DOI:
10.1039/C8CE01580E
(Paper)
CrystEngComm, 2019,
21, 108-117
Modulation of coordination in pincer-type isonicotinohydrazone Schiff base ligands by proton transfer†
Received
14th September 2018
, Accepted 30th October 2018
First published on 30th October 2018
Introduction
Crystal engineering is not a trivial problem and still many aspects of controlling crystal growth need to be studied and it is not easy to predict the outcome of crystallization.1–6 In fact, a major challenge using polydentate chelating–bridging building blocks in the synthesis of metal coordination compounds is the unpredictability of the structure of the products which may depend on several factors such as the nature of the ligands used, the oxidation state, geometry, and size of the metal centers, the metal-to-ligand ratio, etc.7–9 Hydrazone–pyridine based chelating ligands are known N,N,O pincer type ligands.10 They are usually synthesized by the one-pot Schiff-base condensation reaction in high yields11 and it makes them a good subject for studies because the geometries of the ligands can have a great impact on the structural architecture of coordination compounds.12 Pyridine based ligands containing amide groups generally are coordinated to the metal centers through their pyridyl nitrogen atoms and interact with each other by hydrogen bonds involving the amide groups, and the location of pyridyl substituent at the peripheral part of the main molecular skeleton in the isonicotinohydrazone based ligand may result in interesting supramolecular motifs.13 The chromophore hydrazone functional group is conformationally flexible and it has numerous hydrogen bonding sites including a molecular backbone. Moreover, isonicotinohydrazone based ligand possesses an additional pyridine hydrogen bond acceptor region and the ability to establish π⋯π stacking interactions taking advantage of the aromatic rings located on the two extremities of the molecule.13 The strength of coordination bonding may, however, be tuned by some changes in the electronic structure of the ligand when proton transfer occurs (Scheme 1). In pyridyl groups, the electron lone pair does not delocalize, therefore it is a good proton acceptor. On the other hand, the amide part may undergo keto–enol transformation.14 By tuning the crystallization conditions, the resulting complexes may have different localization of protons within the ligand molecule and different coordination architectures.13c,14f In the present work, we study the differences in the structure of two mercury(II) coordination compounds of hydrazone Schiff bases containing pyridine groups. The change in the electronic structure of the ligand, due to proton transfer, changes its ligating properties and the relative location of orbitals and ligands.
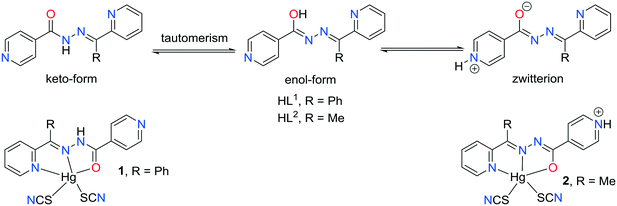 |
| | Scheme 1 Possible proton transfer mechanisms in isonicotinohydrazone based ligands and the structure of complexes 1 and 2. | |
The geometric features and the coordination behavior of the ligands in the crystal structure of compound 2 revealed a preference for the formation of chelate ring stacking along with more conventional pyridine⋯pyridine stacking interactions. In contrast, the steric effect of the phenyl group in the HL1 ligand (compound 1) prevents the formation of chelate ring stacking. To gain insight into these structural features, we employed Hirschfield surface analysis and DFT (M06-2X) calculations of interaction energies, which confirm that chelate ring stacking is a robust synthon to be considered in crystal engineering.
Experimental and theoretical methods
Materials and methods
All chemicals were commercially available and the solvents were used without further purification. The hydrazone–pyridine based ligands were synthesized according to the reported method as described elsewhere.8 In short, pure HL1 and HL2 were obtained by the condensation of ethanolic solutions of isonicotinohydrazide, and 2-benzoylpyridine and 2-acetylpyridine, respectively. Elemental analyses were carried out using a Heraeus CHN-O-Rapid analyzer and FT-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with KBr pellets in the range of 4000–400 cm−1. Melting points of the prepared metal complexes were measured using an Electrothermal 9100 apparatus.
Synthesis of the complexes
Compounds [Hg(SCN)2(HL1)] (1) and [Hg(SCN)2(HL2)] (2) were synthesized using the same method mixing equimolar quantities of mercury(II) salt and the ligand, as explained below. A branched tube was used to obtain suitable crystals for X-ray determination, as detailed in Scheme S1.†
[Hg(SCN)2(HL1)] (1).
Mercury(II) thiocyanate (0.158 g, 0.5 mmol) and 2-benzoylpyridine-isonicotinoylhydrazone (HL1) (0.151 g, 0.5 mmol) were placed in the main arm of a branched tube. Methanol (10 ml) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. After 3 days, crystals of 1 isolated in the cooler arm were filtered off, washed with acetone and ether, and dried in air. The isolated yield was 0.217 g, 70%. m. p. 206 °C. Anal. calcd for C20H14HgN6OS2: C, 38.80; H, 2.28; N, 13.57%. Found: C, 38.49; H, 2.17; N, 13.94%. FT-IR (cm−1) selected bands: 3452 (w), 3093 (w), 2117 (vs), 1663 (m), 1596 (m), 1538 (vs), 1466 (m), 1437 (m), 1282 (vs), 1136 (m), 965 (m), 839 (m), 772 (m) and 661 (m).
[Hg(SCN)2(HL2)] (2).
Mercury(II) thiocyanate (0.158 g, 0.5 mmol) and 2-acetylpyridine-isonicotinoylhydrazone (HL2) (0.120 g, 0.5 mmol) were placed in the main arm of a branched tube. Methanol (10 ml) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. After 3 days, crystals of 2 isolated in the cooler arm were filtered off, washed with acetone and ether, and dried in air. The isolated yield was 0.240 g, 84%. m. p. 180 °C. Anal. calcd for C15H12HgN6OS2: C, 32.34; H, 2.17; N, 15.09%. Found: C, 32.60; H, 2.22; N, 15.29%. FT-IR (cm−1) selected bands: 3423 (w), 3368 (w), 2993 (w), 2061 (vs), 1622 (m), 1593 (m), 1534 (s), 1481 (s), 1438 (m), 1288 (s), 1157 (m), 948 (m), 834 (m), 787 (m) and 699 (m).
Hirshfeld surface analysis
The Hirshfeld surfaces and 2D fingerprints of the studied compounds were generated with the use of the Crystal Explorer package ver. 3.1.15 Crystal structures were imported from CIF files. Hirshfeld surfaces were generated using high resolution and mapped with the dnorm and shape-index functions. 2D fingerprint plots were prepared with the use of the same software.
X-ray diffraction
The diffraction intensities for 1 and 2 were measured at room temperature using the ω-scan technique on Bruker four-circle diffractometers (APEX-II CCD) with graphite-monochromated MoKα radiation (λ = 0.71073 Å). The detector frames were integrated by the use of the program SAINT16 and the empirical absorption corrections were performed using the SADABS program.17 The structures were solved and refined using the SHELXS and olex2.refine refinement package using Gauss–Newton minimization.18,19 All non-hydrogen atoms were refined anisotropically, and the positions of all hydrogen atoms were placed in idealized positions except for the H atom at pyridine in 2 which was found in the difference Fourier maps. Then, all H-atoms were refined as the ‘riding model’ with isotropic displacement parameters set at 1.2 (1.5 for the methyl group) times Ueq of the appropriate carrier atoms. Details of the crystallographic data collection and refinement parameters are given in Table 1.
Table 1 Crystal data and structure refinement data for 1 and 2
| Crystal |
1
|
2
|
| Empirical formula |
C20H14N6OS2Hg |
C15H12N6OS2Hg |
| Formula weight |
619.11 |
557.04 |
| Temperature/K |
296(2) |
296(2) |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
C2/c |
P21/c |
|
a/Å |
28.861(6) |
12.9176(3) |
|
b/Å |
9.191(2) |
7.7756(2) |
|
c/Å |
17.149(3) |
17.4321(4) |
|
β/° |
111.76(3) |
93.02(1) |
| Volume/Å3 |
4224.8(17) |
1748.5(1) |
|
Z
|
8 |
4 |
|
ρ
calc g cm−3 |
1.9465 |
2.1159 |
|
μ/mm−1 |
7.510 |
9.060 |
|
F(000) |
2353.2 |
1048.6 |
| Crystal size/mm3 |
0.20 × 0.22 × 0.40 |
0.08 × 0.22 × 0.24 |
| Radiation |
Mo Kα (λ = 0.71073) |
Mo Kα (λ = 0.71073) |
| 2Θ range for data collection/° |
4.68 to 50.48 |
3.16 to 65.12 |
| Reflections collected |
11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 606 606 |
40853 |
| Independent reflections |
3711 |
6334 |
|
R
int = 0.0433 |
R
int = 0.0285 |
|
R
sigma = 0.0483 |
R
sigma = 0.0248 |
| Data/restraints/parameters |
3711/0/270 |
6334/0/226 |
| Goodness-of-fit on F2 |
0.829 |
0.990 |
| Final R indexes [I ≥ 2σ(I)] |
R
1 = 0.0366, wR2 = 0.0830 |
R
1 = 0.0256, wR2 = 0.0441 |
| Final R indexes [all data] |
R
1 = 0.0561, wR2 = 0.0880 |
R
1 = 0.0460, wR2 = 0.0497 |
| Largest diff. peak/hole/e Å−3 |
1.85/−2.35 |
1.32/−1.55 |
| CCDC no. |
1867818
|
1867819
|
CCDC 1867818 and 1867819 contain the supplementary crystallographic data for this paper.
Theoretical methods
The geometries of the complexes included in this study were computed at the M06-2X/def2-TZVP level of theory using the crystallographic coordinates within the GAUSSIAN-09 program.20 For the Hg atom we have used the LanL2DZ basis set. This level of theory is adequate for studying noncovalent interactions dominated by dispersion effects like π-stacking. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.21 The “atoms-in-molecules” (AIM)22 analysis of the electron density has been performed at the same level of theory using the AIMAll program.23 Molecular electrostatic potential surfaces have been computed at the same level using the GAUSSIAN-09 program.
Results and discussion
Molecular structure
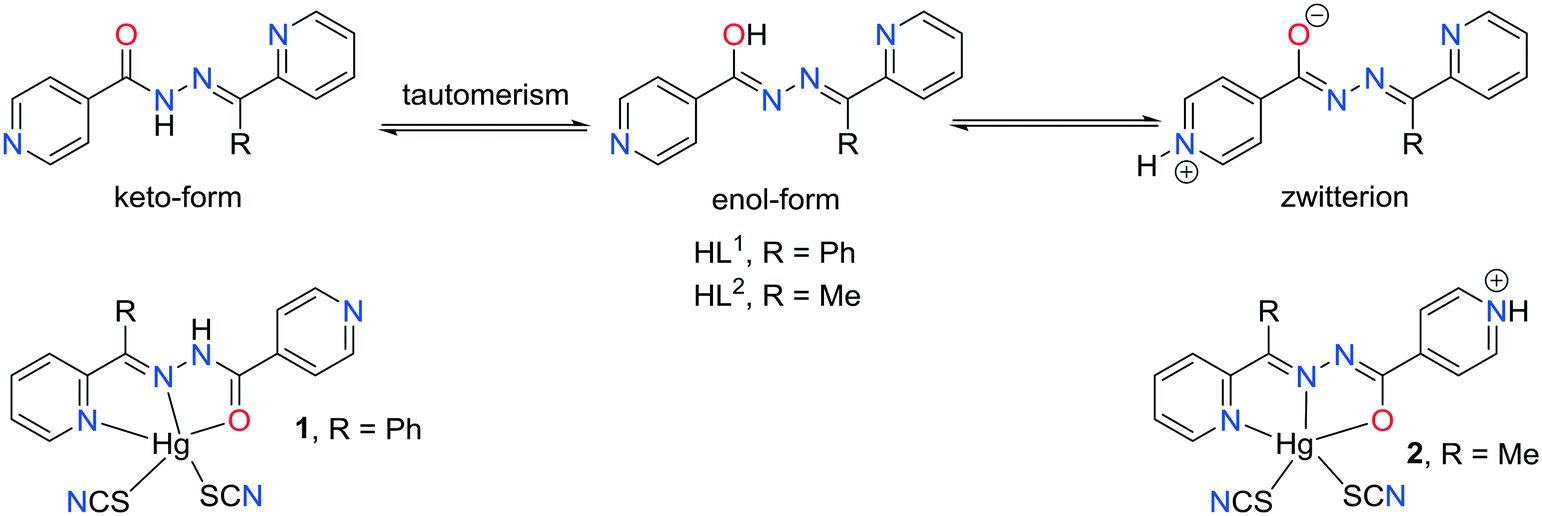
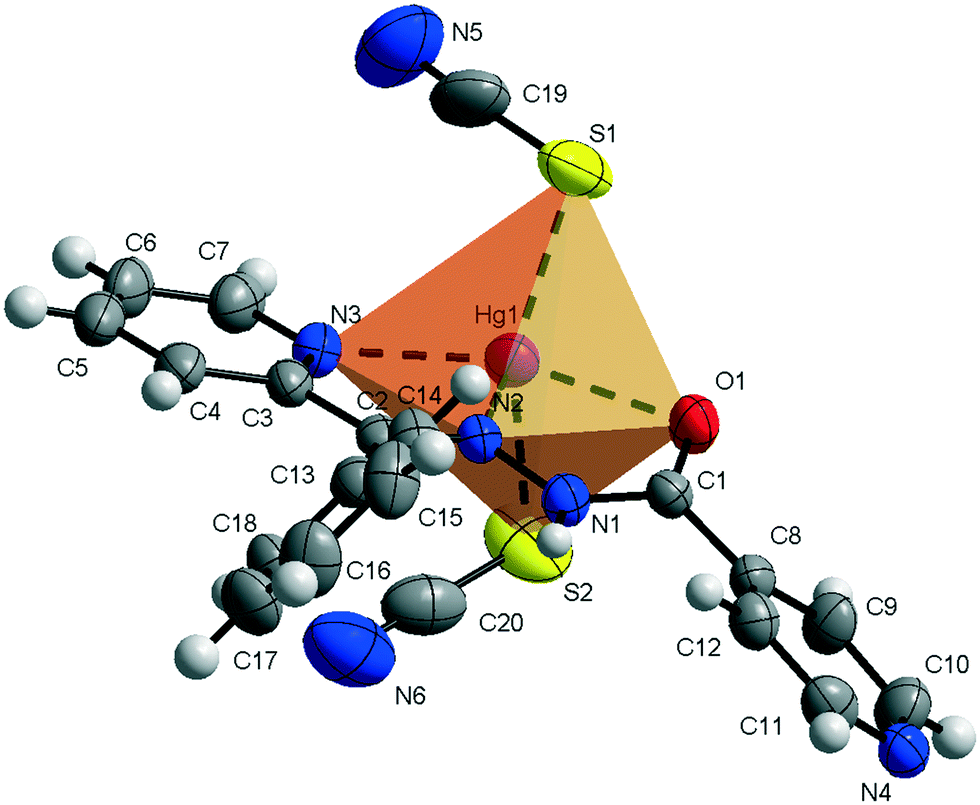
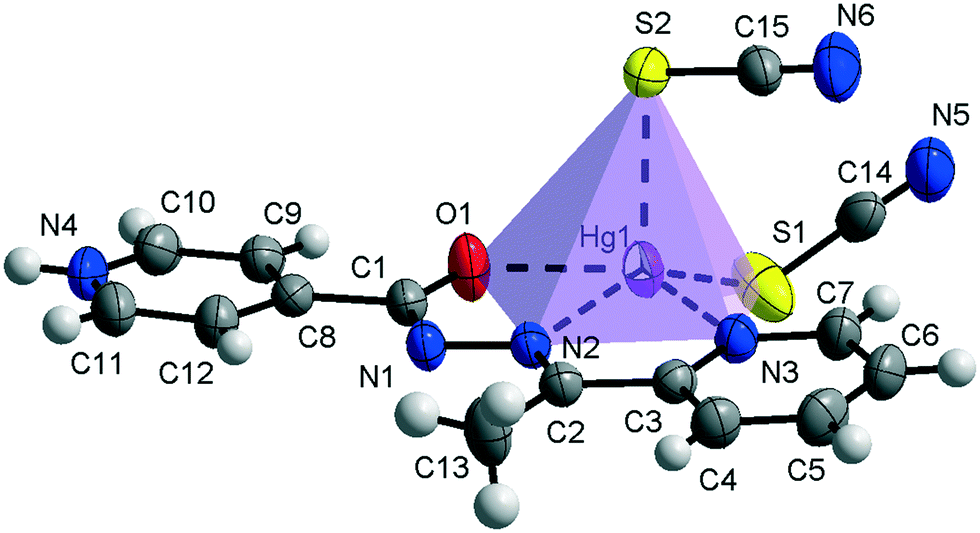
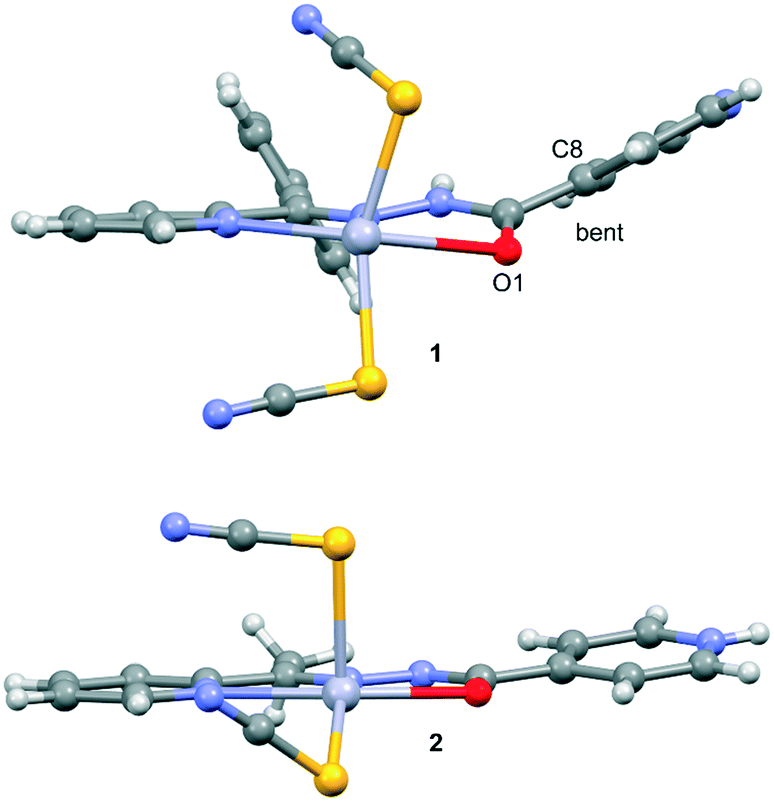
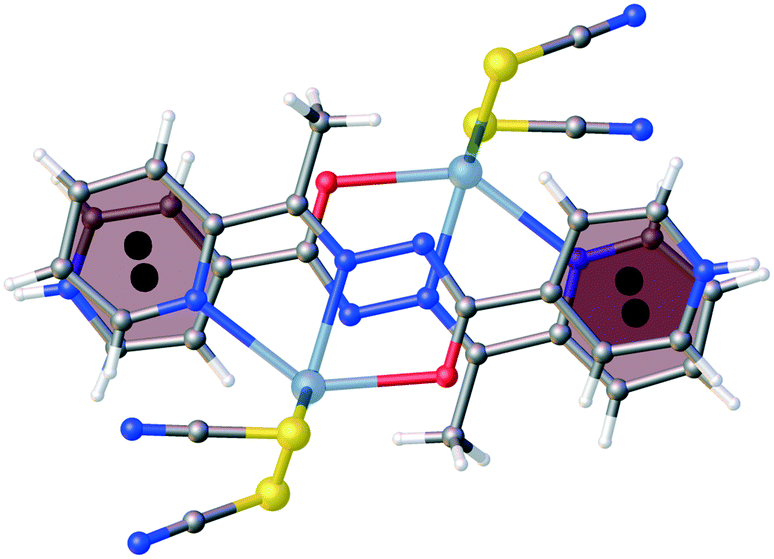
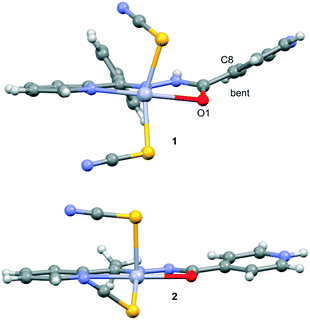
Compound 1 crystallizes in the monoclinic C2/c space group (Table 1) with the amide fragment being in a keto tautomeric form. A voluminous benzyl group is substituted at the C7 atom and it is rotated by ca. 60° in regard to the hydrazone mean plane (Fig. 1). Compound 2 (P21/c) has a methyl substituent instead of a benzyl one. Under the reaction conditions, the amide group undergoes deprotonation. The proton is transferred from the amide N1 to pyridyl N4 atom forming a zwitterion (Fig. 2). The bond lengths (Table 2), as well as the intermolecular hydrogen bond motifs (Table S1, Fig. S1 and S2†), confirm the keto form in 1 and the enol form of the amide group in 2. The C1–N1 bond length in 2 is shorter by 0.02 Å, whereas the C1–O1 and C1–C8 bonds are longer by 0.03 and 0.02 Å, respectively, than that in the keto form in 1. The electronic coupling in the molecule in crystal 2 results in a flat structure (Table 3 and Fig. 3). Conversely, in compound 1 the molecule is bent where the hydrazone fragment is not planar with the O1 and C8 atoms being below (0.21 Å) and above (0.30 Å) the mean C1N2N1 plane, respectively. A comparison of both compounds is provided in Fig. 3.
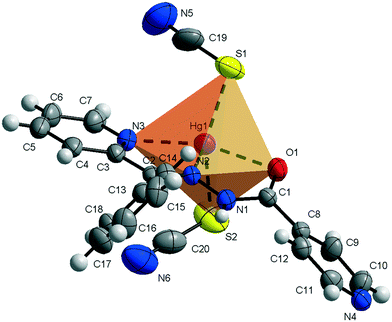 |
| | Fig. 1 Thermal ellipsoids and coordination polyhedron in 1. | |
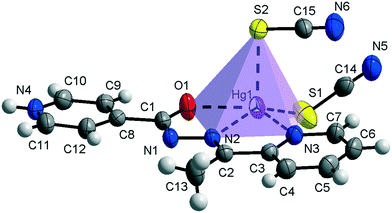 |
| | Fig. 2 Thermal ellipsoids and coordination polyhedron in 2. | |
Table 2 Selected bond lengths for 1 and 2 in Å
| Bond |
1
|
2
|
Bond |
1
|
2
|
| Hg1–N2 |
2.491(5) |
2.279(2) |
C7–N3 |
1.334(9) |
1.332(4) |
| Hg1–N3 |
2.423(6) |
2.459(2) |
C8–C9 |
1.371(10) |
1.389(4) |
| Hg1–S1 |
2.399(2) |
2.428(1) |
C8–C12 |
1.380(10) |
1.385(4) |
| Hg1–S2 |
2.406(2) |
2.640(1) |
C9–C10 |
1.385(10) |
1.372(4) |
| Hg1–O1 |
2.687(5) |
2.392(2) |
C10–N4 |
1.312(10) |
1.324(4) |
| C1–O1 |
1.226(7) |
1.257(3) |
C11–N4 |
1.342(10) |
1.327(4) |
| C1–N1 |
1.346(9) |
1.324(3) |
C11–C12 |
1.377(10) |
1.373(4) |
| C1–C8 |
1.477(9) |
1.499(4) |
N5–C19/or C14 |
1.143(13) |
1.143(4) |
| N1–N2 |
1.382(7) |
1.372(3) |
N6–C20/or C15 |
1.158(14) |
1.148(4) |
| N2–C2 |
1.282(9) |
1.288(3) |
S1–C19/or C14 |
1.643(11) |
1.667(4) |
| C2–C13 |
1.467(9) |
1.496(4) |
S2–C20/or C15 |
1.650(13) |
1.649(3) |
| C3–N3 |
1.346(9) |
1.338(3) |
|
|
|
Table 3 Selected bond angles and torsions in 1 and 2 in deg
| Angle |
1
|
2
|
| S1–Hg1–S2 |
152.1(1) |
107.5(1) |
| O1–Hg1–S1 |
86.1(1) |
108.6(1) |
| O1–Hg1–S2 |
85.5(1) |
96.3(1) |
| N2–Hg1–O1 |
61.8(2) |
69.1(1) |
| N3–Hg1–O1 |
126.7(2) |
138.0(1) |
| S1–Hg1–N2 |
104.6(1) |
148.8(1) |
| S1–Hg1–N3 |
101.4(1) |
105.5(1) |
| S2–Hg1–N2 |
94.7(1) |
103.6(1) |
| S2–Hg1–N3 |
105.0(2) |
96.1(1) |
| N2–Hg1–N3 |
65.4(2) |
69.0(1) |
| Hg1–S1–C19/or C14 |
100.2(4) |
97.6(1) |
| Hg1–S2–C20/or C15 |
96.7(3) |
99.7(1) |
| O1–Hg1–N3–C7 |
171.1(5) |
175.9(2) |
| C1–N1–N2–Hg1 |
13.2(7) |
2.6(3) |
| N1–N2–Hg1–N3 |
169.0(5) |
179.0(2) |
 |
| | Fig. 3 Comparison of the planarity of the ligands in complexes 1 (top) and 2 (bottom). | |
The consequences of proton transfer are also pronounced in the geometry of the coordination spheres. In both complexes the organic ligands serve as tridentate N2O pincer type chelators, but in the zwitterionic structure 2 the distance between Hg1⋯O1 atoms is shorter (by 0.29 Å) than that in 1 (Table 2). Additionally, the ligand to metal orbital relative orientation is different. In 1, the coordination polyhedron is a deformed trigonal bipyramid (Fig. 1). The pincer ligand lies in the base and two thiocyanate ions are in the apical positions at nearly the same distance of ca. 2.4 Å. In 2, the pincer ligand is bonded strongly to the metal center with shorter intramolecular N–Hg and O–Hg distances than that in 1. It forms a distorted square pyramid with the isonicotinohydrazone part of the ligand and with one of the SCN− ions lying in the same plane. The second thiocyanate anion takes the apical position with a longer distance Hg1–S2 of 2.640(1) Å (Fig. 2).
π⋯π interactions
The flattening of the isonicotinohydrazone moiety in 2 is essential for π⋯π intermolecular interactions. In 2, the 2-pyridyl and 4-pyridyl rings (R1 and R2 in Table S2†) interact with each other with nearly full molecular overlay (Fig. 4, Table S2†). In 1, the π⋯π interaction is possible only between two 2-pyridyl fragments of the molecules transformed by inversion (R1 rings in Table S2;†Fig. 5). The second π⋯π contact in 1 is between 4-pyridyl and benzyl rings (R2 and R3 in Table S2†). In crystal of 2, the columns of stacked molecules are the main packing motif, whereas in 1 a zig-zag arrangement is present.
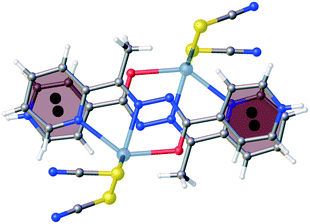 |
| | Fig. 4 The π⋯π intermolecular interactions between 2-pyridyl and 4-pyridyl rings in 2 (nearly full overlay of the molecules). | |
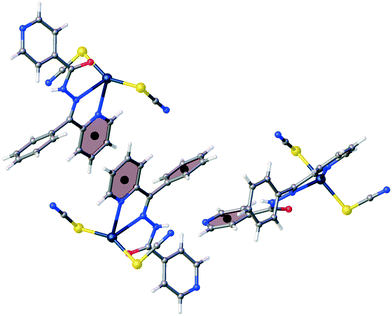 |
| | Fig. 5 The π⋯π intermolecular interactions between two 2-pyridyl rings and between 4-pyridyl and benzyl rings in 1. | |
IR spectroscopy
The IR spectrum of 1, where the amide N-atom is not deprotonated, exhibits both the amide N–H (3452 cm−1) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1663 cm−1) stretching vibrations that are typical for a free ligand24 and there is no peak characteristic of the deprotonated ligand. On the other hand, the free ligand CO stretching vibrations are not observed in the IR spectrum of compound 2 and an absorption band response to the CN–NC–O moiety appearing around 1622 cm−1 indicates that the ligand has lost the amide proton during the tautomerization, and the proton is attached to the pyridyl nitrogen of the isonicotine fragment making a zwitterion.
O (1663 cm−1) stretching vibrations that are typical for a free ligand24 and there is no peak characteristic of the deprotonated ligand. On the other hand, the free ligand CO stretching vibrations are not observed in the IR spectrum of compound 2 and an absorption band response to the CN–NC–O moiety appearing around 1622 cm−1 indicates that the ligand has lost the amide proton during the tautomerization, and the proton is attached to the pyridyl nitrogen of the isonicotine fragment making a zwitterion.
Hirshfeld surface analysis
The Hirshfeld surface analysis25,26 of 1 and 2 gives a clear difference in the molecular structure and crystal packing. The antennas related to the N⋯H contacts in 2 are longer and narrower than those in 1 as a result of shorter N⋯H intermolecular interactions in 2 (Fig. 6). In 1, the additional aromatic substituent shows that the H⋯H contacts are more abundant (21.3 vs. 13.7%), but the aromatic character of the molecule is more pronounced in the N⋯N contacts due to the π⋯π stacks in 2 (0.9 vs. 0.1%). The Hg⋯H contacts show that in 2 the metal core is more hindered than that in 1 (0.2 vs. 1.2%).
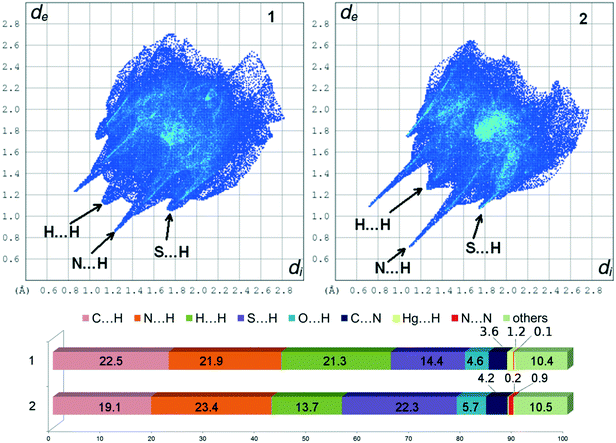 |
| | Fig. 6 2D fingerprint plots and percentage contributions of contacts to the Hirshfeld surface in the structures of 1 and 2 calculated from crystal structures. | |
Theoretical study
The theoretical study reported herein is devoted to comparing the energetic features of the two types of π-stacking interactions (chelate ring–π and π–π) observed in the crystal packing of compounds 1–2 described above (see Fig. 4 and 5). These interactions are crucial to understanding the crystal packing of complexes 1–2. In addition to the classical π-stacking interaction involving aromatic rings, other planar moieties can also participate in more “unconventional” stacking interactions.27 For instance, chelate rings with delocalized π bonds establish stacking27 interactions similar to those of aromatic organic molecules28 in transition-metal complexes. Similar chelate ring π-stacking interactions have also been reported by us in HgX2 and PbX2 (X = Cl, Br and I) coordination compounds.13,14
To study the donor–acceptor ability of HgLn complexes, we have computed the molecular electrostatic potential (MEP) surfaces of compounds 1 and 2, which are shown in Fig. 7. Expectedly, the most negative electrostatic potential corresponds to the region of the SCN ligands and the most positive part is located in the region of the N–H groups. Therefore, the H-bonding interaction between the N–H and SCN groups should be energetically favored. Furthermore, perpendicular to the molecular plane, in compound 2 each 5-membered chelate ring has almost negligible MEP values. Consequently, the stacking interactions between chelate rings will likely be dominated by dispersion effects. The MEP value is large and positive over the protonated pyridine rings and it is negative (−5 kcal mol−1) in the coordinated pyridine ring. Therefore, pyridine–pyridine interactions are expected to be electrostatically greatly favored due to a significant electrostatic attraction. In compound 1, the chelate ring is not accessible to the particular geometry around the Hg metal center and the location of the SCN ligands.
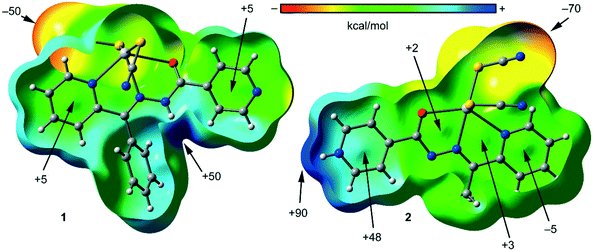 |
| | Fig. 7 MEP surfaces plotted onto the van der Waals surfaces of compound 1 (left) and 2 (right) and the energies at selected points of the surface are given in kcal mol−1. | |
In the crystal packing of compound 1, the mononuclear Hg(II) complex forms self-assembled dimers in the solid state governed by the formation of an antiparallel π–π interactions involving the coordinated pyridine rings, assisted by H-bonding interactions between the aromatic CH bonds and the pseudohalide (see Fig. 8a). These CH⋯N/S hydrogen bonds are expected to be energetically strong due to the enhanced acidity of the H atoms due to the coordination of the pyridide to the Hg(II) metal center. The dimerization energy of this self-assembled dimer is ΔE1 = −27.9 kcal mol−1, which is large due to the contribution of both interactions. In an effort to calculate the contribution of the different forces, we have computed a theoretical model in which one of the pseudoligands (SCN−) has been replaced by a hydrido ligand (see small arrows in Fig. 8b) and consequently the H-bonding interactions are not formed. Consequently, the interaction energy is reduced to ΔE2 = −15.9 kcal mol−1 that can be attributed to the contribution of the antiparallel π-stacking interaction. This binding energy is stronger than that of more conventional π-stacking interactions27 due to the presence of the Hg(II) metal center that increases considerably the dipole moment of the π-system. The contribution of the H-bonding interactions to the formation of the assembly can be evaluated by difference (ΔE1−ΔE2 = −12.0 kcal mol−1), thus revealing a major contribution of the π-stacking.
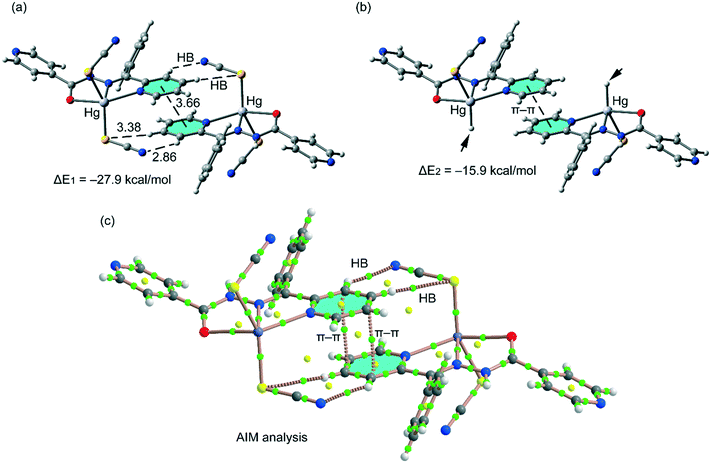 |
| | Fig. 8 (a) Interaction energy of the self-assembled π-stacked dimer observed in the solid state of compound 1. Distances in Å. (b) Interaction energy in the theoretical model of 1. (c) AIM analysis of the self-assembled dimer retrieved from the X-ray structure of compound 1. Bond and ring critical points are represented by green and yellow spheres, respectively. The bond paths connecting the bond critical points are also represented by dashed lines. | |
In order to provide additional evidence for the existence of the noncovalent interactions commented above we have analyzed the self-assembled π-stacked dimer of compound 1 using Bader's theory of “atoms in molecules” (AIM).29 The existence of a bond critical point and bond path connecting two atoms is an unambiguous evidence of interaction. In Fig. 8c, we show the AIM analysis of the dimer of compound 1 and it can be observed that the antiparallel π–π interaction is characterized by the presence of two bond critical points (green spheres) that interconnect two atoms of the pyridine rings, thus confirming the interaction. Furthermore, the distribution of critical points reveals the existence of two symmetrically related C–H⋯N and C–H⋯S H-bonding interactions since two bond critical points connect the N and S atoms of the pseudohalide with the aromatic H-atoms. A ring critical point (yellow sphere) also appears upon complexation due to the formation of a supramolecular ring.
In compound 2, we have computed the interaction energy of the self-assembled π-stacked dimer shown in Fig. 9a, which is stabilized by an intricate combination of noncovalent interactions, that is, H-bonds (red dashed lines), π–π stacking interactions between the coordinated and uncoordinated pyridine rings and chelate ring⋯chelate ring (CR–CR) stacking interactions. Remarkably, opposite to compound 1, the square planar pyramidal geometry of the Hg atom in compound 2 facilitates the approximation of the chelate rings, resulting in shorter CR⋯CR interactions (3.54 Å). The dimerization energy (ΔE3 = −64.6 kcal mol−1) is very large due to the contribution of the three interactions and the strong dipole⋯dipole interactions due to the zwitterionic nature of the ligand. In an effort to roughly estimate the contribution of the different forces that govern the formation of the self-assembled dimer, we have computed a theoretical model in which the methyl groups have been replaced by H atoms (see small arrows in Fig. 9b) and consequently the C–H⋯S H-bonds involving the methyl groups are not formed. As a result, the interaction energy is reduced to ΔE4 = −57.6. Therefore, the contribution of both symmetrically equivalent H-bonds can be estimated by difference (−7 kcal mol−1). Furthermore, we have used an additional dimer, where the pyridinium ring has been replaced by a hydrogen atom, and consequently, the py⋯py interactions and H-bonds are not formed. Since the pyridine rings are protonated, in this model we have added a hydrogen atom to the hydrazone group to keep the model neutral. The resulting interaction energy is further reduced to ΔE5 = −30.5 kcal mol−1 which corresponds to the contribution of the CR–CR π-stacking interaction. The contribution of both py⋯py interactions and C–H⋯S bonds can be estimated by difference (−27.1 kcal mol−1) thus emphasizing the importance of these charge-assisted π-stacking interactions. The CR–CR interaction is unexpectedly strong, which is likely due to the model used for the calculations where an H-atom has been added to the hydrazone group. This H-atom likely forms an extra H-bond with the S atom of the pseudohalide ligand, thus also contributing to the binding energy (see blue dashed line in Fig. 9c). Therefore, the conventional py⋯py interaction is underestimated and the CR–CR interaction is overestimated using this model. Nevertheless, the strong binding energy of the whole assembly indicates that this combination of two types of π-stacking interactions and H-bonds is a robust binding motif in the solid state of compound 2.
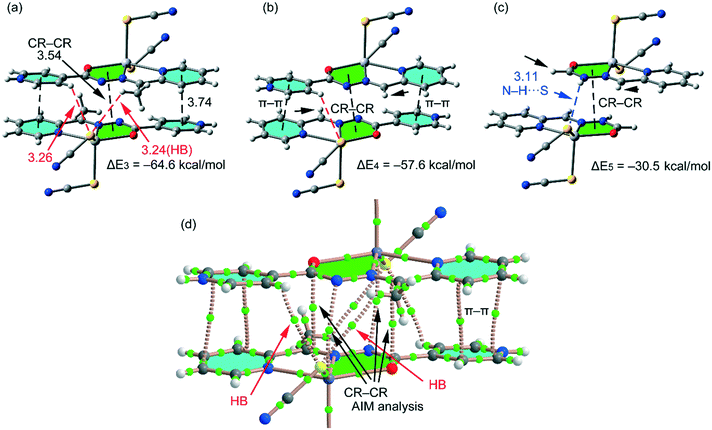 |
| | Fig. 9 (a) Interaction energy of the self-assembled π-stacked dimer observed in the solid state of compound 2. Distances in Å. (b and c) Geometries and interaction energies in the theoretical models of 2. (d) AIM analysis of the self-assembled dimer retrieved from the X-ray structure of compound 1. Bond critical points are represented by green spheres. The bond paths connecting the bond critical points are also represented by dashed lines. Ring and cage critical points have been omitted for clarity. | |
In Fig. 9d, we show the AIM analysis of compound 2. Each conventional π–π interaction (pyridine rings) is characterized by the presence of two bond critical points that interconnect the two atoms of the protonated pyridine ring to the two atoms of the coordinated one, thus confirming the interaction. Furthermore, the distribution of critical points reveals the existence of four symmetrically related C–H⋯S H-bonding interactions. Each one is characterized by a bond critical point and bond path connecting the H atom with the S atom of the pseudohalide. Finally, the unconventional CR⋯CR interaction is confirmed by the presence of two bond critical points interconnecting the Hg atoms to the N atoms and two additional bond critical points connecting the O atoms to the C atoms of the hydrazone group. The value of the Laplacian of the charge density at the bond critical points is positive, as is common in closed-shell interactions.
The binding energies and π-stacking distances (3.54 Å) described above for the CR⋯CR interactions in compound 2 are comparable to those reported for Hg(II) complexes where halide instead of pseudohalides were used as co-ligands (3.30 to 3.67 Å).30 CR⋯CR interactions have been also described recently in Zn(II) and Cd(II) complexes involving (iso)nicotinohydrazide ligands that exhibited similar binding energies (≃28 kcal mol−1) and distances (3.44–3.50 Å).13a Moreover, a recent study has described CR⋯CR interactions in Cu(II) polymers where polydentate (E)-2-(1-(pyridin-2-yl)methyleneamino)terephthalic acid was used as the ligand.31 The solid state X-ray architecture of the polymers was governed by CR⋯CR interactions along with H-bonding. These reports along with the structures reported herein provide strong evidence for preferential formation of chelate ring stacking, which can be considered as a synthon interaction for (iso)nicotinohydrazide metal complexes. Moreover, the interaction energies reported herein confirm that the chelate–chelate interactions are stronger than those reported for classical π–π complexes.28
Conclusions
Herein, we reported the syntheses and characterization of two mercury(II) complexes of two isonicotinohydrazone Schiff base ligands. The ligands acted as pincer type tridentate N2O-donor ligands, in which the isonicotinamide pyridine nitrogen remained uncoordinated. By using thiocyanate ligands along with the organic ligands, the coordination geometry around each mercury center resulted in a distorted trigonal bipyramid and a square-pyramid in 1 and 2, respectively. Compounds 1 and 2 exhibit interesting π–π stacking interactions in their solid state, which have been analyzed using DFT calculations in detail. The combination of conventional π-stacking with more unconventional chelate ring stacking interactions is a robust binding motif in compound 2. The protonation of 4-pyridyl substituent may increase the strength of the pincer type coordination in hydrazone based Schiff base ligands. The presented systems are complementary in the sense of proton donor and proton acceptor properties of the molecular subcomponents. The proton transfer should not be overlooked when designing new structures based on supplementary molecular fragments such as hydrazone based ligands substituted with pyridines. It should be emphasized that steric effects (bulky phenyl vs. methyl) are the origin of the different behaviours of the complexes. The proton transfer in the isonicotinohydrazone methyl substituted ligand (HL2) enables the intramolecular coupling and flattening of the molecule which increases the chances for stronger pincer type coordination. It favors the conventional (π–π) and unconventional chelate ring stacking interactions, which are useful tools in crystal engineering.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the University of Maragheh for the financial support of this research. The publication was also prepared with the support of the “RUDN University Program 5-100”. AF thanks the MINECO/AEI from Spain for a “Juan de la Cierva” contract. We thank the MINECO/AEI from Spain for financial support (project number CTQ2017-85821-R, FEDER funds). We are grateful to the CTI (UIB) for free allocation of computer time.
References
- M. O. Besenhard, P. Neugebauer, O. Scheibelhofer and J. G. Khinast, Cryst. Growth Des., 2017, 17, 6432 CrossRef CAS PubMed.
- P. Erk, H. Hengelsberg, M. F. Haddow and R. van Gelder, CrystEngComm, 2004, 6, 474 RSC.
- N. Tumanova, N. Tumanov, K. Robeyns, F. Fischer, L. Fusaro, F. Morelle, V. Ban, G. Hautier, Y. Filinchuk, J. Wouters, T. Leyssens and F. Emmerling, Cryst. Growth Des., 2018, 18, 954 CrossRef CAS.
- J. R. G. Sander, D.-K. Bučar, R. F. Henry, B. N. Giangiorgi, G. G. Z. Zhang and L. R. MacGillivray, CrystEngComm, 2013, 15, 4816 RSC.
- D.-K. Bučar, Cryst. Growth Des., 2017, 17, 2913 CrossRef.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed.
- A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS.
- M. Sarkar and K. Biradha, Cryst. Growth Des., 2007, 7, 1318 CrossRef CAS.
- A. Beheshti, W. Clegg, V. Nobakht and R. W. Harrington, Cryst. Growth Des., 2013, 13, 1023 CrossRef CAS.
-
K. J. Szabó and O. F. Wendt, Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, by John Wiley & Sons, Inc, 2014 Search PubMed.
-
(a) S. Yumnam and L. Rajkumari, J. Chem. Eng. Data, 2009, 54, 28 CrossRef CAS;
(b) A. A. Khandar, B. K. Ghosh, C. Lampropoulos, M. S. Gargari, V. T. Yilmaz, K. Bhar, S. A. Hosseini-Yazdi, J. M. Cain and G. Mahmoudi, Polyhedron, 2015, 85, 467 CrossRef CAS;
(c) M. Abedi, O. Z. Yesilel, G. Mahmoudi, A. Bauza, S. E. Lofland, Y. Yerli, W. Kaminsky, P. Garczarek, J. K. Zareba, A. Ienco, A. Frontera and M. S. Gargari, Inorg. Chim. Acta, 2016, 443, 101 CrossRef CAS;
(d) G. Mahmoudi, A. Bauzá, A. V. Gurbanov, F. I. Zubkov, W. Maniukiewicz, A. Rodríguez-Diéguez, E. López-Torres and A. Frontera, CrystEngComm, 2016, 18, 9056 RSC;
(e) M. S. Gargari, V. Stilinović, A. Bauzá and A. Frontera, Chem. – Eur. J., 2015, 21, 17951 CrossRef PubMed.
-
(a) M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS;
(b) J. Xie, S. Shen, R. Chen, J. Xu, K. Dong, J. Huang, Q. Lu, W. Zhu, T. Ma, L. Jia, H. Cai and T. Zhu, Org. Lett., 2017, 13, 4413–4419 CAS;
(c) Z. Y. Hao, Q. W. Liu, J. Xu, L. Jia and S. B. Li, Chem. Pharm. Bull., 2010, 58, 1306–1312 CrossRef CAS PubMed.
-
(a) G. Mahmoudi, J. K. Zaręba, A. Bauza, M. Kubicki, A. Bartyzel, A. Keramidas, L. Butusov, B. Miroslaw and A. Frontera, CrystEngComm, 2018, 20, 1065 RSC;
(b) A. A. Khandar, F. A. Afkhami, S. A. Hosseini-Yazdi, J. M. White, S. Kassel, W. G. Dougherty, J. Lipkowski, D. Van Derveer, G. Giester and F. Costantino, Inorg. Chim. Acta, 2015, 427, 87 CrossRef CAS;
(c) A. A. Khandar, F. A. Afkhami, S. A. Hosseini-Yazdi, J. Lipkowski, W. G. Dougherty, W. S. Kassel, H. R. Prieto and S. G. Granda, J. Inorg. Organomet. Polym., 2015, 25, 860 CrossRef CAS;
(d) F. A. Afkhami, A. A. Khandar, J. M. White, A. Guerri, A. Ienco, J. T. Bryant, N. Mhesn and C. Lampropoulos, Inorg. Chim. Acta, 2017, 457, 150 CrossRef CAS;
(e) F. A. Afkhami, A. A. Khandar, G. Mahmoudi, M. Amini, E. Molins, P. Garczarek, J. Lipkowski, J. M. White and A. M. Kirillov, Inorg. Chim. Acta, 2017, 458, 68 CrossRef CAS;
(f) D. Hean, T. Gelbrich, U. J. Griesser, J. P. Michael and A. Lemmerer, CrystEngComm, 2015, 17, 5143–5153 RSC;
(g) J. A. Lessa, G. L. Parrilha and H. Beraldo, Inorg. Chim. Acta, 2012, 393, 53–63 CrossRef CAS;
(h) G. N. Babu, A. R. B. Rao, S. Keesara and S. Pal, J. Organomet. Chem., 2017, 848, 243 CrossRef CAS;
(i) P. Doungdee, S. Sarel, N. Wongvisetsirikul and S. Avramovici-Grisaru, J. Chem. Soc., Perkin Trans. 2, 1995, 319 RSC.
-
(a) S. Yumnam and L. Rajkumari, J. Chem. Eng. Data, 2009, 54, 28 CrossRef CAS;
(b) C. M. Armstrong, P. V. Bernhardt, P. Chin and D. R. Richardson, Eur. J. Inorg. Chem., 2003, 1145 CrossRef CAS;
(c) C. B. Aakeröy, S. Forbes and J. Desper, CrystEngComm, 2012, 14, 2435 RSC;
(d) Z. He, C. He, E. Q. Gao, Z. M. Wang, X. F. Yang, C. S. Liao and C. H. Yan, Inorg. Chem., 2003, 42, 2206 CrossRef CAS PubMed;
(e) K. L. Abboud, R. C. Palenik, G. J. Palenik and R. M. Wood, Inorg. Chim. Acta, 2007, 360, 3642 CrossRef CAS;
(f) F. A. Afkhami, A. Akbar Khandar, G. Mahmoudi, W. Maniukiewicz, J. Lipkowski, J. M. White, R. Waterman, S. G. Granda, E. Zangrando, A. Bauzái and A. Frontera, CrystEngComm, 2016, 18, 4587 RSC.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer package, ver. 3.1, University of Western Australia, Perth, Australia, 2013 Search PubMed.
-
SAINT Plus, Data Reduction and Correction Program, v. 6.01, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
-
SADABS, v.2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
-
T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- T.-F. Zhu, R.-H. Chen, T.-L. Ma, Y. Wang and Z.-Q. Xu, Transition Met. Chem., 2015, 40, 485 CrossRef.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
-
(a) B. D. Ostojić, G. V. Janjić and S. D. Zarić, Chem. Commun., 2008, 6546–6548 RSC;
(b) Z. D. Tomić, S. B. Novaković and S. D. Zarić, Eur. J. Inorg. Chem., 2004, 11, 2215–2218 CrossRef;
(c) D. N. Sredojević, Z. D. Tomić and S. D. Zarić, Cent. Eur. J. Chem., 2007, 5, 20–31 Search PubMed;
(d) Z. D. Tomić, D. N. Sredojević and S. D. Zarić, Cryst. Growth Des., 2006, 6, 29–31 CrossRef;
(e) D. N. Sredojević, G. A. Bogdanović, Z. D. Tomić and S. D. Zarić, CrystEngComm, 2007, 9, 793–798 RSC;
(f) A. Castineiras, A. G. Sicilia-Zafra, J. M. Gonzales-Perez, D. Choquesillo-Lazarte and J. Niclos-Gutierrez, Inorg. Chem., 2002, 41, 6956–6958 CrossRef CAS PubMed;
(g) E. Craven, C. Zhang, C. Janiak, G. Rheinwald and H. Lang, Z. Anorg. Allg. Chem., 2003, 629, 2282–2290 CrossRef CAS;
(h) U. Mukhopadhyay, D. Choquesillo-Lazarte, J. Niclos-Gutierrez and I. Bernal, CrystEngComm, 2004, 6, 627–632 RSC;
(i) D. Pucci, V. Albertini, R. Bloise, A. Bellusci, A. Cataldi, C. V. Catapano, M. Ghedini and A. Crispini, J. Inorg. Biochem., 2006, 100, 1575–1578 CrossRef CAS PubMed;
(j) S. P. Mosae, E. Suresh and P. S. Subramanian, Polyhedron, 2009, 28, 245–252 CrossRef;
(k) X. J. Wang, H. X. Jian, Z. P. Liu, Q. L. Ni, L. C. Gui and L. H. Tang, Polyhedron, 2008, 27, 2634–2642 CrossRef CAS;
(l) S. Chowdhury, M. G. B. Drew and D. Datta, Inorg. Chem. Commun., 2003, 6, 1014–1016 CrossRef CAS;
(m) X. Wang, O. V. Sarycheva, B. D. Koivisto, A. H. McKie and F. Hof, Org. Lett., 2008, 100, 297–300 CrossRef PubMed.
-
(a) A. K. Tewari and R. Dubey, Bioorg. Med. Chem., 2008, 16, 126–143 CrossRef CAS PubMed;
(b) P. Mignon, S. Loverix, J. Steyaert and P. Geerlings, Nucleic Acids Res., 2005, 33, 1779–1789 CrossRef CAS PubMed;
(c) J. Sponer, K. E. Riley and P. Hobza, Phys. Chem. Chem. Phys., 2008, 10, 2595–2610 RSC;
(d) X. J. Wang, L. C. Gui, Q. L. Ni, Y. F. Liao, X. F. Jiang, L. H. Tang, L. H. Zhang and Q. Wu, CrystEngComm, 2008, 10, 1003–1010 RSC;
(e) S. L. Cockroft, C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Am. Chem. Soc., 2005, 127, 8594–8595 CrossRef CAS PubMed;
(f) T. Sato, T. Tsuneda and K. Hirao, J. Chem. Phys., 2005, 123, 104307–104317 CrossRef PubMed;
(g) S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430–3434 CrossRef CAS PubMed;
(h) M. Rubeš, O. Bludsky and P. Nachtigall, ChemPhysChem, 2008, 9, 1702–1708 CrossRef PubMed;
(i) E. C. Lee, D. Kim, P. Jurecka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446–3457 CrossRef CAS PubMed;
(j) M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2006, 110, 1065610668 CrossRef PubMed;
(k) R. Podeszwa, R. Bukowski and K. Szalewicz, J. Phys. Chem. A, 2006, 110, 10345–10354 CrossRef CAS PubMed;
(l) M. Pitonak, P. Neogrady, J. Rezac, P. Jurecka, M. Urban and P. Hobza, J. Chem. Theory Comput., 2008, 4, 1829–1834 CrossRef CAS PubMed.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- F. A. Afkhami, A. A. Khandar, G. Mahmoudi, W. Maniukiewicz, A. V. Gurbanov, F. I. Zubkov, O. Sahin, O. Z. Yesilel and A. Frontera, CrystEngComm, 2017, 19, 1389 RSC.
- S. Saha, N. Biswas, A. Sasmal, C. J. Gómez-García, E. Garribba, A. Bauza, A. Frontera, G. Pilet, G. M. Rosair, S. Mitra and C. Roy Choudhury, Dalton Trans., 2018 10.1039/C8DT02417K.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1867818 and 1867819. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01580e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Ali Akbar
Khandar
b,
Farhad Akbari
Afkhami
b,
Barbara
Miroslaw
*a,
Ali Akbar
Khandar
b,
Farhad Akbari
Afkhami
b,
Barbara
Miroslaw