
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Degradation of ZIF-8 in phosphate buffered saline media†
Miriam de J.
Velásquez-Hernández
 a,
Raffaele
Ricco
a,
Francesco
Carraro
a,
F. Ted
Limpoco
f,
Mercedes
Linares-Moreau
a,
Erich
Leitner
c,
Helmar
Wiltsche
c,
Johannes
Rattenberger
d,
Hartmuth
Schröttner
d,
Philipp
Frühwirt
a,
Eduard M.
Stadler
a,
Georg
Gescheidt
a,
Heinz
Amenitsch
e,
Christian J.
Doonan
*b and
Paolo
Falcaro
*ab
a,
Raffaele
Ricco
a,
Francesco
Carraro
a,
F. Ted
Limpoco
f,
Mercedes
Linares-Moreau
a,
Erich
Leitner
c,
Helmar
Wiltsche
c,
Johannes
Rattenberger
d,
Hartmuth
Schröttner
d,
Philipp
Frühwirt
a,
Eduard M.
Stadler
a,
Georg
Gescheidt
a,
Heinz
Amenitsch
e,
Christian J.
Doonan
*b and
Paolo
Falcaro
*ab
aInstitute of Physical and Theoretical Chemistry, Graz University of Technology, 8010 Graz, Austria. E-mail: paolo.falcaro@tugraz.at
bDepartment of Chemistry, The University of Adelaide, 5005 Adelaide, South Australia, Australia. E-mail: christian.doonan@adelaide.edu.au
cInstitute of Analytical Chemistry and Food Chemistry, Graz University of Technology, 8010, Graz, Austria
dGraz Centre for Electron Microscopy (ZFE), 8010 Graz, Austria
eInstitute of Inorganic Chemistry, Graz University of Technology, 8010 Graz, Austria
fOxford Instruments GmbH Asylum Research, 65205 Wiesbaden, Germany
First published on 8th June 2019
Abstract
Understanding the stability of zeolitic imidazolate framework-8 (ZIF-8) under physiological conditions is critical in biotechnology and biomedicine for biosensing, biocatalysis, and drug delivery. The use of ZIF-8 has shown that this metal organic framework (MOF) and its derived bio-composites can degrade in presence of buffer solutions. Here we present an in-depth analysis of the structural and chemical changes of pure ZIF-8 particles exposed to phosphate buffered saline (PBS) media. Two different particle sizes (2 μm and 250 nm) were selected and the decomposition operated by 10 mM PBS (aka 1X) was studied using powder X-ray diffraction (PXRD), Fourier transformed infrared spectroscopy (FTIR), time resolved atomic force microscopy (AFM), in situ small angle X-ray scattering (SAXS), and 31P NMR.
Introduction
Metal–organic frameworks (MOFs) are constructed via connecting metal ions or clusters coordinated with organic ligands into an extended network.1,2 The chemical mutability of the constituent building blocks allows for the properties of MOFs to be tailored for a variety of different applications including gas storage, catalysis, sensing, and optics.3–5 Recently, MOFs have been explored for their potential use in drug delivery, bio-storage and bio-catalysis.6–10 For example, MOFs can act as protective hosts to preserve enzymes and active pharmaceutical ingredients (APIs) from biological degradation induced by temperature, solvents and proteolytic agents.11–15 Zeolitic imidazolate framework-8 (ZIF-8), composed of tetrahedral Zn2+ ions linked by 2-methylimidazolate (mIM) ligands, is one of the most studied MOF carriers.16–20 The widespread interest in ZIF-8 can be attributed the following reasons: 1) encapsulation of APIs and biomolecules within ZIF-8 can be carried out in aqueous media,16,20 2) the loading and release efficiency can reach 100%,20 3) the protection of biomolecules in ZIF-8 can be extended to conditions that would typically result in their degradation (e.g. high temperature),21 and 4) the release of APIs and biomolecules can be controlled either by lowering the pH below 6.5 or adding chelating agents (e.g. ethylenediaminotetraacetic acid, EDTA).20These properties encouraged the investigation of ZIF-8 as a drug delivery system from small to large therapeutics (e.g. doxorubicin and insulin) for treatment of diseases ranging from cancer to diabetes.22,23 The use of ZIF-8 was extended to bio-catalytic application with ZIF particles encapsulating and protecting enzymes such as horseradish peroxidase (HRP), catalase, lipase from inhospitable environments.9 Then, protective properties of ZIF-8 were tested for the bio-preservation of viruses and yeast cells showing that the ZIF-8 coatings can be controllably assembled and dissolved.24,25 In all these examples, the use of ZIF-8 with APIs, enzymes, viruses and cells, involved MOF crystals ranging from hundreds of nanometers to micrometers.9,16,24–29 For example, enzyme@MOF particles produced via biomimetic mineralization are in the micrometer range (e.g. 2 μm).9,16,26,27
However, for drug delivery applications, particles around 250 nm are preferable as they present long blood-circulation time, and high cellular uptake, which help with the drug delivery efficiency in intravenous drug administration.10 Another important property that needs to be considered for the drug release performance of MOFs is how the material degrades under physiological conditions.30–33 Ruyra et al. demonstrated that several nanoMOFs (e.g. UiO-67, ZIF-7, HKUST-1) become amorphous or decompose releasing cations when exposed to the cell culture medium (Dulbecco's modified Eagle's medium).31 Li et al. showed that the particle size of MIL-100(Fe), and MIL-101(Fe) determined the stability and biodegradability of the MOF when exposed to phosphate buffered saline (PBS).32 The mechanism of the MOF degradation was attributed to the affinity of phosphate groups for polyvalent cations. This hypothesis was supported by the detection of insoluble metal phosphates.32 Additionally, Liang et al. proposed the degradation of ZIF-8 in PBS to explain the enzymatic activity of solutions containing catalase@ZIF-8 micrometric particles.9 In another study, Luzuriaga et al. showed that ZIF-8 can degrade in several buffer solutions suggesting that this aspect should be taken into account for biotechnological applications.33 However, the mechanism of degradation of ZIF-8 particles in PBS has not been fully elucidated and the related kinetics of decomposition remain unknown.
To shed light on the kinetics of biodegradation of ZIF-8 we present a comparative study on the stability of pure micro- and nano-ZIF-8 particles (2 μm and 250 nm) using a PBS (10 mM, aka 1X) medium under physiological pH (7.4) and temperature (37 °C). We focus on PBS as it is one of the most frequently used media to maintain physiological pH and osmolarity for biomedical dilutions in cell culture and protein chemistry.34–37 We first examined the chemical and structural behavior of pure micro-ZIF-8 with an average size of ca. 2 μm, as this is the typical size of ZIF-8-based biocatalysts.9,16,26–28 The stability of these particles was evaluated in terms of crystallinity, morphology, and chemical composition, using powder X-ray diffraction (PXRD), infrared spectroscopy (IR), and scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM/EDS). Further, the decomposition of micro-ZIF-8 particles was monitored by in situ atomic force microscopy (AFM) experiments, 31P NMR, and quantitatively determined by monitoring the concentration of 2-methylimidazole released over time, using gas chromatography-mass spectrometry (GC-MS) as analytical technique. We then focused on examining how the nano-ZIF-8 particles (average size of 250 nm) behave when exposed to PBS. Our results indicate that the decomposition of ZIF-8 particles is size-dependent (degradation was found to be faster for these smaller particles) and thus suggests that the particle dimensions must be carefully considered when designing ZIF-8-based composites for applications where controlled release is required.
Results and discussion
Synthesis of micro- and nano-ZIF-8 particles
Micro-ZIF-8 particles, ca. 2 μm, were prepared by mixing aqueous solutions of Zn(OAc)2·2H2O (15 mL, 120 mM) and 2-methylimidazole (HmIM) (30 mL, 960 mM) (see ESI† for details). Based on our previous study on how precursor stoichiometry influences the ZIF structure,38 we employed a HmIM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn2+ molar ratio of 16:1 to yield pure sod-ZIF-8. Furthermore, the crystal size was controlled by modifying the concentration of the MOF precursors in the reaction mixture. Thus, nano-ZIF-8 particles, ca. 250 nm, were achieved by increasing the concentration of Zn(OAc)2·2H2O (10 mL, 240 mM) and HmIM (20 mL, 1920 mM). Close analysis of the SEM reveals that both micro- and nano-ZIF-8 crystals present a rhombic dodecahedron morphology typical of sod-ZIF-8 (Fig. S3a and b, ESI†). The phase purity of both samples was confirmed by powder X-ray diffraction (PXRD) analysis (Fig. S3c and d, ESI†).
:Zn2+ molar ratio of 16:1 to yield pure sod-ZIF-8. Furthermore, the crystal size was controlled by modifying the concentration of the MOF precursors in the reaction mixture. Thus, nano-ZIF-8 particles, ca. 250 nm, were achieved by increasing the concentration of Zn(OAc)2·2H2O (10 mL, 240 mM) and HmIM (20 mL, 1920 mM). Close analysis of the SEM reveals that both micro- and nano-ZIF-8 crystals present a rhombic dodecahedron morphology typical of sod-ZIF-8 (Fig. S3a and b, ESI†). The phase purity of both samples was confirmed by powder X-ray diffraction (PXRD) analysis (Fig. S3c and d, ESI†).
Analysis of the stability of micro-ZIF-8 and nano-ZIF-8 particles
PBS (1X) closely mimics the pH, osmolarity, and ion concentrations of the human body. Therefore, this buffer is widely used as a medium in biological research.34–37 In this realm, PBS has been widely employed in therapeutic in vitro trials to evaluate the suitability of MOFs as drug carriers,22,39–41 and has also been used for standard enzymatic tests applied to active MOF biocomposites.42–49 Accordingly, a thorough understanding of how PBS buffer affects the stability of ZIF-8, including the kinetics of decomposition and resulting by-products, is important not only for the design of drug carriers with controlled release of their corresponding cargo, but also for the interpretation of bio-catalytic activity data for enzyme@MOF composites. To this end, micro-ZIF-8 (0.5 mg mL−1) particles were incubated in PBS (1X) buffer media, under physiological conditions of pH (7.4) and temperature (37 °C), for 1 h, 3 h, 6 h, and 24 h (Fig. 1). The concentration of the micro-ZIF-8 particles used in this study was based on: (1) the cytotoxicity threshold concentration of ZIF-8 (30 μg mL−1),50 (2) the minimum amount required to enable the recollection of the remaining ZIF-8 particles upon 24 h of treatment with PBS (see ESI† for details). The effect of the PBS was monitored by recovering the solid, via filtration, at different incubation time points (see ESI† for details). The solid collected after the incubation of micro-ZIF-8 particles for 24 hours in PBS was analyzed by SEM. The data revealed that the initial rhombic dodecahedron morphology was transformed into clusters of spherical shaped nanoparticles (Fig. 1a and b).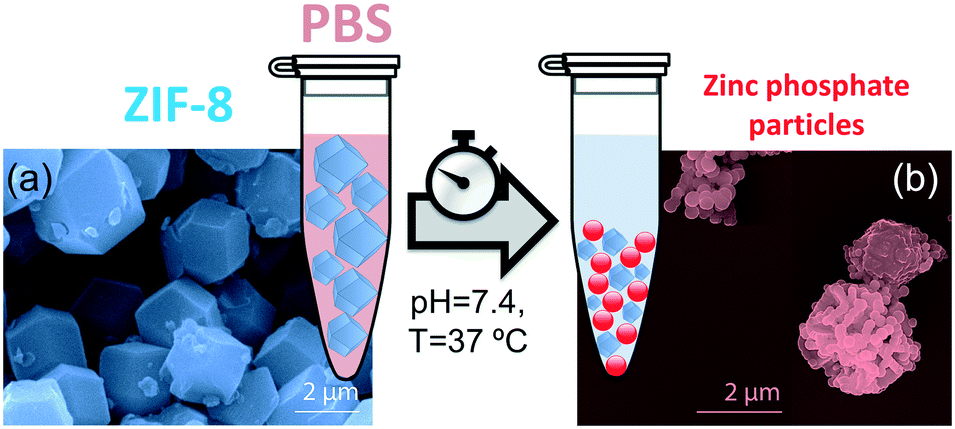 | ||
| Fig. 1 Incubation process of ZIF-8 particles in phosphate buffered saline media (PBS) under physiological conditions of pH (7.4) and temperature (37 °C). (a) SEM image of as-synthesized micro-ZIF-8 particles. (b) SEM image of micro-ZIF-8 particles after being soaked in PBS for 24 h. | ||
Then, PXRD technique was employed to evaluate the relative crystallinity of the micro-ZIF-8 particles upon their treatment in PBS. The measurements were performed adding a known quantity (20% wt) of commercial TiO2 (anatase) as internal standard (Fig. 2a). This method allows us to exclude the possibility that the decrease in intensity of the diffraction peaks resulted from fluctuations in the mass of the crystalline material from sample to sample. The diffraction patterns were baseline corrected. Then, the peak corresponding to the (101) anatase (2θ = 25.3°)51 was fitted with a Gaussian curve, and the intensity normalized (see ESI† for details).
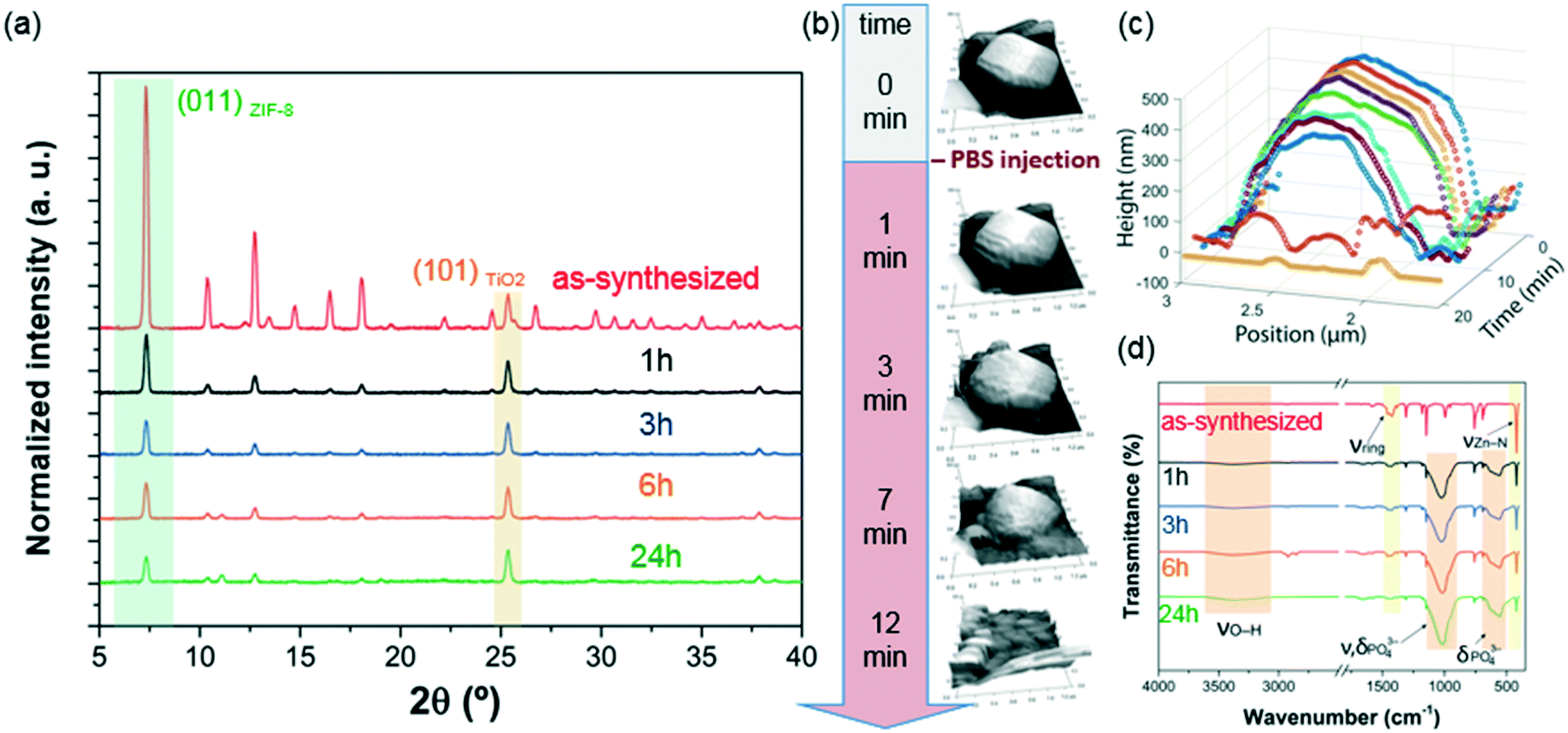 | ||
| Fig. 2 (a) PXRD patterns illustrating the structural evolution of micro-ZIF-8 particles before and after the incubation process in PBS. (b) Evaluation of the degradation process followed by in situ AFM experiments. (c) Height profiles extracted from (b), showing the morphological evolution of a single micrometric ZIF-8 particle upon the incubation in PBS (the first 6 min the measurements were performed in deionized (DI) water as incubation media). (d) Comparative FTIR spectroscopic analysis of the micro-ZIF-8 particles before and after the treatment with PBS. | ||
We found that the normalized intensity of the peak arising from the (011) plane of sod-micro-ZIF-8 crystal decreased with incubation time in PBS (Fig. 2). Specifically, after 1 h the intensity drops to 26% of its original value, and only 11% after 24 h (Table S2, ESI†). This loss in peak intensity suggests that, under these conditions, the long range order is lost as MOF crystals are decomposing.52 The PXRD patterns collected after the incubation process do not present new diffraction peaks suggesting that by-products are amorphous.
To monitor the morphological changes of micro-ZIF-8 particles during immersion in PBS, a polycrystalline ZIF-8 coating was fluxed with 50 μL min−1 of buffer solution within a fluidic cell. An in situ real-time AFM study was performed to examine the changes in morphology (ESI† Movie.mp4). Selected 3D AFM images are shown in Fig. 2b where a single micro-ZIF-8 particle is tracked over time. A change in morphology is clearly evident within the first 3 minutes of incubation; the initial sharp edges of the rhombic dodecahedron crystal became progressively rounded. Within 9 minutes, a 20% reduction in the original height of the ZIF-8 crystal can be observed, and in less than 15 minutes the crystal is almost completely dissolved (Fig. 2c). This qualitative measurement demonstrates the rapid decomposition of micro-ZIF-8 particles once exposed to 10 mM PBS media.
Then, we performed FTIR spectroscopy to assess changes in the atomic connectivity upon exposure to PBS. Fig. 2d shows that significant changes in the vibrational bands occur when the as-synthesized micro-ZIF-8 particles are immersed in PBS. For example, a progressive decrease in the peak intensity of vibration modes related to HmIM (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, 1584 cm−1; νring, 1500–1350 cm−1; and δout-of-plane bending, 800–650 cm−1) is evident. Furthermore, the spectra show a progressive reduction in the intensity of the band attributed to the Zn–N stretching mode (421 cm−1). These data suggest that the degradation process of ZIF-8 involves the release of the ligand with a change in the coordination environment of the Zn2+ ions. In addition, new vibrational modes were detected at 1160–900 cm−1 and 660–530 cm−1 with increasing intensity over time. Given the composition of the solution, and previously reported infrared studies of amorphous zinc phosphate nanospheres,53 the broad bands observed at 1160–900 cm−1 could be ascribed to the antisymmetric stretching mode of PO43− with the shoulder peak at 956 cm−1 originating from the P–O bending mode, and the broad band at 660–530 cm−1 is engendered by bending of PO43− groups. Considering the high affinity of phosphate groups for polyvalent cations (e.g. Zn2+),54 it may be proposed that the new bands in the FTIR spectra are the result of zinc phosphates formed as degradation by-products of the micro-ZIF-8 crystals.
N, 1584 cm−1; νring, 1500–1350 cm−1; and δout-of-plane bending, 800–650 cm−1) is evident. Furthermore, the spectra show a progressive reduction in the intensity of the band attributed to the Zn–N stretching mode (421 cm−1). These data suggest that the degradation process of ZIF-8 involves the release of the ligand with a change in the coordination environment of the Zn2+ ions. In addition, new vibrational modes were detected at 1160–900 cm−1 and 660–530 cm−1 with increasing intensity over time. Given the composition of the solution, and previously reported infrared studies of amorphous zinc phosphate nanospheres,53 the broad bands observed at 1160–900 cm−1 could be ascribed to the antisymmetric stretching mode of PO43− with the shoulder peak at 956 cm−1 originating from the P–O bending mode, and the broad band at 660–530 cm−1 is engendered by bending of PO43− groups. Considering the high affinity of phosphate groups for polyvalent cations (e.g. Zn2+),54 it may be proposed that the new bands in the FTIR spectra are the result of zinc phosphates formed as degradation by-products of the micro-ZIF-8 crystals.
To ascertain the presence of zinc phosphates, we performed EDX analysis (Fig. 3a and b). The elemental maps of the samples before and after exposure to PBS show a significant change in the elemental composition and distribution. The as-synthesized micro-ZIF-8 particles present a uniform distribution of Zn and N within the micro-sized particles and no evidence of P (Fig. 3a). However, after the incubation in PBS, we found that while the N signal was negligible, Zn was present as a constituent of solid aggregates. Furthermore, these aggregates showed a local concentration of O co-localized with P (Fig. 3b).
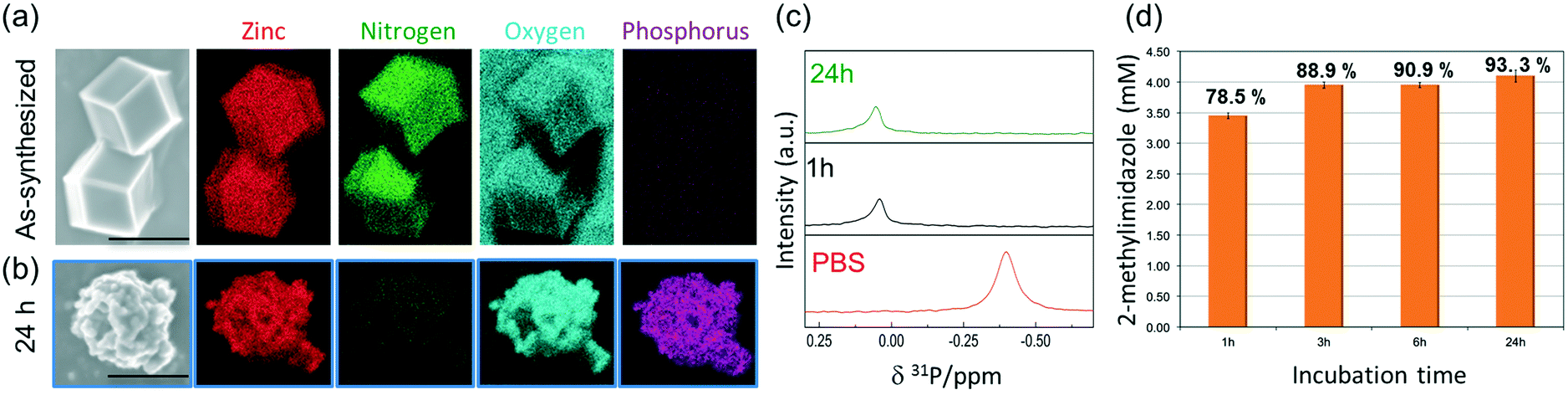 | ||
| Fig. 3 (a) EDX elemental maps of the as-synthesized ZIF-8 microcrystalline powder. (b) EDX elemental maps of the powder recovered after the incubation process in PBS for 24 h. (c) 31P NMR of PBS prepared in D2O before (lowest trace) and after adding micro-ZIF-8 particles (0.5 mg mL−1, 1 and 24 h, middle and upper trace, resp.). (d) Quantitative determination of 2-methylimidazole released after the incubation process in PBS (1 h, 3 h, 6 h and 24 h). | ||
To gain an insight into the degradation process we monitored the 31P NMR resonance of a PBS solution (prepared with D2O) after adding ZIF-8 (Fig. 3c). The intensity of the signal associated to phosphate in PBS decreases upon the addition of ZIF-8, accompanied by a small downfield shift of 0.44 ppm. Importantly, no other 31P peaks appeared in spectra recorded after 1 and 24 h (Fig. 3c). This observation can be explained by the formation of insoluble zinc phosphates, which escape from NMR detection (see Fig. S1†).54 Insoluble zinc phosphates, that are formed during the degradation, can be removed by filtration, as confirmed by the low concentration of Zn2+ in the supernatant determined by ICP-OES (see Table S1†).
These findings clarify the mechanism of the degradation process, in which the coordination equilibrium, between Zn2+ ions and HmIM in solution, is altered by the presence of phosphate species. Such phosphates have a high affinity for Lewis metal centers shifting the equilibrium towards the formation of insoluble inorganic by-products. Our data suggest that the competition of phosphate species for the metallic centers progressively releases HmIM in solution. This process might be also favored by the pH conditions imposed by the buffer media (pH = 7.4). Indeed, under these conditions, the ligand exists in solution as a protonated specie (pKa1 = 7.85; pKa2 = 15.1),55 which reduces the complexing power of the linker toward the cation.
A quantitative determination of the degradation process was achieved by measuring the concentration of HmIM released in the mother liquors by gas chromatography-mass spectrometry (GC-MS) (Fig. 3d) after 1, 3, 6 and 24 h exposure of ZIF-8 to PBS. The collected data show that the most significant changes occur within the first hour of treatment with PBS, as around 79% of the HmIM is released into the incubation media. The release of HmIM after 3 h and 6 h, respectively, of treatment in PBS is comparatively less significant. After 24 h incubation, ca. 93% of HmIM is present in the mother liquors thus supporting the interpretation of the PXRD and 31P NMR data. Given the relevance of nanoMOFs for biomedical applications,10 we extended the analysis to evaluate the stability of the nano-ZIF-8 particles with average size of 250 nm, incubated in PBS, following a procedure analogous to that described for micro-ZIF-8 samples (see ESI† for details). The degradation process was monitored up to 72 h, which is a suitable period to assess the feasibility of these systems for long-term drug delivery applications.10
The PXRD patterns and FTIR data indicate that the nano-ZIF-8 particles experienced a faster degradation than micro-ZIF-8 particles. This is evidenced by the rapid decrease in the intensity of both the Bragg peaks of sod-ZIF-8, and the bands associated to Zn–N stretch mode (421 cm−1) (Fig. 4a and b). Furthermore, FTIR confirms the formation of zinc phosphate species for nano-ZIF-8 exposed to PBS. We performed time-resolved SAXS experiments to follow the rapid degradation of the nano-sized particles, by monitoring a nano-ZIF-8 dispersion in PBS (10 mM) and in DI water as a control. The evolution of the diffraction peak attributed to the plane (011) sod-ZIF-8 was recorded with a 3 min time resolution (Fig. 4c). These data, together with the simultaneous decrease of the scattering intensity at low angles, confirm the instability of nano-ZIF-8 particles in the PBS solution: the crystallinity is completely lost within 40 min (see ESI† for details). Conversely, the intensity of the (011) diffraction peak of sod-ZIF-8 particles dispersed in DI water is almost unchanged after a 5 h exposure (Fig. S2†). These observations were quantitatively corroborated by GC-MS to determine the concentration of HmIM in the mother liquors (Fig. 4d). Importantly, the comparative results between the micro- and nano-ZIF-8 particles show that the concentration of ligand in solution for the nano-ZIF-8 is 10% larger for 1 h and 24 h incubation. This is supported by the AFM images, micro-ZIF-8 particles that show etching of the external surface after only one minute. Thus, the faster degradation of nano-ZIF-8 particles can be explained by their larger surface area exposed to the PBS media. We also investigated the morphology of nano-ZIF-8 before and after 24 h of treatment in PBS. SEM micrographs show a transformation from original rhombic dodecahedron morphology to clusters of spherical nanoparticles (Fig. 4e and f). The EDX elemental maps analysis confirmed the presence of Zn, O, and P in such clusters; however, N was not detected. These experiments evidenced that nano-ZIF-8 degrades into zinc phosphate species as in the case of micro-ZIF-8. However, we note that the kinetics of degradation was slower for large particles and faster for smaller ZIF-8 crystals.
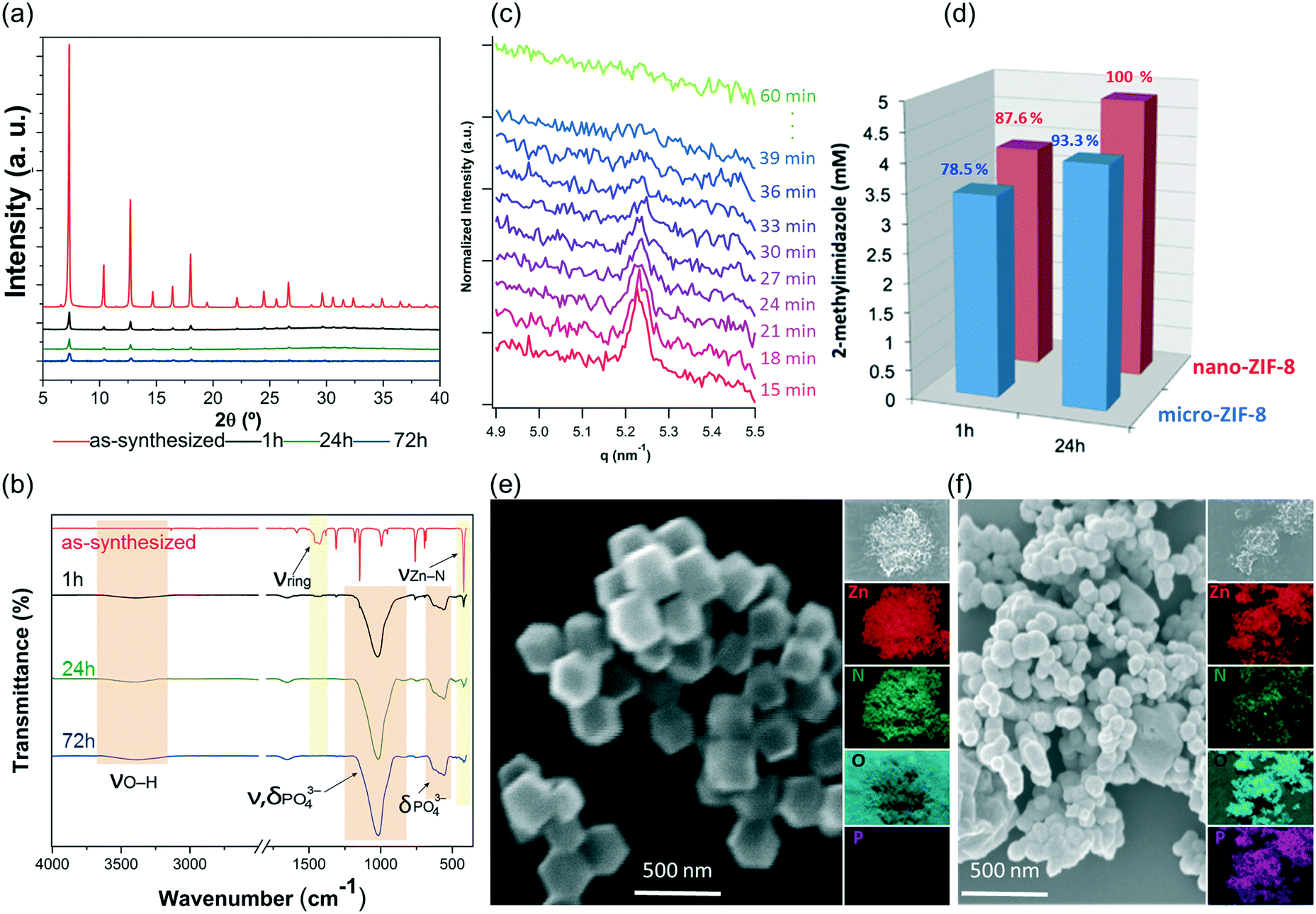 | ||
| Fig. 4 (a) PXRD diffractograms of nano-ZIF-8 particles before and after the incubation process in PBS (1 h, 24 h, and 72 h). (b) FTIR spectroscopy of the micro-ZIF-8 particles before and after the treatment with PBS. (c) Time-resolved SAXS of nano-ZIF-8 particles dispersed in PBS (1X) solution. The reported q range (4.9–5.5 nm−1) highlights the time-evolution of the intensity of the (011) diffraction peak of sod-ZIF-8. (d) Quantitative comparison of the degradation process suffered by nano- and micro-ZIF-8 particles by the determination of the concentration of 2-methylimidaloze in the mother liquors. (e) SEM micrographs, and EDX elemental mapping (Zn, N, O, and P) images of the as-synthesized nano-ZIF-8 particles. (f) SEM and EDX images of the powder recovered upon the treatment with PBS for 24 h. | ||
Conclusions
In this study, we demonstrated that ZIF-8 particles can rapidly decompose when immersed in PBS (10 mM). The decomposition of the sod-Zn(mIM)2 crystalline network leads to the formation of zinc phosphate insoluble particles. The kinetic of decomposition depends on ZIF-8 particles size and is faster for smaller particles. The current elucidation of the decomposition mechanism and by-products can be relevant for biochemical processes. In case of bio-composites for bio-catalysis (e.g. enzyme@MOF), the potential release of enzymes in buffer solution should be taken into consideration, especially when the precise quantification of the enzymatic activity is needed (e.g. recyclability of the biocatalyst). In case of drug release, phosphates are essential components in intravascular, interstitial and intracellular fluids.34,56,57 Therefore, it is possible to take advantage of the biodegradability of ZIF-8, induced by the presence of phosphates, to stimulate the release of APIs incorporated within the porous network.Conflicts of interest
The authors confirm that there are no conflicts to declare.Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Union's Horizon 2020 Programme (FP/2014-2020)/ERC Grant Agreement no. 771834 – POPCRYSTAL. The authors acknowledge support from the European Union's Horizon 2020 FETOPEN-1-2016-2017 research and innovation program under grant agreement 801464. M. J. V. H. acknowledges The National Council of Science and Technology (CONACyT, México) for the postdoctoral scholarship (CVU 419210). R. R. acknowledges the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement #748649 (project “MNEMONIC”). P. F. acknowledges TU Graz for the Lead Project (LP-03). Dr. Florian Johann (Oxford Instruments GmbH) is acknowledged for his technical assistance with the AFM measurements. The authors acknowledge the CERIC-ERIC Consortium for the access to experimental facilities and financial support. P. F., E. M. S., G. G. acknowledge NAWI Graz for support.References
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- C. S. Diercks, M. J. Kalmutzki, N. J. Diercks and O. M. Yaghi, ACS Cent. Sci., 2018, 4, 1457–1464 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed.
- W. Chen and C. Wu, Dalton Trans., 2018, 47, 2114–2133 RSC.
- Z. Zhao, J. Pang, W. Liu, T. Lin, F. Ye and S. Zhao, Microchim. Acta, 2019, 186, 295 CrossRef PubMed.
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed.
- Y. Chen, P. Li, J. A. Modica, R. J. Drout and O. K. Farha, J. Am. Chem. Soc., 2018, 140, 5678–5681 CrossRef CAS PubMed.
- C. Doonan, R. Riccò, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed.
- M. Giménez-Marqués, T. Hidalgo, C. Serre and P. Horcajada, Coord. Chem. Rev., 2016, 307, 342–360 CrossRef.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J.-S. Chang, Y. K. Hwang, V. Marsaud, P.-N. Bories, L. Cynober, S. Gil, G. Férey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- M. H. Teplensky, M. Fantham, P. Li, T. C. Wang, J. P. Mehta, L. J. Young, P. Z. Moghadam, J. T. Hupp, O. K. Farha, C. F. Kaminski and D. Fairen-Jimenez, J. Am. Chem. Soc., 2017, 139, 7522–7532 CrossRef CAS PubMed.
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed.
- C. Wang, G. Sudlow, Z. Wang, S. Cao, Q. Jiang, A. Neiner, J. J. Morrissey, E. D. Kharasch, S. Achilefu and S. Singamaneni, Adv. Healthcare Mater., 2018, 7, 1800950 CrossRef PubMed.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed.
- F.-S. Liao, W.-S. Lo, Y.-S. Hsu, C.-C. Wu, S.-C. Wang, F.-K. Shieh, J. V. Morabito, L.-Y. Chou, K. C.-W. Wu and C.-K. Tsung, J. Am. Chem. Soc., 2017, 139, 6530–6533 CrossRef CAS PubMed.
- E. Astria, M. Thonhofer, R. Ricco, W. Liang, A. Chemelli, A. Tarzia, K. Alt, C. E. Hagemeyer, J. Rattenberger, H. Schroettner, T. Wrodnigg, H. Amenitsch, D. M. Huang, C. J. Doonan and P. Falcaro, Mater. Horiz., 2019, 6, 969–977 RSC.
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- H. Zheng, Y. Zhang, L. Liu, W. Wan, P. Guo, A. M. Nyström and X. Zou, J. Am. Chem. Soc., 2016, 138, 962–968 CrossRef CAS PubMed.
- W.-H. Chen, G.-F. Luo, M. Vázquez-González, R. Cazelles, Y. S. Sohn, R. Nechushtai, Y. Mandel and I. Willner, ACS Nano, 2018, 12, 7538–7545 CrossRef CAS PubMed.
- S. Li, M. Dharmarwardana, R. P. Welch, C. E. Benjamin, A. M. Shamir, S. O. Nielsen and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2018, 10, 18161–18169 CrossRef CAS PubMed.
- R. Riccò, W. Liang, S. Li, J. J. Gassensmith, F. Caruso, C. Doonan and P. Falcaro, ACS Nano, 2018, 12, 13–23 CrossRef PubMed.
- Q. Wang, X. Zhang, L. Huang, Z. Zhang and S. Dong, Angew. Chem., Int. Ed., 2017, 56, 16082–16085 CrossRef CAS PubMed.
- F. Pitzalis, C. Carucci, M. Naseri, L. Fotouhi, E. Magner and A. Salis, ChemCatChem, 2018, 10, 1578–1585 CrossRef CAS.
- W. Liang, R. Ricco, N. K. Maddigan, R. P. Dickinson, H. Xu, Q. Li, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Mater., 2018, 30, 1069–1077 CrossRef CAS.
- M. A. Luzuriaga, R. P. Welch, M. Dharmarwardana, C. E. Benjamin, S. Li, A. Shahrivarkevishahi, S. Popal, L. H. Tuong, C. T. Creswell and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2019, 11, 9740–9746 CrossRef CAS PubMed.
- E. Bellido, M. Guillevic, T. Hidalgo, M. J. Santander-Ortega, C. Serre and P. Horcajada, Langmuir, 2014, 30, 5911–5920 CrossRef CAS PubMed.
- À. Ruyra, A. Yazdi, J. Espín, A. Carné-Sánchez, N. Roher, J. Lorenzo, I. Imaz and D. Maspoch, Chem. – Eur. J., 2015, 21, 2508–2518 CrossRef PubMed.
- X. Li, L. Lachmanski, S. Safi, S. Sene, C. Serre, J. M. Grenèche, J. Zhang and R. Gref, Sci. Rep., 2017, 7, 13142 CrossRef CAS PubMed.
- M. A. Luzuriaga, C. E. Benjamin, M. W. Gaertner, H. Lee, F. C. Herbert, S. Mallick and J. J. Gassensmith, Supramol. Chem., 2019, 0, 1–6 CrossRef CAS.
- D. G. Fasman, Handbook of biochemistry and molecular biology. Physical and Chemical Data, CRC Press, Boca Raton, Florida, 3rd edn, 1975, vol. 1 Search PubMed.
- Cell biology: a laboratory handbook, ed. J. E. Celis, Academic Press, San Diego, 2nd edn, 1998 Search PubMed.
- M. N. Gupta, Methods for affinity-based separations of enzymes and proteins, 2002 Search PubMed.
- D. C. White, Proteins, Peptides, and Amino Acids SourceBook, Springer, 2002 Search PubMed.
- W. Liang, R. Ricco, N. K. Maddigan, R. P. Dickinson, H. Xu, Q. Li, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Mater., 2018, 30, 1069–1077 CrossRef CAS.
- E. Bellido, T. Hidalgo, M. V. Lozano, M. Guillevic, R. Simón-Vázquez, M. J. Santander-Ortega, Á. González-Fernández, C. Serre, M. J. Alonso and P. Horcajada, Adv. Healthcare Mater., 2015, 4, 1246–1257 CrossRef CAS PubMed.
- Y. Li, Y. Zheng, X. Lai, Y. Chu and Y. Chen, RSC Adv., 2018, 8, 23623–23628 RSC.
- F. Shu, D. Lv, X.-L. Song, B. Huang, C. Wang, Y. Yu and S.-C. Zhao, RSC Adv., 2018, 8, 6581–6589 RSC.
- S. Jung, Y. Kim, S.-J. Kim, T.-H. Kwon, S. Huh and S. Park, Chem. Commun., 2011, 47, 2904–2906 RSC.
- F.-X. Qin, S.-Y. Jia, F.-F. Wang, S.-H. Wu, J. Song and Y. Liu, Catal. Sci. Technol., 2013, 3, 2761–2768 RSC.
- S.-L. Cao, H. Xu, X.-H. Li, W.-Y. Lou and M.-H. Zong, ACS Sustainable Chem. Eng., 2015, 3, 1589–1599 CrossRef CAS.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC.
- Y. Yin, C. Gao, Q. Xiao, G. Lin, Z. Lin, Z. Cai and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 29052–29061 CrossRef CAS PubMed.
- C. Zhang, X. Wang, M. Hou, X. Li, X. Wu and J. Ge, ACS Appl. Mater. Interfaces, 2017, 9, 13831–13836 CrossRef CAS PubMed.
- W.-H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS.
- J. Chen, L. Huang, Q. Wang, W. Wu, H. Zhang, Y. Fang and S. Dong, Nanoscale, 2019, 11, 5960–5966 RSC.
- M. Hoop, C. F. Walde, R. Riccò, F. Mushtaq, A. Terzopoulou, X.-Z. Chen, A. J. deMello, C. J. Doonan, P. Falcaro, B. J. Nelson, J. Puigmartí-Luis and S. Pané, Appl. Mater. Today, 2018, 11, 13–21 CrossRef.
- R. G. Freitas, F. W. S. Lucas, M. A. Santanna, R. A. Mendes, A. J. Terezo, G. L. C. de Souza, L. H. Mascaro and E. C. Pereira, Phys. Chem. Chem. Phys., 2016, 18, 26885–26893 RSC.
- J. B. DeCoste, M. S. Denny, Jr., G. W. Peterson, J. J. Mahle and S. M. Cohen, Chem. Sci., 2016, 7, 2711–2716 RSC.
- S.-H. Jung, E. Oh, D. Shim, D.-H. Park, S. Cho, B. R. Lee, Y. U. Jeong, K.-H. Lee and S.-H. Jeong, Bull. Korean Chem. Soc., 2009, 30, 2280–2282 CrossRef CAS.
- D. H. Nies, Metallomics, 2016, 8, 481–507 RSC.
- K. Kida, M. Okita, K. Fujita, S. Tanaka and Y. Miyake, CrystEngComm, 2013, 15, 1794 RSC.
- T. Kokubo and H. Takadama, Biomaterials, 2006, 27, 2907–2915 CrossRef CAS PubMed.
- T. Kokubo and H. Takadama, in Handbook of Biomineralization, ed. E. Buerlein, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2007, pp. 97–109 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional figures and in situ real-time AFM movie. See DOI: 10.1039/c9ce00757a |
| This journal is © The Royal Society of Chemistry 2019 |