 a) ORTEP representation of the asymmetric unit of the TBR·3HBA cocrystal (thermal ellipsoids were plotted with the 50% probability level); b) molecular ribbon composed of structural units R44(26) connected by N–H⋯O hydrogen bonds, which in turn take part in R22(8) cyclic array formation; c) C–H⋯O interactions between acid molecules occurring on the sheet bend (Fig. 5a). | ||
The components of this cocrystal are arranged in ribbons parallel to the (52) and (15![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ) crystallographic planes, which are inclined by γ = 100.6(1)° and form the “zigzag” sheet (Fig. 5a) by C5–H5⋯O3i interactions between 3HBA molecules (Fig. 4c). The 1D ribbons form stacks, which are sustained by π(TBR)⋯π(TBR) and π(TBR)⋯π(3HBA) forces (Fig. 5b and Table 5). The average distance between neighboring layers is equal to 3.159(1) Å. The distance between equivalent TBR:3HBA is equal to 11.875(1) Å, determined as a half (1/2 W) of the O1⋯O1x,−1 + y, z distance (Fig. 5a).
) crystallographic planes, which are inclined by γ = 100.6(1)° and form the “zigzag” sheet (Fig. 5a) by C5–H5⋯O3i interactions between 3HBA molecules (Fig. 4c). The 1D ribbons form stacks, which are sustained by π(TBR)⋯π(TBR) and π(TBR)⋯π(3HBA) forces (Fig. 5b and Table 5). The average distance between neighboring layers is equal to 3.159(1) Å. The distance between equivalent TBR:3HBA is equal to 11.875(1) Å, determined as a half (1/2 W) of the O1⋯O1x,−1 + y, z distance (Fig. 5a).
 | ||
| Fig. 5 a) The “zigzag” sheet of TBR·3HBA formed by C–H⋯O interactions between ribbons, which are stacked by π⋯π interactions, b) representation of stacking interaction in TBR·3HBA (green and blue colors represent TBR and 3HBA molecules, respectively). | ||
. The asymmetric unit contains two hydrate systems (I and II, Fig. 6a). Each of the hydrates forms a 1D ribbon with the same hydrogen bond architecture (Fig. 6b). In this system, we can distinguish TBR–TBR and 4HBA–4HBA dimers R22(8) held by N–H⋯O (synthon I, Fig. 3) and O–H⋯O hydrogen bonds, respectively (Table 4). The hydroxyl group of one 4HBA molecule is connected with the imidazole nitrogen atom of TBR through O–H⋯N interaction (synthon IV), and the hydroxyl group of the second 4HBA molecule is a proton donor for the oxygen atom from the water molecule (O–H⋯O interaction). Hydrogen atoms of the solvent are hydrogen bonded to the oxygen atom from the exo-carbonyl group of one TBR molecule (synthon VIII) and from the endo-carbonyl group of the second TBR molecule (synthon VII) via O–H⋯O hydrogen bonds, respectively. In this way, together with N–H⋯O forces, motifs R23(8) are formed. In the 1D ribbon, C–H⋯O interactions are present, where aromatic carbon atoms of 4HBA molecules are proton donors for two oxygen atoms of the neighboring acid–acid dimer.
 | ||
| Fig. 6 a) ORTEP representation of the TBR·2(K4HB)·H2O asymmetric unit, which is composed of two hydrate systems (I and II; thermal ellipsoids were drawn with the 50% probability level); b) molecular layer of hydrate I formed by C–H⋯O interactions (green color) between ribbons; in ribbons C–H⋯O hydrogen bonds were identified between aromatic carbon atoms, which are proton donors for oxygen atoms participating in acid–acid dimer formation. | ||
By means of C–H⋯O hydrogen bonds, neighboring 1D polymers in TBR·2(4HBA)·H2O are arranged in layers (Fig. 6b). The methyl groups at the imidazole ring are proton donors for oxygen atoms of the exo-carbonyl groups in TBR. In the second interaction, the imidazole carbon atom is a proton donor for the 4HBA hydroxyl group connected to the water molecule. π(TBR)⋯π(K4HB) interactions (Fig. 7b and Table 5) are responsible for creating stacks (Fig. 7a).
 | ||
| Fig. 7 a) Molecular layers of TBR·2(4HBA)·H2O, which are held by π⋯π interactions (blue and green colors indicate the layers composed of I and II hydrate systems, respectively); b) representation of π(TBR)⋯π(K4HB) interactions in the TBR·2(4HBA)·H2O cocrystal hydrate. | ||
3.2. Comparison of supramolecular synthons found in theobromine, caffeine and theophylline cocrystals with monohydroxybenzoic acids
Molecules of theobromine (3,7-dimethylxanthine), theophylline (1,3-dimethylxanthine) and caffeine (1,3,7-trimethylxanthine) differ by the number and the position of the methyl group(s).Three nitrogen atoms of the caffeine molecule are substituted by methyl groups, therefore this purine molecule cannot form any CAF–CAF homodimer by strong classical hydrogen bonds. In turn, in theophylline one kind of TPH–TPH homodimer R22(10) with the participation of CO(endo-carbonyl) and N–H(imidazole) groups can be observed (Fig. 8E). However, in the theobromine molecule, there are two possibilities of TBR–TBR homosynthon R22(8) amide–amide formation via CO(exo-carbonyl) or CO(endo-carbonyl) together with the N–H(pyrimidine) group, respectively (Fig. 8B). The carboxylic acid–carboxylic acid homosynthon is popular37 (Fig. 8A), but in the presence of the alkaline imidazole nitrogen atom this hydrogen-bonded moiety is uncommon.38 Below, the supramolecular synthons in theobromine cocrystals with monohydroxybenzoic acids are discussed and compared with theophylline and caffeine cocrystals with the same coformers (Table 6).
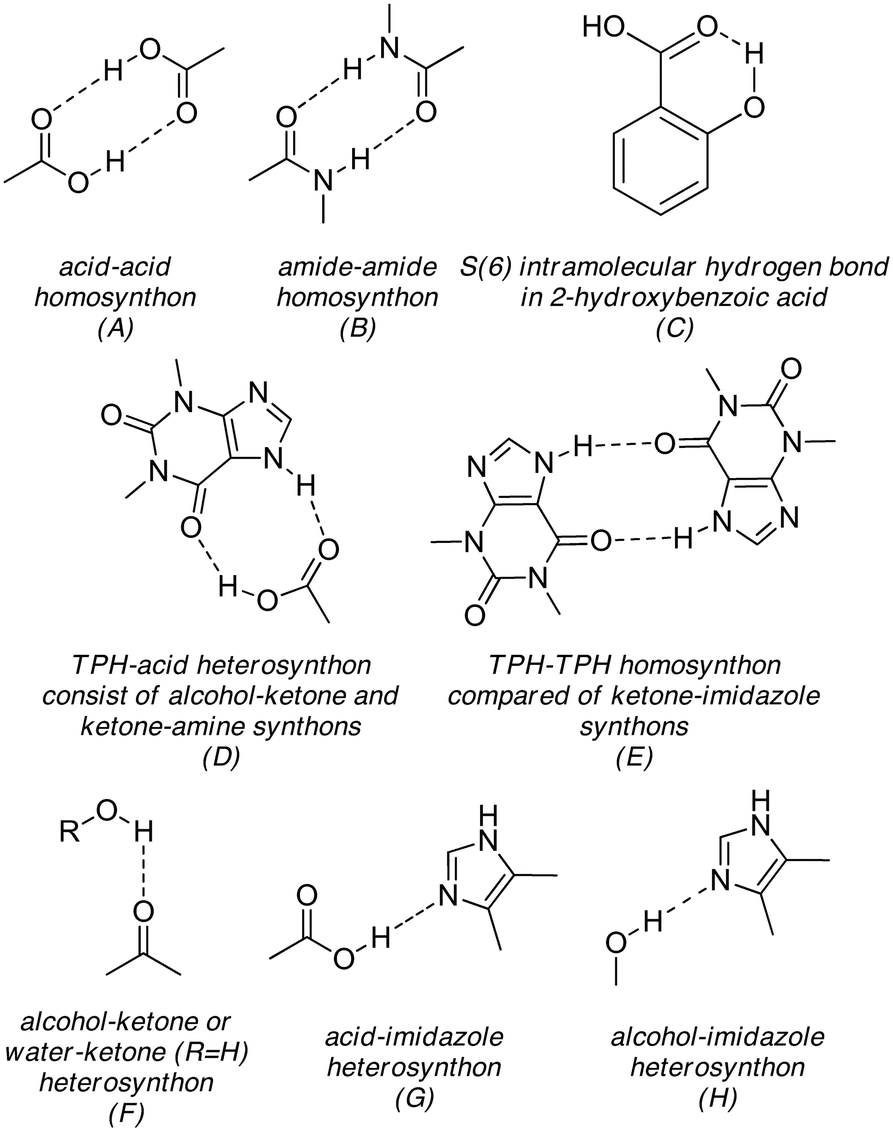 | ||
| Fig. 8 (A–H) Supramolecular synthons present in the described theobromine, theophylline and caffeine cocrystals with monohydroxybenzoic acids as coformers. A, B, F and G motifs are some of the commonly studied and used synthons in crystal design.37 | ||
| 2HBA | 3HBA | 4HBA | |
|---|---|---|---|
| Theobromine (TBR) | TBR·2HBA (RUTHEV,35 this work) | TBR·3HBA (this work) | TBR·2(4HBA)·H2O (this work) |
| Theophylline (TPH) | TPH·2HBA (KIGLES01)24 | TPH·3HBA (DOPMUS)24 | TPH·4HBA (DOPNAZ)24 |
| Caffeine (CAF) | CAF·2HBA (XOBCAT01)25 | CAF·3HBA (MOZCOU)25 | 2(CAF)·4HBA (MOZCUA)25 |
| CAF·2(4HBA) (MOZDAH)25 | |||
| CAF·4HBA·H2O (LATBIT)36 |
In the CAF·2HBA cocrystal, the alkaloid and acid molecule form a two-molecular complex by a COOH⋯N hydrogen bond (Fig. 8G). The remaining interactions are weak stacking interactions (1D and 2D structures) and C–H⋯O hydrogen bonds (3D structure).25 In the case of the TPH·2HBA cocrystal, the COOH⋯Nimidazole heterosynthon, the TPH–TPH homosynthon R22(10) (Fig. 8E) and the acid–acid heterosynthon held through O–H(o-hydroxyl)⋯OC(carboxyl) hydrogen bonds (Fig. 8F) are responsible for the 1D molecular ribbon formation. In turn, 2D and 3D architectures of TPH·2HBA are stabilized by C–H⋯O and π-stacking interactions, respectively.24 In both structures, the intramolecular O–H⋯O interaction in salicylic acid was observed. (Fig. 8C).
In the TBR·2HBA crystal lattice, TBR–TBR R22(8) dimers hydrogen bonded via N–H(pyrimidine)⋯OC(exo-carbonyl) interactions (Fig. 8B), TBR–2HBA dimers sustained by COOH⋯Nimidazole hydrogen bonds (Fig. 8G) and the S11(6) cyclic array in the 2HBA acid (Fig. 8C) are in line with expectations. These synthons are some of the most common hydrogen-bonded motifs present in organic cocrystals.37
O(exo-carbonyl)⋯H–N(imidazole) hydrogen bonds (Fig. 8E).
The structural motifs in TBR·3HBA are similar to those aforementioned in CAF and TPH analogues. The theobromine molecules form homosynthons held by N–H(pyrimidine)⋯OC(exo-carbonyl) hydrogen bonds (Fig. 8B), TBR–3HBA systems are held by COOH⋯Nimidazole interactions (Fig. 8G) and the meta-hydroxyl group in the acid is a proton donor for the oxygen atom in the endo-carbonyl group of this xanthine molecule (Fig. 8F). So, based on the caffeine and theophylline complexes with 3-hydroxybenzoic acid, it was possible to predict which synthons would be responsible for the crystal lattice arrangement in the theobromine cocrystal with the investigated coformer. The 2D and 3D networks of these cocrystals are sustained by C–H⋯O and stacking interactions.
C(carboxyl) hydrogen bonds. In TPH·4HBA, theophylline molecules are connected with one acid molecule by Nimidazole⋯HO interaction (Fig. 8H) and with the second one by O–H(carboxyl)⋯OC(exo-carbonyl) and CO(carboxyl)⋯H–N(imidazole) hydrogen bonds (R22(9) motif, Fig. 8D). Homosynthons are not observed in these cocrystals. Thus, it was difficult to predict whether the combination of theobromine and 4-hydroxybenzoic acid would result in a cocrystal or cocrystal hydrate, and what stoichiometry of substrates and which synthons would be responsible for the arrangement of these components in complex.
Theobromine and 4-hydroxybenzoic acid cocrystallize in a 1:2:1 stoichiometric ratio together with a water molecule and form a cocrystal hydrate. In this structure, we recognized two types of homosynthons, i.e. the TBR–TBR homodimer, which are held by CO(endo-carbonyl)⋯H–N(pyrimidine) hydrogen bonding (Fig. 8B) and the acid–acid homosynthon between two carboxylic groups (Fig. 8A). The imidazole nitrogen atom accepts a proton from the hydroxyl group of one acid molecule (Fig. 8H). In purine cocrystals with 2HBA and 3HBA, this synthon is not observed. The hydroxyl group of the second acid molecule is a proton donor to water molecule, which is in turn the donor of two protons to endo- and exo-carbonyl oxygen atoms of two different theobromine molecules (Fig. 8F). The last motif was observed in CAF·4HBA·H2O.36
3.3. Powder X-ray diffraction
The powder diffractograms of theobromine (API), coformers (2-, 3- and 4-hydroxybenzoic acid) and the studied cocrystals were obtained (Fig. 9–11). The differences between the diffractograms of the substrates and the cocrystals are clearly visible. What is more, the similarity of the powder patterns for the cocrystal samples obtained by crystallization from the solution and the milling samples indicates that grinding is an alternative method for the preparation of the studied theobromine derivatives. | ||
| Fig. 9 Comparison of powder X-ray diffraction patterns for theobromine, 2-hydroxybenzoic acid and the TBR·2HBA cocrystal. | ||
 | ||
| Fig. 10 Comparison of powder X-ray diffraction patterns for theobromine, 3-hydroxybenzoic acid and the TBR·3HBA cocrystal. | ||
 | ||
| Fig. 11 Comparison of powder X-ray diffraction patterns for theobromine, 4-hydroxybenzoic acid and the TBR·2(4HBA)·H2O cocrystal hydrate. | ||
3.4. Steady-state UV-vis spectroscopy
and P21/c), their architecture is comparable. In TBR·2HBA and TBR·3HBA cocrystals, an identical R22(8) synthon is observed. Additionally, similar structural motifs R44(22) and R66(22) in TBR·2HBA and R44(26) in TBR·3HBA are recognized. The main difference between the crystal structures of these cocrystals is the strength of intermolecular interactions. In the first, more soluble cocrystal, the presented motifs are formed by O–H⋯N and weak C–H⋯O forces, whereas R44(26) systems in TBR·3HBA are stabilized by strong classical O–H⋯O/N hydrogen bonds. The theobromine solubility in the TBR·2(4HBA)·H2O cocrystal hydrate is lower than that in TBR·2HBA and greater than that in TBR·3HBA. This may be due to the presence of a water molecule in the crystal lattice and more impact of strong hydrogen bonds on the molecular arrangement compared to TBR·2HBA.
3.5. Simultaneous thermal analysis (STA)
Fig. 12 shows the TGA and DSC curves of TBR·2HBA, TBR·3HBA and TBR·2(4HBA)·H2O. | ||
| Fig. 12 The simultaneous thermal analysis (STA) curves of TBR·2HBA (top), TBR·3HBA (center) and TBR·2(4HBA)·H2O (bottom). TG and DSC curves are represented by black and red colors, respectively. | ||
The presence of water in the TBR·2(4HBA)·H2O crystal structure is evident from the DSC measurements. The first signal of the cocrystal appears at a temperature of about 113 °C. It can be assumed that the water molecules from the crystal structure are released. The second signal, at 215 °C, refers to the complete decomposition of the 4HBA molecules, and is in agreement with the melting point of pure 4HBA.42 The signal at 315 °C is attributed to the decomposition of the TBR molecules. Because of the low mass content of water in the crystal structure, the weight loss in the TGA curve at about 113 °C is nearly unnoticeable. In the 104–250 °C temperature range, the loss of material reaches 32%. The second mass loss takes place in the temperature range of 250–325 °C.
In the TBR·2HBA cocrystal, the first signal is observed at around 194 °C and in the TBR·3HBA cocrystal, it is at 223 °C. At these temperatures, the appropriate monohydroxybenzoic acids decompose. Since their decomposition temperature is higher than that of the pure acids (melting temperature of pure 2HBA – 158 °C and pure 3HBA – 202 °C).42 It can be concluded that the 2HBA and 3HBA molecules are stabilized in the cocrystals. At these temperatures, a TBR·2HBA mass loss of approximately 30% can be observed in the TGA curve and the TBR·3HBA mass loss is higher and reaches about 45%, respectively. The second DSC signal is related to the theobromine decomposition.
4. Conclusions
Three solids consisting of theobromine and monohydroxybenzoic acids (ortho, meta and para) were synthesized. Good quality single crystals of TBR·2HBA, TBR·3HBA and TBR·2(4HBA)·H2O were obtained by slow evaporation from solution and were characterized by the single-crystal X-ray diffraction method. It allowed us to prove the crystallochemical nature of the new cocrystals and redetermine the TBR·2HBA crystal structure.35 Powder X-ray diffraction studies confirmed the successful green chemistry synthesis by grinding in a ball mill. Cocrystallization of theobromine improves its solubility in water approximately 6 times for the TBR·2HBA cocrystal, 5 times for the TBR·2(4HBA)·H2O cocrystal hydrate and 3 times for the TBR·3HBA cocrystal. Additionally thermal analysis confirms the presence of water molecules in the crystal lattice of TBR·2(4HBA)·H2O. In all of the compounds, the acids decompose first, followed by theobromine.Structural analysis showed that strong hydrogen bonds play a key role in the molecular arrangement in the crystal lattice of the described theobromine derivatives. In this work, the supramolecular synthons observed in the theobromine cocrystals with monohydroxybenzoic acids were discussed and they were also compared to the structural motifs in theophylline and caffeine cocrystals with the same coformers. Generally, supramolecular heterosynthons are more preferred than homosynthons.34 Our studies showed that in all of the investigated theobromine cocrystals with monohydroxybenzoic acids, the amide–amide homosynthon is present. Additionally, in the TBR·4HBA·H2O cocrystal hydrate, the acid–acid homosynthon is formed. In comparison to the theophylline cocrystals with 2HBA and 3HBA as coformers, the alkaloid–alkaloid homosynthon (TPH–TPH) is observed. Caffeine molecules do not form homosynthons and only heterosynthons are present in their cocrystals with monohydroxybenzoic acids.
The oxygen atoms of the exo- and endo-carbonyl groups are good proton acceptors. In all of the described alkaloid complexes with monohydroxybenzoic acids, at least one of these groups is a proton acceptor from the hydroxyl group or water molecule in the case of the cocrystal hydrate. The imidazole nitrogen atom accepts a proton from the carboxyl group and only in two cases (TPH·4HBA and TBR·4HBA·H2O) from the hydroxyl group. In all of the investigated structures, weak C–H⋯O hydrogen bonds and π-stacking interactions stabilize the 2D and 3D networks. The knowledge (based on the CSD data) about theophylline and caffeine cocrystals with monohydroxybenzoic acids allowed us to predict partially which supramolecular synthons would be responsible for the arrangement of theobromine and these acid molecules, when they form cocrystals.
Conflicts of interest
There are no conflicts to declare.Acknowledgement
This work was supported by grant no. POWR.03.02.00-00-I026/16 co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development.References
- F. Wöhler, Justus Liebigs Ann. Chem., 1844, 51, 145–163 CrossRef.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Commun., 2016, 52, 640–655 RSC.
- N. J. Babu and A. Nangia, Cryst. Growth Des., 2011, 11, 2662–2679 CrossRef CAS.
- S. Y. K. Fong, A. Ibisogly and A. Bauer-Brandl, Int. J. Pharm., 2015, 496, 382–391 CrossRef CAS PubMed.
- F. Kesisoglou, S. Panmai and Y. Wu, Adv. Drug Delivery Rev., 2007, 59, 631–644 CrossRef CAS PubMed.
- J. C. Chaumeil, Methods Find. Exp. Clin. Pharmacol., 1998, 20(3), 211–215 CAS.
- P. Sheth, H. Sandhu, D. Singhal, W. Malick, N. Shah and M. Serpil Kislalioglu, Curr. Drug Delivery, 2012, 9, 269–284 CrossRef CAS.
- H. Griesser, A. Schwenger and C. Richert, ChemMedChem, 2017, 12, 1759–1767 CrossRef CAS PubMed.
- C. Stillhart and M. Kuentz, J. Pharm. Biomed. Anal., 2012, 59, 29–37 CrossRef CAS PubMed.
- D. Gupta, D. Bhatia, V. Dave, V. Sutariya and S. Varghese Gupta, Molecules, 2018, 23, 1719 CrossRef PubMed.
- A. Ascenso, R. Guedes, R. Bernardino, H. Diogo, F. A. Carvalho, N. C. Santos, A. M. Silva and H. C. Marques, AAPS PharmSciTech, 2011, 12, 553–563 CrossRef CAS PubMed.
- O. N. Kavanagh, D. M. Croker, G. M. Walker and M. J. Zaworotko, Drug Discovery Today, 2019, 24, 796–804 CrossRef CAS PubMed.
- Pharmaceutical Society of Great Britain, The British Pharmaceutical Codex, Pharmaceutical Society of Great Britain, 1907 Search PubMed.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharmaceutics, 2007, 4, 347–354 CrossRef CAS PubMed.
- P. Bowles, S. J. Brenek, S. Caron, N. M. Do, M. T. Drexler, S. Duan, P. Dubé, E. C. Hansen, B. P. Jones, K. N. Jones, T. A. Ljubicic, T. W. Makowski, J. Mustakis, J. D. Nelson, M. Olivier, Z. Peng, H. H. Perfect, D. W. Place, J. A. Ragan, J. J. Salisbury, C. L. Stanchina, B. C. Vanderplas, M. E. Webster and R. M. Weekly, Org. Process Res. Dev., 2014, 18, 66–81 CrossRef CAS.
- R. Santra, B. K. R. Bhogala, C. H. Khanduri and Sun Pharmaceutical Industries Limited, US Pat. App. 15/576452, 2018 Search PubMed.
- L. Peikova, M. Manova, S. Georgieva and G. Petrova, Biotechnol. Biotechnol. Equip., 2013, 27, 4044–4047 CrossRef.
- W. T. A. Harrison, H. S. Yathirajan, S. Bindya, H. G. Anilkumar and Missing Devaraju, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63(2), o129–o131 CrossRef CAS PubMed.
- Science, http://xocoatl.org/science.htm (accessed on 17 June, 2019) Search PubMed.
- H. R. Ha, F. Follath, J. Chen and S. Krähenbühl, Eur. J. Clin. Pharmacol., 1996, 49, 309–315 CrossRef CAS PubMed.
- K. Izawa, Y. Amino, M. Kohmura, Y. Ueda and M. Kuroda, In Comprehensive Natural Products II, Elsevier, 2010, pp. 631–671 Search PubMed.
- J. K. Guillory, J. Med. Chem., 2003, 46, 4213–4213 CrossRef CAS.
- D.-K. Bučar, R. F. Henry, G. G. Z. Zhang and L. R. MacGillivray, Cryst. Growth Des., 2014, 14, 5318–5328 CrossRef.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, L. R. MacGillivray and G. G. Z. Zhang, Cryst. Growth Des., 2009, 9, 1932–1943 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- Agilent ( 2014). CrysAlis PRO. Agilent Technologies Ltd, Yarnton, Oxfordshire, England Search PubMed.
- Oxford Diffraction ( 2006). CrysAlis RED. Oxford Diffraction Ltd, Abingdon, Oxfordshire, England Search PubMed.
- G. M. Sheldrick, ( 1996). SADABS. Program for Empirical Absorption Correction, University of Gottingen, Germany Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- https://www.fzu.cz/~knizek/kalvados/ (accessed on 21 June, 2019).
- S. Kumar and A. Nanda, Indian J. Pharm. Sci., 2017, 79(6), 858–871 Search PubMed.
- F. Fischer, M. Joester, K. Rademann and F. Emmerling, Chem. – Eur. J., 2015, 21, 14969–14974 CrossRef CAS.
- S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2012, 14, 2381 RSC.
- M. K. Corpinot and D.-K. Bučar, Cryst. Growth Des., 2018, 19, 1426–1453 CrossRef.
- D.-K. Bučar, R. F. Henry, X. Lou, R. W. Duerst, T. B. Borchardt, L. R. MacGillivray and G. G. Z. Zhang, Mol. Pharmaceutics, 2007, 4, 339–346 CrossRef PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- P. Sanphui and A. Nangia, J. Chem. Sci., 2014, 126(5), 1249–1264 CrossRef CAS.
- R. M. Dannenfelser and S. H. Yalkowsky, Sci. Total Environ., 1991, 109–110(C), 625–628 CrossRef CAS.
- The Human Metabolome Database (HMDB) (accessed on 23 June, 2019) Search PubMed.
- S. H. Yalkowsky and Y. He, Handbook of Aqueous Solubility Data, CRC Press, Boca Raton, 2003, p. 377 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Steady-state absorption calibration curves of TBR·2HBA, TBR·3HBA and TBR·2(4HBA)·H2O. CCDC 1934751, 1934758 and 1934759. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce01020c |
| This journal is © The Royal Society of Chemistry 2019 |