
EDTA-assisted hydrothermal synthesis of cubic SrF2 particles and their catalytic performance for the pyrolysis of 1-chloro-1,1-difluoroethane to vinylidene fluoride†
Zhikun
Wang
,
Wenfeng
Han
 * and
Huazhang
Liu
* and
Huazhang
Liu
Institute of Industrial Catalysis, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou 310032, Zhejiang, PR China. E-mail: hanwf@zjut.edu.cn
First published on 21st December 2018
Abstract
Uniform, free-standing and cubic SrF2 microparticles were successfully fabricated by a facile hydrothermal method with ethylenediaminetetraacetic acid (EDTA) as the chelating agent. The influences of preparation conditions, such as the pH value, amount of EDTA and hydrothermal time, on the formation of SrF2 crystals were investigated. The formation mechanism of cubic SrF2 particles was proposed based on the experimental results. Following calcination in air at 500 °C, SrF2 particles were evaluated as the catalyst for the pyrolysis of 1-chloro-1,1-difluoroethane (HCFC-142b, CH3CClF2) to vinylidene fluoride (VDF, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) at 350 °C and a space velocity of 600 h−1. The results indicate that SrF2 cubes exhibit high catalytic activity with a HCFC-142b conversion of about 70% and a selectivity to VDF of 80–87%. No significant deactivation was observed within the time on stream of 30 h. With the reaction temperature increased to 450 °C, the conversion of HCFC-142b is close to 94%, while the selectivity to VDF remains almost unchanged. Although the SrF2 catalyst prepared by the conventional precipitation method also shows high conversion, its selectivity to VDF is only around 50–70%. We suggest that the surface acidity and specific surface area play major roles in the catalytic performance. Compared with the temperatures for industrial manufacture of VDF of 650–700 °C, the SrF2 catalysts provide a promising pathway to produce VDF at much lower temperatures.
CF2) at 350 °C and a space velocity of 600 h−1. The results indicate that SrF2 cubes exhibit high catalytic activity with a HCFC-142b conversion of about 70% and a selectivity to VDF of 80–87%. No significant deactivation was observed within the time on stream of 30 h. With the reaction temperature increased to 450 °C, the conversion of HCFC-142b is close to 94%, while the selectivity to VDF remains almost unchanged. Although the SrF2 catalyst prepared by the conventional precipitation method also shows high conversion, its selectivity to VDF is only around 50–70%. We suggest that the surface acidity and specific surface area play major roles in the catalytic performance. Compared with the temperatures for industrial manufacture of VDF of 650–700 °C, the SrF2 catalysts provide a promising pathway to produce VDF at much lower temperatures.
Introduction
Alkaline earth fluorides are important photonic materials because of their exclusive and diverse features.1,2 SrF2 is one of the typical earth fluorides. Nanomaterials of SrF2 with various morphologies and sizes have been successfully prepared by various methods, majorly for their luminescence. Doped with trivalent rare earth elements, such as Yb3+, Er3+, Tb3+ and so on, they function as satisfactory up-conversion luminescent phosphors.3–7In addition, homogeneous and monodispersed SrF2 nanocrystals were obtained in a water, ethanol and oleic acid system.8 Thermal decomposition of trifluoroacetate in a mixture of oleic acid, oleylamine and 1-octadecene also leads to the formation of SrF2 nanocrystals.9 The concentration of trifluoroacetate, decomposition temperature and decomposition time play major roles in the shapes of SrF2 crystals (from one dimension to three dimensions) and their corresponding luminescence performance. Hydrothermal synthesis was confirmed to be an effective route for the preparation of hollow micro-spheres, solid microspheres and hexagonal flakes of SrF2 and SrF2:Ln3+ (Ln = Eu, Ce, Tb).10 In addition, in the presence of different surfactants or chelating agents, micro and cubic SrF2 nanocrystals were achieved.11 However, there are very few reports on the application of SrF2 particles as catalysts, except as the promoter of catalysts for oxidative coupling of methane.12–14
Vinylidene fluoride (VDF, CH2CF2)15,16 is one of the most important fluorinated alkenes and monomers and is applied in various fields, such as the feedstock for the production of polyvinylidene fluoride (PVDF).17,18 In addition, it is also copolymerized with other monomers (such as hexafluoropropylene and vinylidene chloride) for the preparation of various co-polymers.19,20 It is also the key component of fluorine-containing resins and fluorine-containing rubbers.21–23 Due to its excellent performance,24 the demand for VDF is expected to increase. At present, thermal dehydrochlorination of HCFC-142b (1-chloro-1,1-difluoroethane, CH3CClF2) is the main method to manufacture VDF in the PVDF industry, although other routes such as co-pyrolysis of CHF3 and CH4 were reported.25–27 The pyrolysis of HCFC-142b takes place through reactions (1) and (2),15,28 leading to the formation of VDF and VCF (CH2CClF) at elevated temperatures. Industrially, the pyrolysis temperatures are usually between 650 °C and 700 °C. A HCFC-142b conversion of about 80–100% and a selectivity to VDF of about 85–95% are achieved under optimized conditions. However, the production suffers from significant coke deposition in the tubular reactors and has to be shut down for the removal of coke periodically (usually every two weeks).
| CH3CClF2(HCFC-142b) → CH2CF2(VDF) + HCl | (1) |
| CH3CClF2(HCFC-142b) → CH2CClF(VCF) + HF | (2) |
In addition, high reaction temperatures also lead to high energy consumption of the pyrolysis process. Furthermore, elevated temperatures usually result in significant amounts of by-products. Therefore, a catalyst is highly desired and it is an effective and environmentally friendly route for the pyrolysis of HCFC-142b and production of VDF. However, following dehydrochlorination, HCl is formed as a major by-product which poses a significant challenge for the survival of catalysts. To survive in a highly corrosive HCl atmosphere, the use of metal chlorides, metal fluorides, metal oxides, activated carbon (AC), and metal salts impregnated on activated carbon as catalysts was attempted. Herbert et al.29 adopted NiCl2 as the catalyst for HCFC-142b dehydrochlorination, achieving 80% conversion of HCFC-142b and 100% selectivity to VDF. R. Geetha et al.30 reported that BaCl2/AC catalyst suppresses side reactions significantly on HCFC-142b dehydrochlorination. Wang et al.31 suggested metal chlorides (FeCl3, CuCl2, and NiCl2) impregnated on activated carbon as catalysts, achieving 10–90% conversion of HCFC-142b and 60–90% selectivity to VDF at 400–630 °C. Unfortunately, the stability of the catalyst is far from industrial application. In our previous work,32 we found that N-doped carbon materials (NAC) are efficient catalysts in dehydrochlorination of HCFC-142b. Unfortunately, the NAC catalysts are difficult to be recovered after deactivation.
In the present work, uniform, free-standing and cubic SrF2 particles were prepared by a hydrothermal method with ethylenediaminetetraacetic acid (EDTA) as the chelating agent. The effect of preparation conditions on the formation of SrF2 cubic particles was investigated. Following calcination in air at 500 °C, SrF2 cubic particles were evaluated as the catalyst for pyrolysis of HCFC-142b for the first time. For comparison, a SrF2 catalyst prepared by the precipitation method was also included. SrF2 prepared by the hydrothermal method presents satisfactory performance for the pyrolysis of HCFC-142b. Hence, it provides a promising pathway to produce VDF through a low-temperature and energy-saving process (catalytic dehydrochlorination of HCFC-142b).
Experimental
Reagents and materials
Strontium chloride hexahydrate (≥99.5%), ethylenediaminetetraacetic acid, potassium hydroxide (≥95%), ammonium fluoroborate (≥99.5%) and ammonium fluoride (≥98%) were purchased from Aladdin Industrial Corporation (Shanghai, China). Ethanol (≥99.5%) and calcium chloride anhydrous were obtained from Sinopharm Chemical Reagent Co. LTD in China. HCFC-142b (99.8%), from Juhua Group Corporation (Quzhou, China), was adopted as the feed gas without further purification.Preparation of SrF2 cubic particles
Catalytic activity evaluation
The performance of the catalysts was evaluated using a fixed-bed reactor (nickel tube with an i.d. of 22 mm). The system was first purged with nitrogen to remove water vapour and air before each evaluation experiment at reaction temperatures. Then the gas phase of HCFC-142b with a GHSV (gas hourly space velocity) of 600 h−1, mixed with equivalent flow of nitrogen, was passed through the reactor. The effluent gas from the reactor passed into a scrubber containing about 1 M KOH solution (450 mL) to remove the acid gases of HCl and HF, followed by composition analysis with a Jie Dao GC-1690 gas chromatograph equipped with a thermal conductivity detector (TCD).Catalyst characterization
For morphology investigation, SEM (scanning electron microscopy) images were obtained from a FESEM, Hitachi S-4700 at an accelerating voltage of 15 kV, equipped with an X-ray energy spectrometer (EDS). The X-ray diffraction (XRD) patterns of the catalysts were recorded on a Kratos AXIS Ultra DLD analytical instrument. A monochromatic Al K radiation source (1486.6 eV) with an analyzer pass energy of 80 eV was operated at 3 mA and 15 kV. X-ray photoelectron spectroscopy (XPS) measurements of the catalysts were performed using a Thermo ESCALAB 250XI, with monochromatised Al Kα X-ray as the excitation source (24.2 W), with an analyser pass energy of 187.85 eV for survey scans and 46.95 eV for detailed elemental scans. In order to subtract the surface charging effect, binding energies were referenced to C1s binding energy of carbon, taken to be 284.6 eV. The XPS spectra were analysed by the XPS peak software. TEM (transmission electron microscopy) was adopted to further explore the microstructure of the catalysts using a JEOL 2100F transmission electron microscope at an accelerating voltage of 200 kV. The surface area and total pore volume of the catalysts were measured by N2 adsorption–desorption at −196 °C with a Quantachrome autosorb automated gas sorption system from USA. Prior to the measurement, the catalyst samples were degassed at 200 °C for 6 h under vacuum. Thermogravimetric analysis (TGA) was performed on a TA TGA Q500 instrument. The samples were heated in platinum crucibles up to 800 °C from room temperature with a heating rate of 10 °C min−1 in an atmosphere of 30 mL min−1 air. NH3-TPD was carried out in a self-made instrument and a thermal conductivity detector (TCD) was used for detecting the desorption of NH3.Results and discussion
Synthesis, morphology, and structure of SrF2 particles prepared by the hydrothermal method
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sr2+ = 1:1). The phase structures of synthesized SrF2 samples by the hydrothermal route were examined by XRD. Fig. 1 presents their XRD patterns.
:Sr2+ = 1:1). The phase structures of synthesized SrF2 samples by the hydrothermal route were examined by XRD. Fig. 1 presents their XRD patterns.
 | ||
| Fig. 1 XRD patterns of the as-prepared SrF2 samples with EDTA as the chelating agent (EDTA:Sr2+ = 1:1) at 160 °C for 24 h. During the hydrothermal synthesis, pH values were controlled at (1) pH = 5, (2) pH = 7, (3) pH = 9, and (4) pH = 11, respectively. The standard profile of SrF2 (PDF# 88-2294) was included as a reference. | ||
As indicated in Fig. 1, the XRD patterns of SrF2 obtained from hydrothermal synthesis agree well with that of the standard SrF2 pattern (PDF# 88-2294, space group: Fm3m (225)), indicating that the samples obtained at different pH values possess the cubic phase of the SrF2 crystal. No other impurities were identified. The strong, sharp diffraction peaks imply that the as-obtained samples are highly crystallized. In addition, with the variation of the pH value, nearly the same intensities based on the crystal diffraction peaks of (111), (220) and (311) of all the SrF2 samples indicate that the samples are composed of the same primary particles.
Although no significant difference was observed from the XRD patterns, the pH value of the solution during hydrothermal synthesis plays an important role in the size and the morphology of the synthesized samples. The pH dependent morphology evolution of SrF2 was investigated by SEM as illustrated in Fig. 2. With the pH value of 5, the SrF2 sample is mainly composed of aggregated particles (Fig. 2a). The cubic particles start to form when the pH value of the hydrothermal solution is higher than 7 (Fig. 2b). With the further increase of the pH value to 9, regular and well-defined cubic particles with an average size of 133.75 nm are obtained (Fig. 2c and e4). As revealed by the HRTEM images in Fig. 2e1 and e2, clear and solid SrF2 crystals are identified. As displayed in Fig. 2e3, the inter-planar distances between the adjacent lattice fringes are determined to be 0.335 and 0.290 nm, respectively, which are indexed to the d-spacing values of the (111) and (100) planes in the cubic SrF2 crystal. Clearly, more facets of (111) and (100) are exposed over these samples. The corresponding selected area electron diffraction (SAED) pattern suggests the poly-crystalline nature of the SrF2 cubic particles, indicating that the particles are not composed of individual crystals (Fig. 2e3). The diffraction dots can be indexed to (220), (311) and (420), corresponding to the (110), (311) and (210) planes of the SrF2 crystal, respectively. It is also proved by TEM images (Fig. 2e1 and e2) that cubic SrF2 is actually a multi-layer structure. The chemistry involved in the SrF2 formation process is illustrated in reactions (3)–(7).33,34 As demonstrated in reaction (3), the hydrolysis of BF4− releases F− which then combines with Sr2+ leading to the formation of SrF2 (reaction 4). According to the SEM images, when the pH value is lower than 9, aggregates of cubic particles are obtained. In particular, with the pH value of 5, no cubic SrF2 is derived. Clearly, in addition to the above two reactions, EDTA also plays an important role in the formation of SrF2 particles. It is well accepted that EDTA (H4Y for short) exhibits ionization equilibrium in the solution as expressed in reaction (5).35 When the pH value is low, the equilibrium shifts toward left for reaction (5) and majorly it is in the form of H4Y, which is only slightly soluble in water. Therefore, with a low pH value, EDTA shows weak chelating ability (reactions 6 and 7).36 As displayed in reaction (3), the hydrolysis of BF4− releases H+ and it leads to the decrease in the pH value to around 3 after the hydrothermal process. Hence, low pH values are not favourable for the formation of cubic SrF2 particles.
| [BF4]− + 3H2O → 4F− + 3H+ + H3BO3 | (3) |
| Sr2+ + 2F− → SrF2 | (4) |
H4Y ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [HxY]x−4 + (4 − x)H+ [HxY]x−4 + (4 − x)H+ | (5) |
| [HxY]x−4 + Sr2+ → [HxY]x−4 − Sr2+ | (6) |
| [HxY]x−4 − Sr2+ + 2F− → SrF2 + [HxY]x−4 | (7) |
 | ||
| Fig. 2 SEM images of SrF2 samples prepared at different pH values: (a) pH = 5, (b) pH = 7, (c) pH = 9 and (d) pH = 11; (e1) and (e2) TEM and inset showing the corresponding SAED pattern; (e3) HRTEM image of the SrF2 sample prepared at pH = 9; (e4) width distribution determined from the SEM image in (c). | ||
Fig. 3 illustrates the representative SEM images of the as-prepared SrF2 samples with different molar ratios of EDTA to Sr2+ at 160 °C for 24 h. Clearly, the morphology and size of SrF2 particles significantly change with the addition of EDTA. In the absence of EDTA in solution, the morphology with irregular and mixed-size SrF2 particles is derived (Fig. 3a). Following the introduction of EDTA, the morphologies and sizes differ dramatically. In the presence of a relatively small amount of EDTA (with an EDTA to Sr2+ ratio of 0.5:1), aggregates of fine powders were formed (Fig. 3b). Hence, the introduction of EDTA with an EDTA to Sr2+ ratio of 0.5:1 is not favourable for the formation of SrF2 cubic particles. On further increasing the ratio to 1:1.5, relatively small, ordered and uniform SrF2 cubes were fabricated (as shown in Fig. 2c). This indicates that the chelating agent, EDTA, plays an important role in the formation of uniform SrF2 cubes and prevents the particles from aggregating. However, with the molar ratio of EDTA to Sr2+ of 2:1, the excessive EDTA seems to facilitate the growth of SrF2. The size of the cubes increases to about 2 μm (Fig. 3d). In addition, with the ratio of 2:1, it is disclosed that SrF2 cubes are attached to SrF2 sheets. This reinforces the role of EDTA in the formation of SrF2 cubes which is consistent with the results mentioned previously.
 | ||
| Fig. 3 SEM images of the SrF2 samples obtained with different molar ratios of EDTA to Sr2+: (a) 0:1, (b) 0.5:1, (c) 1.5:1, and (d) 2:1. | ||
Based on the above results, we suggest that the formation of [HxY]x−4via dissociation of EDTA is inhibited by H+ released via the hydrolysis of BF4− (reaction 3) with low EDTA to Sr2+ ratios. Consequently, it is not favourable for EDTA to function as a chelating agent (reactions 6 and 7). However, with excessive amounts of EDTA, in addition to the chelating ability, it also serves as the layer growth agent. Hence, the EDTA to Sr2+ ratio of 1:1 is the most favourable for the formation of SrF2 microcubes.
:1. These experiments facilitate the disclosure of the formation and development of SrF2 particles.
Fig. 4 displays the SEM images of the samples obtained after hydrothermal treatment at different times. At the beginning (30 minutes of reaction, Fig. 4a), a large number of irregular nanoparticles with a diameter of 40–50 nm are found. After 45 minutes (Fig. 4b), large amounts of cubic SrF2 particles start to appear. The average length of the cube is about 60 nm. In addition, the surface of the cubic SrF2 particles is rough and covered with very small particles. After 1 hour of hydrothermal treatment (Fig. 4c), only cubic SrF2 particles are observed. The size and morphology of the cubes are similar to those of the particles shown in Fig. 3c. In addition, the cubic SrF2 particles continue to grow. The average length of the cubic SrF2 particles becomes 80 nm and the surface of the cubic SrF2 particles becomes smooth, as shown in Fig. 4c. When the hydrothermal treatment time is prolonged to 2 hours (Fig. 4d), the cubic SrF2 particles begin to aggregate as lengths higher than 200 nm are detected. After 4 hours (Fig. 4e), the cubic SrF2 particles further grow. After hydrothermal treatment of 8 hours (Fig. 4f), large SrF2 cubes are formed. However, small amounts of fine and irregular sheets are still observed in addition to the SrF2 cubes. Furthermore, the edges of the cubic particles start to become rounded, indicating Ostwald ripening with extended hydrothermal treatment.37 With further extending the treatment time to 12 hours and 24 hours, no significant change in morphology is observed except for agglomeration (Fig. 4g and h).
 | ||
| Fig. 4 SEM images of morphology evolution as a function of hydrothermal treatment time: (a) 30 min, (b) 45 min and the inset showing the corresponding high magnification image, (c) 1 h, (d) 2 h, (e) 4 h, (f) 8 h, (g) 12 h and (h) 24 h following hydrothermal treatment at 160 °C. | ||
 | ||
| Fig. 5 Schematic illustration of the morphological evolution process of the SrF2 cube architecture. | ||
The leaving [HxY]x−4 ions still surrounded the particle and some of the [HxY]x−4 ions were adsorbed over the particle surface function as the surfactant.38,39 The adsorption of [HxY]x−4 is not favourable for the aggregation of SrF2 cubes with several cube-like SrF2 particles.40,41 The surface elements of SrF2 obtained with hydrothermal treatment for 24 hours at a pH of 9, a temperature of 160 °C and an EDTA to Sr2+ ratio of 1:1 include C, N and O, in addition to Sr and F determined by XPS (Table 1). This indicates that the particles adsorbed significant amounts of [HxY]x−4 during preparation. In the formation process, similar to the surfactant, the chelating agent EDTA also plays a role in preventing the particles from aggregating and the evolution of the SrF2 shape. In addition, the above discussion is supported by the following results. Firstly, in Fig. 3a, it is observed that the SrF2 particles are irregular and larger in the absence of EDTA. Secondly, with low pH of the solution, the cubic SrF2 particles are not observed (Fig. 2a). Clearly, EDTA plays an important role in obtaining cubic SrF2 particles. Thirdly, as indicated in Fig. 2, cubic SrF2 particles possess clear multi-layer structures. Fig. 3d reinforces this structure. Hence, as illustrated in Fig. 5, we suggest that the initial grains of SrF2 particles (nuclei) with high surface energy adsorb the unsaturated [HxY]x−4–Sr2+. Then [HxY]x−4–Sr2+ bonds to fluorine ions deposited on the surface of the initial grains. In this way, numerous initial grains obtained through deposition of [HxY]x−4–Sr2+ form cubic SrF2 particles after several hours.
:1 (determined by XPS)
| Samples | Atomic contents (atom%) | ||||
|---|---|---|---|---|---|
| C | N | O | F | Sr | |
| SrF2 | 10.6 | 2.6 | 13.9 | 51.1 | 21.8 |
Although the adsorption of [HxY]x−4 prevents the cubes from agglomerating, a combination of cubes is still observed with extension of the hydrothermal treatment to longer than 2 h (Fig. 4e–h). As demonstrated in Fig. 4c, with a hydrothermal treatment of 1 h, the particle size of the cubes is around 80 nm. However, sizes of 150 nm to 300 nm are detected in Fig. 4e–h. We suggest that 2 to 4 cubes agglomerate into large cubes (Fig. 5).42
Catalytic activity and characterization of SrF2 catalysts prepared by precipitation and hydrothermal synthesis
The SrF2 catalysts prepared by precipitation and hydrothermal synthesis were first calcined in air at 500 °C for 4 h with a flow rate of 50 mL min−1. The derived catalysts were denoted as SrF2-P for precipitation and SrF2-H for hydrothermal synthesis, respectively. Pyrolysis of HCFC-142b (CH3CClF2) was adopted as a model reaction for the evaluation of the catalytic activity.
Fig. 6 displays the catalytic activities of the SrF2 catalysts for the pyrolysis of HCFC-142b to vinylidene fluoride as a function of time on stream (TOS). The reactions were carried out at 350 °C, a pressure of 1 bar and a GHSV (HCFC-142b) of 600 h−1. In addition to the major product VDF, the by-products obtained during the catalyst activity evaluation include CH2CClF (VCF, dehydrofluorination product of 1,1-chlorofluoroethane) and trace amounts of 1,1,1-trifluoroethane (HFC-143a, CH3CF3). As expected, the activity increases with reaction temperature (Fig. 6a). SrF2 starts to catalyse the decomposition of HCFC-142b at temperatures below 250 °C. It is worth noting that the SrF2 catalyst exhibits high activity as the conversion levels of HCFC-142b are about 70% and close to 100% at reaction temperatures of 350 °C and 450 °C, respectively. These temperatures are significantly lower than the temperatures for industrial manufacture of VDF of 650–700 °C. As demonstrated in Fig. 6a, the activity of SrF2-P is slightly higher than that of the SrF2-H catalyst within the temperature range investigated. However, at temperatures above 300 °C, the activities of both catalysts are close. The selectivity to the target product, VDF, over these two catalysts differs dramatically (Fig. 6b). A selectivity of 90–97% is achieved over the SrF2-H catalyst and it only changes with reaction temperature slightly. By contrast, the selectivity to VDF over the SrF2-P catalyst is significantly lower than that over SrF2-H. Although the SrF2 catalyst (SrF2-P) prepared by the conventional precipitation method shows relatively high conversion, its selectivity to VDF is only around 50–70%. As demonstrated in Fig. 6c and d, both catalysts exhibit stable activity at time on stream of 30 h at 350 °C.
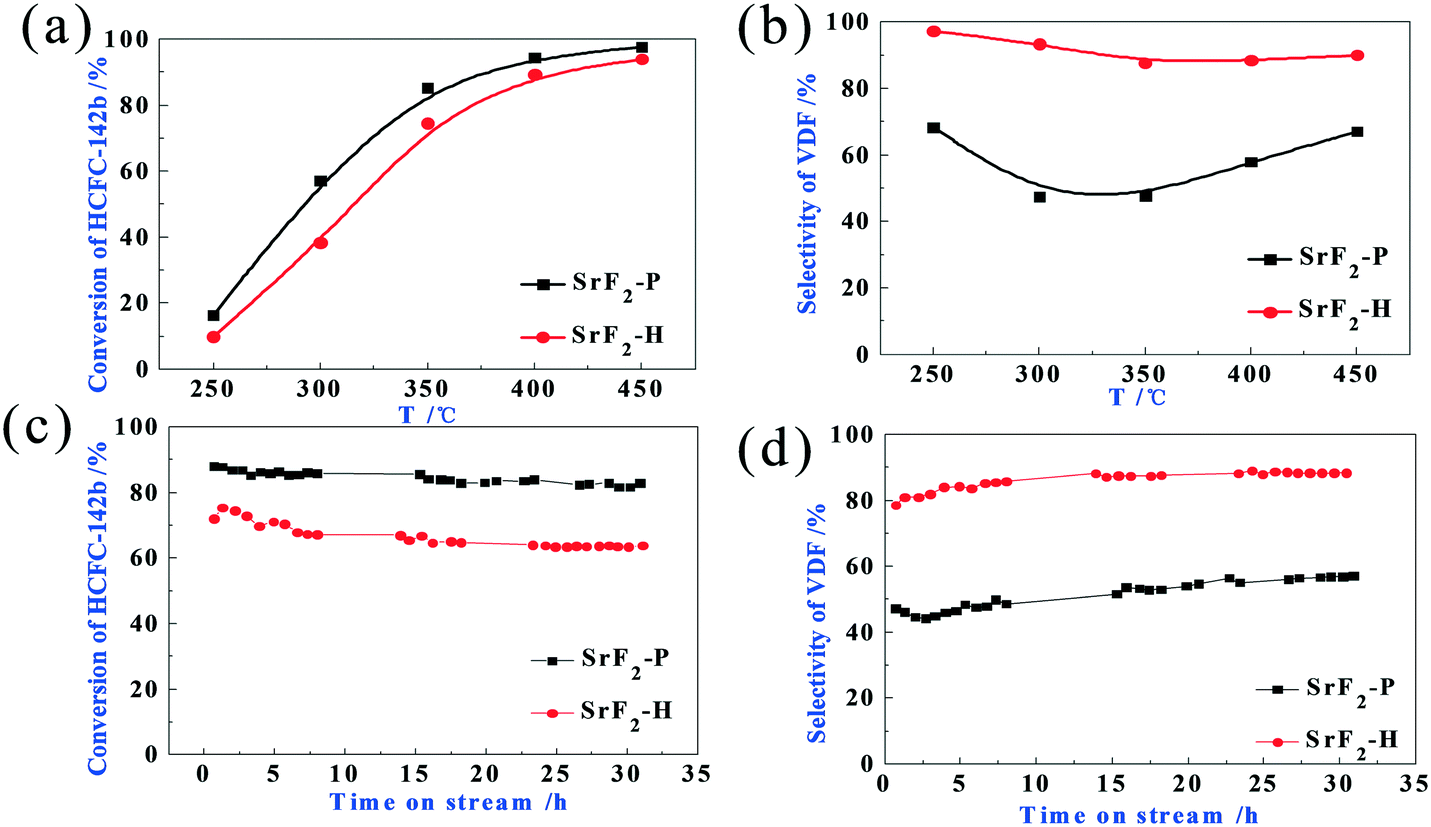 | ||
| Fig. 6 The performance of SrF2 catalysts prepared by precipitation (P) and the hydrothermal (H) method for the pyrolysis of HCFC-142b to vinylidene fluoride. (a) The conversion of HCFC-142b and (b) the selectivity to VDF as a function of reaction temperature. (c) The conversion of HCFC-142b and (d) the selectivity to VDF as a function of time on stream at 350 °C. Reaction conditions: 1 bar, N2:HCFC-142b of 1:1, GHSV (HCFC-142b) of 600 h−1. | ||
To elucidate the difference in catalytic activity between SrF2-P and SrF2-H catalysts, both catalysts were characterized by XRD and N2 adsorption–desorption isotherms. As shown in Fig. 7a, it is clearly observed that all the diffraction peaks of both catalysts are perfectly consistent with those of the standard SrF2 pattern (PDF# 88-2294) following calcination in air at 500 °C for 4 h. SrF2-P and SrF2-H are composed of the same crystalline grains. The strong, sharp diffraction peaks reveal that both catalysts are highly crystallized. However, the intensity of SrF2-P is a little higher than that of SrF2-H, indicating that the SrF2-P catalyst has higher crystallinity and possesses larger crystal particles. This agrees well with the SEM images which show that the particles of the SrF2-P catalyst are larger than those of SrF2-H, as shown in Fig. 8a and b. It is reasonable to infer that SrF2-P should possess a lower specific surface area than SrF2-H.
 | ||
| Fig. 7 (a) X-ray diffraction patterns and (b) N2 adsorption–desorption isotherms of SrF2-H and SrF2-P catalysts. | ||
 | ||
| Fig. 8 SEM images of (a) calcined SrF2-P and (b) calcined SrF2-H catalysts. Low and high magnification TEM images of the calcined SrF2-H catalyst (c1, c2 and c3). HRTEM image of the calcined SrF2-H catalyst (c4) and the inset showing the corresponding SAED pattern. | ||
The specific surface area was determined by N2 adsorption–desorption. The isotherms of both catalysts are illustrated in Fig. 7b. Clearly, both catalysts reveal typical type III isotherms and H3 hysteresis loops which are usually observed with aggregates of particles giving rise to slit-shape pores.43 The results of N2 adsorption indicate that the porous structures of both catalysts result from the aggregation of particles which agrees well with the observations of SEM and TEM. The surface area of SrF2-H and SrF2-P was determined to be 15.0 m2 g−1 and 5.5 m2 g−1, respectively (Table S1†). During the catalytic reaction of HCFC-142b pyrolysis, the metal fluoride serves as the active site for the dehydrochlorination and dehydrofluorination reactions of HCFC-142b.44 This suggests that the catalyst with a higher surface area usually has higher catalytic activity. However, the SrF2-H catalyst with a higher surface area and smaller crystal size does not lead to higher conversion of HCFC-142 compared with the SrF2-P catalyst. Hence, the crystal size and the surface area are not the key factors affecting the catalytic activity although they play important roles in the conversion of HCFC-142b.
Following calcination in air at 500 °C for 4 h, aggregation of fine particles is noted for the SrF2-P catalyst (Fig. 8a). By contrast, no obvious change in morphology is found over the SrF2-H catalyst (Fig. 8b). As shown in Fig. 8b and c1, c2, c3, the SrF2-H catalyst still maintains the cube-like particles after treatment at 500 °C for 4 h. It is observed that slight aggregation takes place. As indicated by the arrow in Fig. 8c4, it is observed that the surface of SrF2-H was capped by the carbon-containing compound derived from the calcination of adsorbed EDTA. As mentioned previously, the particles of SrF2 prepared by the hydrothermal method adsorb small amounts of EDTA. The adsorption sites prefer to locate at sites with large surface energy, more specifically the Lewis acid sites. Hence, the carbon-containing compound reduces the adsorption of the feed gas over the SrF2-H catalyst during the catalytic reaction.
The surface element content of the calcined SrF2-H catalyst determined by XPS is listed in Table 2, and the whole XPS spectrum of SrF2-H is shown in Fig. S1.† The results indicate that the surface of the SrF2-H catalyst after treatment at 500 °C still contains certain amounts of carbonaceous species. This further confirms that part of the SrF2-H surface is covered by the carbonaceous compound.
| Samples | Atomic contents (atom%) | ||||
|---|---|---|---|---|---|
| C | N | O | F | Sr | |
| SrF2 | 6.2 | 2.5 | 11.2 | 56.8 | 23.3 |
Generally, the acidity, more specifically, the Lewis acidic site of the catalyst plays a key role in dehydrochlorination and dehydrofluorination reactions.45 Teinz44 demonstrated that catalysts possessing strong Lewis acidity exhibit enhanced catalytic activity. However, strong Lewis acidity usually facilitates selective dehydrofluorination rather than dehydrochlorination leading to the poor selectivity to the dehydrochlorination product.46,47
As exhibited in Fig. 9a, the surface acidity of SrF2-P is significantly higher than that of SrF2-H. In addition, the SrF2-P catalyst displays relatively uniform acidic sites with an ammonia desorption temperature of around 520 °C. By contrast, NH3-TPD of the SrF2-H catalyst shows a weak and broad peak with desorption temperature between 100 and 600 °C. This indicates that acidic sites over SrF2-H are much fewer and weaker than those over SrF2-P. Consequently, it leads to the low conversion of HCFC-142b over SrF2-H. Although parts of the surface, especially the strong Lewis acidic sites, are covered by carbonaceous species, SrF2-H possesses a high specific surface area. Consequently, conversion of HCFC-142b close to SrF2-P is approached. Furthermore, as the strong acidic sites over SrF2-H are covered by the carbonaceous species, the SrF2-H catalyst exhibits a much higher selectivity to VDF (by dehydrochlorination) than VCF (dehydrofluorination).
 | ||
| Fig. 9 (a) The profiles of temperature-programmed desorption of ammonia on SrF2-H and SrF2-P catalysts and (b) DTG spectra of SrF2-H and SrF2-P catalysts. | ||
It is generally accepted that the Lewis acidic sites are coke formation centres.48–50 As a result, the catalyst with large amounts of Lewis acidic sites is favourable for carbon deposition. The coke formation over the catalysts was characterized by DTG as shown in Fig. 9b. It is observed that the weight loss rate of both catalysts is very small, indicating that there are very small amounts of coke after catalytic reaction for 30 h. Furthermore, SrF2-P has a higher weight loss than the SrF2-H catalyst, indicating that SrF2-P suffers from greater coke formation. In addition, a weight loss over 700 °C is noted for the spent SrF2-H catalyst. It is reasonable to infer that the weight loss is ascribed to the carbonaceous species covering the surface of the SrF2-H catalyst.
Conclusions
In summary, cube-like architectures of SrF2 particles were synthesized through a convenient and facile hydrothermal synthesis route with ethylenediaminetetraacetic acid as the chelating agent. During the hydrothermal synthesis of SrF2, the pH of the solution plays an important role in the morphology and size of SrF2 particles. The suitable synthesis conditions for cube-like SrF2 were determined to be a pH of 9 and an EDTA:Sr2+ of 1:1 at 160 °C. The formation process of the free-standing and uniform cube-like sub-micro particles was proposed based on the above results.
The obtained SrF2 samples were adopted as efficient catalysts for the pyrolysis of 1-chloro-1,1-difluoroethane to vinylidene fluoride. The SrF2-P catalyst prepared by the precipitation method is included as a reference. The results indicate that both SrF2 catalysts exhibit high catalytic activity with high HCFC-142b conversion and VDF selectivity at temperatures between 250 °C and 450 °C. No deactivation was observed following time on stream of 30 h at 350 °C. The selectivity to the target product, VDF, over the SrF2-H catalyst is much higher than that over SrF2-P. The surface acidity, the size of particles and the specific surface area of SrF2 play important roles in VDF selectivity and catalytic performance. The carbonaceous species capping the surface of the SrF2-H catalyst are responsible for the high selectivity to VDF.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Zhejiang Provincial Natural Science Foundation of China (LY19B060009), the Qianjiang Talent Project B in Zhejiang Province (2013R10056), and Special Programs for Research Institutes in Zhejiang (2015F50031) is acknowledged.References
- P. P. Fedorov, A. A. Luginina, S. V. Kuznetsov and V. V. Osiko, J. Fluorine Chem., 2011, 132, 1012–1039 CrossRef CAS.
- P. P. Fedorov, S. V. Kuznetsov and V. V. Osiko, in Photonic and Electronic Properties of Fluoride Materials, ed. A. Tressaud and K. Poeppelmeier, Elsevier, Boston, 2016, pp. 7–31 Search PubMed.
- C. M. Zhang, Z. Y. Hou, R. T. Chai, Z. Y. Cheng, Z. H. Xu, C. X. Li, L. Huang and J. Lin, J. Phys. Chem. C, 2010, 114, 6928–6936 CrossRef CAS.
- P. P. Fedorov, A. A. Luginina, J. A. Ermakova, S. V. Kuznetsov, V. V. Voronov, O. V. Uvarov, A. A. Pynenkov and K. N. Nishchev, J. Fluorine Chem., 2017, 194, 8–15 CrossRef CAS.
- S. Kuznetsov, Y. Ermakova, V. Voronov, P. Fedorov, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, J. Mater. Chem. C, 2018, 6, 598–604 RSC.
- A. A. Lyapin, S. V. Gushchin, A. S. Ermakov, S. V. Kuznetsov, P. A. Ryabochkina, V. Y. Proydakova, V. V. Voronov, P. P. Fedorov and M. V. Chernov, Chin. Opt. Lett., 2018, 16, 091901 CrossRef.
- Y. A. Rozhnova, S. V. Kuznetsov, A. A. Luginina, V. V. Voronov, A. V. Ryabova, D. V. Pominova, R. P. Ermakov, V. A. Usachev, N. E. Kononenko and A. E. Baranchikov, Mater. Chem. Phys., 2016, 172, 150–157 CrossRef CAS.
- X. Zhang, Z. Quan, J. Yang, P. Yang, H. Lian and J. Lin, Nanotechnology, 2008, 19, 075603 CrossRef PubMed.
- Y. P. Du, X. Sun, Y. W. Zhang, Z. G. Yan, L. D. Sun and C. H. Yan, Cryst. Growth Des., 2009, 9, 2013–2019 CrossRef CAS.
- Y. P. Sun and P. Y. Jia, J. Nanosci. Nanotechnol., 2014, 14, 3957–3960 CrossRef CAS PubMed.
- J. Y. Sun, J. B. Xian, X. Y. Zhang and H. Y. Du, J. Rare Earths, 2011, 29, 32–38 CrossRef CAS.
- R. Q. Long and H. L. Wan, J. Chem. Soc., Faraday Trans., 1998, 94, 1129–1135 RSC.
- L. Wang, X. Yi, W. Weng, C. Zhang, X. Xu and H. Wan, Catal. Lett., 2007, 118, 238–243 CrossRef CAS.
- L. H. Wang, X. D. Yi, W. Z. Weng and H. L. Wan, Catal. Today, 2008, 131, 135–139 CrossRef CAS.
- C. L. Yaws, McGraw-Hill handbooks, 1999, vol. 4, pp. 48–52 Search PubMed.
- B. Ameduri, Chem. Rev., 2009, 109, 6632–6686 CrossRef CAS PubMed.
- B. Hu, C. Lei, R. Xu, W. Shi, Q. Cai, H. Mo and C. Chen, J. Plast. Film Sheeting, 2014, 30, 300–313 CrossRef CAS.
- T. H. Han, R. Nirmala, T. W. Kim, R. Navamathavan, H. Y. Kim and S. J. Park, J. Nanosci. Nanotechnol., 2016, 16, 595–600 CrossRef CAS PubMed.
- C. G. Zhang, W. W. Liu, C. Cao, F. Y. Zhang, Q. M. Tang, S. Q. Ma, J. J. Zhao, L. Hu, Y. Shen and L. L. Chen, Adv. Healthcare Mater., 2018, 7, e1701466 CrossRef PubMed.
- J. X. Zhang, X. Y. Du, C. C. Wang and K. L. Ren, J. Phys. D: Appl. Phys., 2018, 51 Search PubMed.
- J. Chen, S. B. Tan, G. X. Gao, H. Y. Li and Z. C. Zhang, Polym. Chem., 2014, 5, 2130–2141 RSC.
- S. B. Tan, J. J. Li, G. X. Gao, H. Y. Li and Z. C. Zhang, J. Mater. Chem., 2012, 22, 18496–18504 RSC.
- Y. Kano, S. Akiyama, T. Kasemura and S. Takahashi, Polym. Networks Blends, 1996, 6, 153–159 CAS.
- T. Soulestin, V. Ladmiral, F. D. Dos Santos and B. Améduri, Prog. Polym. Sci., 2017, 72, 16–60 CrossRef CAS.
- Y. M. Cheng, J. C. Wang, W. F. Han, Y. Y. Song, W. C. Liu, L. T. Yang, S. C. Wang, Z. X. Wu, H. D. Tang, J. J. Zhang, M. Stockenhuber and E. M. Kennedy, Greenhouse Gases: Sci. Technol., 2018, 8, 587–602 CrossRef CAS.
- W. F. Han, Y. Y. Song, W. C. Liu, L. T. Yang, H. D. Tang, S. C. Wang, Z. X. Wu and J. J. Zhang, Greenhouse Gases: Sci. Technol., 2017, 7, 891–902 CrossRef CAS.
- W. F. Han, Y. Li, H. D. Tang and H. Z. Liu, J. Fluorine Chem., 2012, 140, 7–16 CrossRef CAS.
- G. Wang, H. F. Zheng, H. Yin, S. F. Yuan and Z. R. Chen, J. Zhejiang Univ., Sci., A, 2015, 49, 1812–1816 Search PubMed.
- M. Herbert, E. Gerhard and H. Hanns, Chem. Ing. Tech., 1984, 56, 626–628 CrossRef.
- R. Geetha, Pop. Plast. Packag., 1998, 43(9), 67–70 CAS.
- G. Wang, H. Zheng, H. Yin, S. Yuan and Z. Chen, Chem. React. Eng. Technol., 2016, 32, 73–77 CAS.
- Z. Wang, W. Han, C. Zhang, S. Zhou, H. Wang, H. Tang and H. Liu, ChemistrySelect, 2018, 3 Search PubMed.
- H. Guo, Y. Guo, H. M. Noh, B. K. Moon, S. H. Park, J. H. Jeong and K. H. Kim, J. Nanosci. Nanotechnol., 2016, 16, 1146–1150 CrossRef CAS PubMed.
- B. L. Gersten, M. M. Lencka and R. E. Riman, J. Am. Ceram. Soc., 2004, 87, 2025–2032 CrossRef CAS.
- R. S. Juang, Y. J. Chen and I. P. Huang, Sep. Sci. Technol., 1999, 34, 3099–3112 CrossRef CAS.
- Y. Zhang, Q. Zhao, B. Shao, W. Lu, X. Dong and H. You, RSC Adv., 2014, 4, 35750–35756 RSC.
- Y. K. Bai, R. T. Zheng, Q. Gu, J. J. Wang, B. S. Wang, G. A. Cheng and G. Chen, J. Mater. Chem. A, 2014, 2, 12770–12775 RSC.
- M. Chatti, S. Sarkar and V. Mahalingam, Microchim. Acta, 2016, 183, 133–140 CrossRef CAS.
- K. Ariga, J. P. Hill and Q. M. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC.
- R. L. Penn and J. F. Banfield, Science, 1998, 281, 969–971 CrossRef CAS PubMed.
- J. J. De Yoreo, P. Gilbert, N. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Z. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Colfen and P. M. Dove, Science, 2015, 349, aaa6760 CrossRef PubMed.
- H. Suo, Z. Yang, C. F. Guo, X. Q. Zhao and L. L. Zhang, CrystEngComm, 2016, 18, 3566–3573 RSC.
- G. C. Bond, K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Annexes, Wiley-VCH Verlag GmbH, 2008 Search PubMed.
- K. Teinz, S. Wuttke, F. Börno, J. Eicher and E. Kemnitz, J. Catal., 2011, 282, 175–182 CrossRef CAS.
- L. E. Manzer and V. N. M. Rao, Adv. Catal., 1993, 39, 329–350 CAS.
- W. Mao, Y. Bai, W. Wang, B. Wang, Q. Xu, L. Shi, C. Li and J. Lu, ChemCatChem, 2017, 9, 824–832 CrossRef CAS.
- K. Teinz, S. R. Manuel, B. Bin Chen, A. Pigamo, N. Doucet and E. Kemnitz, Appl. Catal., B, 2015, 165, 200–208 CrossRef CAS.
- R. M. Navarro, M. C. Alvarez-Galvan, M. C. Sanchez-Sanchez, F. Rosa and J. L. G. Fierro, Appl. Catal., B, 2005, 55, 229–241 CrossRef CAS.
- J. Ni, L. Chen, J. Lin and S. Kawi, Nano Energy, 2012, 1, 674–686 CrossRef CAS.
- W. F. Han, C. P. Zhang, H. L. Wang, S. L. Zhou, H. D. Tang, L. T. Yang and Z. K. Wang, Catal. Sci. Technol., 2017, 7, 6000–6012 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Textural parameters of SrF2 samples, XPS spectra, and SEM images obtained with different solution concentrations. See DOI: 10.1039/c8ce01546e |
| This journal is © The Royal Society of Chemistry 2019 |