DOI:
10.1039/C8CE01015C
(Paper)
CrystEngComm, 2019,
21, 2286-2302
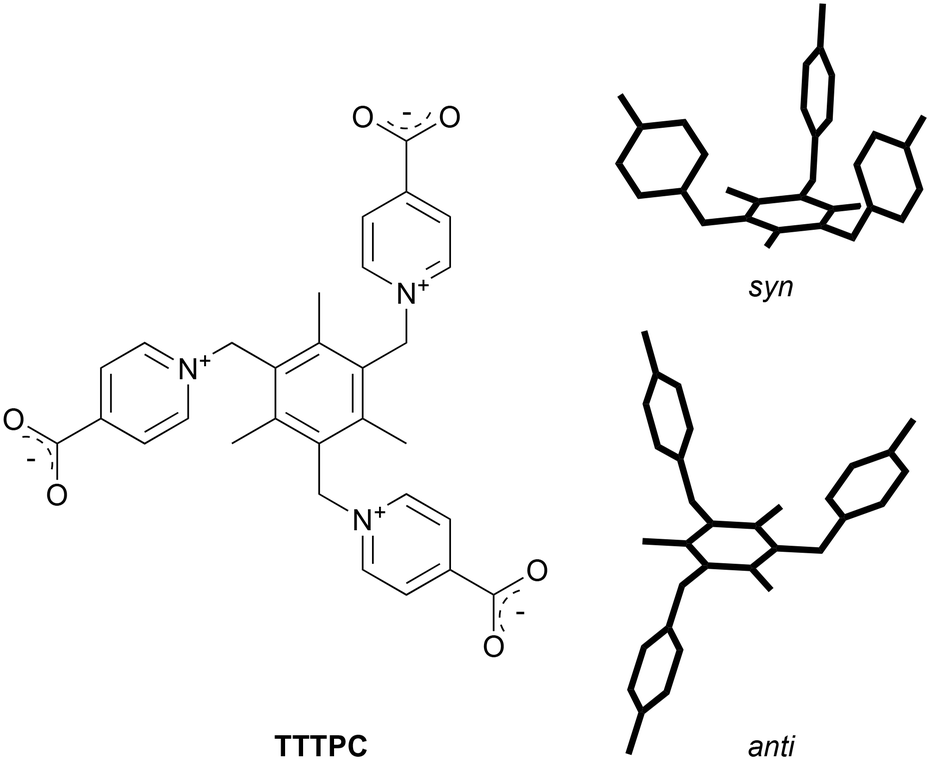
In-depth structural analysis of lanthanoid coordination networks based on a flexible tripodal zwitterionic isonicotinate ligand†
Received
19th June 2018
, Accepted 16th February 2019
First published on 18th February 2019
Abstract
Crystallizing metal–organic frameworks (MOFs) has been studied using a tripodal pyridinecarboxylic acid derivative ligand and selected lanthanoid salts. The zwitterionic ligand, 1,1′,1′′-((2,4,6-trimethylbenzene-1,3,5-triyl)tris(methylene))tris(pyridin-1-ium-4-carboxylate) (TTTPC) introduced as a bromide salt, forms coordination networks in aqueous environments and under ambient conditions with neodymium bromide, trifluoromethanesulfonate (OTf) or acetate (OAc). Seven structures are elucidated in detail using single crystal X-ray crystallography. TTTPC NdBr3, TTTPC NdBr2OTf, TTTPC NdBr(OTf)2 and TTTPC Nd(OTf)3 are porous 3D networks with similar ligand–metal and ligand–anion interactions, but with different anion distributions, coordination modes and/or crystal systems. Freshly prepared TTTPC NdBr2OAc is a porous 2D network, and otherwise has the same attributes, but it transforms into a 3D network upon drying. All network solids crystallize in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (# 2), except TTTPC NdBr(OTf)2 which crystallizes in P21/c (# 14). Compounds retain their crystallinity under vacuum, and a crystal structure for an evacuated sample of TTTPC NdBr2OTf is presented. Thermal analysis of network solids shows that upon heating, all solids exhibit solvent loss and withstand decomposition up to or over 300 °C. In addition to Nd networks, synthesis and crystal structures of several exactly or almost isostructural systems with other lanthanoids are presented. These include TTTPC YbBr3, TTTPC LnBr2OTf (Ln = La, Sm, Eu, Gd and Tb), TTTPC LnBr(OTf)2 (Ln = Sm, Eu, and Tb), TTTPC Yb(OTf)3 and TTTPC2Sm2(OTf)6. The synthesis and crystal structures of Ln(NO3)3 (Ln = La, Nd, and Y) are also briefly discussed as a separate, yet similar, system. Finally, a revised crystal structure of the protonated bromide TTTPC ligand, H2.5TTTPC, is suggested.
(# 2), except TTTPC NdBr(OTf)2 which crystallizes in P21/c (# 14). Compounds retain their crystallinity under vacuum, and a crystal structure for an evacuated sample of TTTPC NdBr2OTf is presented. Thermal analysis of network solids shows that upon heating, all solids exhibit solvent loss and withstand decomposition up to or over 300 °C. In addition to Nd networks, synthesis and crystal structures of several exactly or almost isostructural systems with other lanthanoids are presented. These include TTTPC YbBr3, TTTPC LnBr2OTf (Ln = La, Sm, Eu, Gd and Tb), TTTPC LnBr(OTf)2 (Ln = Sm, Eu, and Tb), TTTPC Yb(OTf)3 and TTTPC2Sm2(OTf)6. The synthesis and crystal structures of Ln(NO3)3 (Ln = La, Nd, and Y) are also briefly discussed as a separate, yet similar, system. Finally, a revised crystal structure of the protonated bromide TTTPC ligand, H2.5TTTPC, is suggested.
1 Introduction
Metal–organic coordination polymers and network solids are widespread and interesting groups of hybrid materials that have plenty of engineerable properties suitable for different applications. By a recommended definition, coordination polymers as a general term include coordination networks, which in turn contain metal–organic frameworks as specific subsets.1 Network solids are used as a generalization to represent the latter two groups in this paper. Metal–organic frameworks (MOFs) are especially well studied and highly sophisticated porous 3D networks, which are about to see broad use in small molecule capture, separation and storage, catalysis, sensing, drug delivery, magnetic and luminescence applications, etc.2,3 Conventionally MOFs are constructed using small rigid organic linker molecules between stringently coordinating metal nodes, such as aromatic carboxylates and transition metals. However, stable systems can also be designed using larger multitopic ligands and coordinatively less demanding metals.4,5 Lanthanoids (also widely known as lanthanides, nowadays a non-IUPAC recommended name) are a group of fifteen f-block metals from La to Lu, and they are well suited as nodes for the flexible subgroup of MOFs, because their coordination environments are geometrically less strict and their coordination numbers tend to be larger than those of conventional d-block metals, owing to their larger ionic radii.6 In addition, their ligand–metal interactions are mostly electrostatic, their ligand field effect is small and their coordination modes are mainly dictated by steric effects of the ligands. According to the hard–soft acid–base (HSAB) theory, lanthanoids are hard acids and readily form coordination compounds with hard O-donor bases, of which carboxylates are by far the most widely studied and reported ligand group.7,8 Owing to the large enthalpy of hydration of lanthanoids, complexes in aqueous environments can be favorably stabilized by entropic effects when using multidentate and multitopic ligands, if desolvation can be overcome first.6 Overall, it is a rational approach to engineer lanthanoid MOFs (Ln-MOFs) in water by implementing carboxylate functional groups in a suitable flexible multipodal ligand with a prominent ionic character.
A well-reported tripodal zwitterionic pyridiniumcarboxylate ligand, namely 1,1′,1′′-((2,4,6-trimethylbenzene-1,3,5-triyl)tris(methylene))tris(pyridin-1-ium-4-carboxylate) (TTTPC, Scheme 1), has been a subject of study in previous publications as a component of coordination polymers with both d-block metals9–15 and f-block metals.16–20TTTPC can adopt several conformers and it can act as a versatile bridging ligand through its three carboxylate groups, and therefore its coordination chemistry with metals is very diverse. Also a somewhat interesting property of TTTPC is that it can readily bind anions with its zwitterionic pyridinium rings by two types of attractive forces that can be described as C–H⋯X− and charge transfer interactions.21 Essentially, the C–H⋯X− interaction is a hydrogen bond (HB) between a soft acid and a hard base (according to the HSAB theory), in this case the hard base being a lone electron pair of an anion.7,22 Because of several different types of ligand interactions, an approach reviewed by Li et al. regarding multi-functional sites in MOFs applies when using TTTPC.3 First d-block metal polymers were reported by Kong and Wu, utilizing Cu, Zn and Cd.9 These polymers consist of TTTPC molecules in a syn “bowl” conformation. A few years passed until Zhuang et al. reported a 2D network structure with Mn and TTTPC, with the ligand adopting again its syn conformation.10 Afterwards, Wen et al.16 published the first 3D Ln-MOF structure with Eu and the TTTPC ligand in an anti “chair” conformation, soon followed by Bag et al.17 with similar structures synthesized on a larger scale and composed of a series of lanthanoids. Zhao et al. expanded the 3D framework engineering by publishing a diamondoid structure with Cd, TTTPC and 1,4-benzenedicarboxylic acid as an auxiliary ligand.11 Respectively, Su et al. published a 2D network with M3L2 cages as repeating units, also with Cd as the node, and 1,3,5-benzenetricarboxylic acid acting as an auxiliary ligand.12 An et al. has worked with Cd as well and published a MOF structure where the ligand was in an exceptional twisted anti conformation.13 As the sole species of actinoids related to the ligand TTTPC, uranium has been used successfully in two studies so far, namely by Bai et al. and by Liang et al.18,19 The latest studies in recent years have been conducted by Lian et al.20 using lanthanoids, by Zhang et al.14 with Cd and by Zhou et al.15 using Cu as node metals.
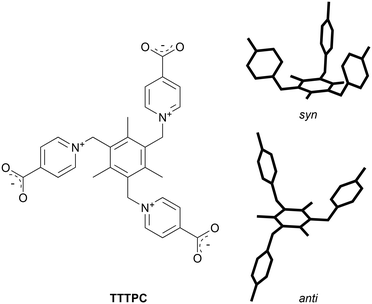 |
| | Scheme 1 Structure of the zwitterionic ligand TTTPC and its two conformers (right), the bowl-shaped syn and the chair-shaped anti conformer (carboxylate moieties and chemical information removed for illustrative purposes). | |
Here we report additional crystal structures that expand the field studied most closely by Wen et al. and Bag et al.16,17 Being a versatile ligand, different structures can be obtained with TTTPC by broadening the scope of conditions under which the network solid synthesis (crystallization) takes place. A change in these conditions either initially or gradually during the crystallization process clearly affects the product outcome. Initially, we wanted to make a series of Ln-MOFs by reacting the ligand bromide with different metal triflate salts, namely La, Nd, Sm, Eu, Gd, Tb and Yb. We wanted to see if the ionic radius of the metal has any driving force towards a specific structure type, and we supposed that it has a minor role at most. However, eventually we experimented with more variables rather than only this aspect and later concentrated on a single metal (Nd) with emphasis on ligand–anion interactions. Specifically, we synthesized and determined the single crystal structures of a complete bromide–triflate bianionic series of a general LMXnYm type, where L is the TTTPC ligand, M is Nd3+ or another lanthanoid(III) and X and Y are the anions (n = 3 − m). In addition to making the TTTPC neodymium Br–OTf series, we also experimented with a directly metal coordinating anion with carboxylate functionalization, to see whether it would impose a change in the aforementioned structures by competing with the tripodal ligand in a controlled fashion. Acetate was chosen because of its availability and simplicity, and it resulted in two unprecedented network solid structures. A freshly prepared single crystal structure was found to differ when its simulated powder diffraction pattern was compared to experimental dry bulk powder samples. Eventually, however, the dry single crystal structure was determined after some effort, and the phase transition event from the fresh to dry form was recorded with a powder diffractometer. A brief survey with homoanionic nitrate systems was made as well, first by exchanging the ligand bromide to nitrate, followed by crystallizing Ln-MOFs with lanthanoid nitrates. All acquired TTTPC network solid structures turned out to be anisotropically porous systems with solvent (water) filled channels running through the material. By exposing bulk samples to a medium vacuum, the solvent could be partially removed without the crystal structure collapsing, and a single crystal structure was also obtained from one said sample. Various physicochemical analyses were conducted with the dry bulk samples obtained in this way. The morphology and elemental distribution were studied using scanning electron microscopy (SEM) imaging and built-in energy-dispersive X-ray spectroscopy (EDX). Solid state UV-vis spectrophotometry was applied in bulk sample light absorption studies. Thermogravimetric analysis was conducted on both fresh network solid samples and evacuated samples, and a comparison was made between these regarding solvent removal and structure stability. Brunauer, Emmett and Teller (BET) surface area analyses were conducted to assess the material porosity and gas adsorption properties. Lastly, we felt a need to revise the first reported TTTPC ligand crystal structure reported by Kong and Wu9 and it will be discussed in accordance with our characterization results.
2 Experimental
For the details of the syntheses, calculations of theoretical yields and characterization of individual intermediates and products not presented in the main text, see the ESI.† Also see the ESI† for all the physicochemical analysis results and their discussion (elemental analyses, powder diffraction, thermogravimetry, electron microscopy, spectroscopy and BET).
2.1 Materials
All commercial reagents, acids and solvents were used as obtained without further purification. 2,4,6-tris(bromomethyl)mesitylene (Aldrich, 98% purity), ethyl isonicotinate (Aldrich, 98% purity), neodymium acetate hydrate (Aldrich, 99.9% purity) and potassium trifluoromethanesulfonate (potassium triflate, Aldrich, 98% purity) were purchased from Sigma-Aldrich. Triflate salts of neodymium and other lanthanoids were purchased from Strem Chemicals Ltd. Neodymium oxide (Fluka, 99.999% purity) was converted to the respective bromide by dissolving it in an excess aqueous solution of 62% (v/v) hydrogen bromide, crystallizing the crude hydrate salt from the acidic solution and finally recrystallizing from deionized water.
2.2 Preparation of H2.5TTTPC
Ligand synthesis was conducted by following literature methods of preparing a TTTPC ethyl ester by Menschutkin reaction, followed by acidic de-esterification reaction and recrystallization from water.9Scheme 2 represents the overall ligand synthesis procedure. See the ESI† for full details and the discussion related to a previously somewhat inadequately reported9 single crystal structure of this compound, to which we present an improved interpretation. Also see the ESI† for nitrate anion exchange experiment details of H2.5TTTPC.
 |
| | Scheme 2 Overall synthesis of H2.5TTTPC. | |
2.3 Preparation of coordination network solids
Synthesis of coordination network solids was conducted under ambient conditions by crystallization in 20 ml borosilicate glass jars, where the starting materials were measured and dissolved in deionized water. Ligand to metal (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M) amounts generally followed a 1:1 ratio, but to obtain the acetate structure, a ratio of 1:2 had to be used. Crystallization attempts were carried out using a vapor diffusion technique. Through free acetone (anti-solvent) vapor diffusion into the crystallization vessels, initial crystals suitable for X-ray diffraction measurements were obtained usually within a week, but the process was carried out for a longer time to enhance the product yield. Nd containing products were generally colored in varying shades of pink or purple under natural light, but under fluorescent light they were colorless, or in the case of the tribromide, pale yellow. See the ESI† for additional experimental details for smaller scale syntheses of other lanthanoids, and additional obtained network solid structures, namely TTTPC YbBr3, TTTPC LnBr2OTf (Ln = La, Sm, Eu, Gd and Tb), TTTPC LnBr(OTf)2 (Ln = Sm, Eu, and Tb), TTTPC Yb(OTf)3, TTTPC2Sm2(OTf)6 and Ln(NO3)3 (Ln = La, Nd, and Y).
:M) amounts generally followed a 1:1 ratio, but to obtain the acetate structure, a ratio of 1:2 had to be used. Crystallization attempts were carried out using a vapor diffusion technique. Through free acetone (anti-solvent) vapor diffusion into the crystallization vessels, initial crystals suitable for X-ray diffraction measurements were obtained usually within a week, but the process was carried out for a longer time to enhance the product yield. Nd containing products were generally colored in varying shades of pink or purple under natural light, but under fluorescent light they were colorless, or in the case of the tribromide, pale yellow. See the ESI† for additional experimental details for smaller scale syntheses of other lanthanoids, and additional obtained network solid structures, namely TTTPC YbBr3, TTTPC LnBr2OTf (Ln = La, Sm, Eu, Gd and Tb), TTTPC LnBr(OTf)2 (Ln = Sm, Eu, and Tb), TTTPC Yb(OTf)3, TTTPC2Sm2(OTf)6 and Ln(NO3)3 (Ln = La, Nd, and Y).
2.3.1 TTTPC NdBr3.
250.0 mg (0.32 mmol) of H2.5TTTPC and 160.9 mg (0.33 mmol, calculated as hexahydrate) of NdBr3 were dissolved in 25 ml of deionized H2O and separated evenly in four 20 ml glass crystallization vessels (roughly 6–7 ml of solution per vessel). Acetone diffusion over a week yielded small, irregular crystal clusters. A sample of sufficient quality for single crystal experiments was found amidst a multitude of chunks from taking a larger crystal cluster apart. After acetone diffusion for several weeks, 305.0 mg (87.0% yield) of TTTPC NdBr3 was obtained after a gentle wash with a water and acetone mixture and keeping the product in a desiccator overnight. The bulk product appearance was a crystalline, dry (non-hygroscopic) and light purple solid.
2.3.2 TTTPC NdBr2OTf.
251.7 mg (0.33 mmol) of H2.5TTTPC and 194.9 mg (0.33 mmol) of Nd(OTf)3 were dissolved in 25 ml of deionized H2O and separated evenly as above. Acetone diffusion over four days yielded thin rods and blocks suitable for diffraction experiments. After vapor diffusion for several weeks, 256.7 mg of the TTTPC NdBr2OTf product was obtained after a gentle wash with water and acetone followed by acetone and keeping the product in a desiccator overnight. The bulk product appearance was a crystalline, dry (non-hygroscopic) and pale pink solid. Phase purity was assessed using FT-IR and powder diffraction, and the bulk product was identified as a mixture of products similar to the next crystallization result. However, this product was kept parallel in the characterization for comparison.
2.3.3
TTTPC NdBr(OTf)
2
and TTTPC Nd(OTf)3.
259.8 mg (0.34 mmol, 1 eq.) of H2.5TTTPC, 201.0 mg (0.34 mmol, 1 eq.) of Nd(OTf)3 and 383.7 mg (2.04 mmol, 6 eq.) of KOTf were dissolved in 25 ml of deionized H2O and separated evenly as above. Acetone diffusion over a week yielded needle-like crystals of sufficient size for single crystal experiments. After crystallization for an additional week, interpenetrated plate-like crystals could be found amidst the needles, and there were some amounts of block-like crystals on the vessel walls as well. The crystal structure of the first identified component was determined, with the needle-like crystals being TTTPC Nd(OTf)3. The structure of the second component, TTTPC NdBr(OTf)2 (plates) was also determined, and finally the blocks were identified as the aforementioned TTTPC NdBr2OTf. The vessel was left to crystallize for a few weeks more, and a compound mixture (TTTPC NdBrOTfMix) was collected after washing with water and acetone and drying in a desiccator overnight. 249.7 mg of this mixture was obtained. The product appeared to be similar to the previously crystallized TTTPC NdBr2OTf, and concordantly their characteristics were observed to be almost identical in the conducted analyses. Some of the bulk product was evacuated afterwards (see section 2.5), and an exceptionally good crystal was found from the mixture, so that a single crystal structure of TTTPC NdBr2OTf (vac) could be obtained (see section 3.2).
2.3.4 TTTPC NdBr2OAc.
254.7 mg (0.33 mmol) of H2.5TTTPC and 224.9 mg (0.70 mmol, calculated as anhydrous) of Nd(OAc)3 were dissolved in 25 ml of deionized H2O and separated evenly as above. Acetone diffusion over a few days yielded rods and planks suitable for initial diffraction experiments. After vapor diffusion for several weeks, 280.3 mg (74.9% yield) of TTTPC NdBr2OAc was obtained after a gentle wash with a water and acetone mixture followed by acetone and drying in a desiccator overnight. The bulk product appearance was a dry (non-hygroscopic) and pink solid that eroded to a fine crystalline powder upon drying. A second batch was also made later, and after sufficient crystallization for several weeks, the mother liquor was removed and the products were washed as before, but drying took place in the crystallization vessels under ambient conditions instead. This yielded dry, but more intact crystals, and a slightly different crystal structure of TTTPC NdBr2OAc (dry) was obtained (see section 3.3), which was a better representative of the dry bulk powder than the fresh system.
2.4 Crystallography
Single crystal diffraction data were collected using a Rigaku Oxford Diffraction SuperNova Dual-source X-ray diffractometer, with Mo and Cu radiation hi-flux microfocus sources (Mo Kα, λ = 0.71073 Å and Cu Kα, λ = 1.54184 Å) and an Atlas CCD detector installed. Mo radiation was used for collecting the data of the H2.5TTTPC ligand, and Cu was used respectively for all the other compounds throughout this work. Crystals were held at a constant temperature of 123 or 120 K during data collection using a liquid nitrogen cooled CryoStream device. The CrysAlisPro software package (ver. no. 171.38.43 and 171.39.43c) was used to conduct data collection and reduction and to apply Gaussian absorption correction during data finalization, based on experimental crystal faces modelled with a recorded crystal movie. Within the OLEX2 structure solution and refinement program23 (ver. no. 1.2.9), all structures were solved using SUPERFLIP charge flipping and EDMA electron density map analysis programs24–26 and refined using SHELXL27 least-squares full-matrix minimization on |F|2. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were calculated isotropically using the standard OLEX2 riding model (for aromatic hydrogen atoms and –CH2– groups: Uiso = 1.2× of the parent atom, and for other groups: Uiso = 1.5× of the parent atom). Occupancies of disordered moieties were determined using free variables in the refinement. Electron density from severely disordered solvent (water) was first examined and then removed on the final refinement of the structure using the OLEX2 solvent mask (SM) tool. Each removed electron density of 10 e− was estimated to correspond to one water molecule, which was taken into account in the chemical formula. Selected crystal structure information and refinement indicators are listed in Table 1 for the Nd network solids discussed in the main text, and additional information can be found in the ESI† (Tables S2, S4, S5, S7, S9, S11, S12, S14, S16 and S17). The Cambridge structural database (CSD) was referred when comparing results to published structures.28 Crystallographic data correspond to the CSD reference numbers 1833428–1833446, 1842357, 1842358 and 1894870. Crystal structures were visualized using either OLEX2 or Mercury (ver. no. 3.10.1).23,29
Table 1 Crystal structure and refinement information for the network solids
| Name |
TTTPC NdBr
3
|
TTTPC NdBr
2
OTf
|
TTTPC NdBr
2
OTf (vac)
|
TTTPC NdBr(OTf)
2
|
TTTPC Nd(OTf)
3
|
TTTPC NdBr
2
OAc
|
TTTPC NdBr
2
OAc (dry)
|
|
Solvent water removed with the OLEX2 solvent mask has been included in the calculations.
|
| Empirical formulaa |
C30H45Br3N3NdO15 |
C30.68H53Br2.31F2.06N3NdO21.06S0.69 |
C31H31Br2F3N3NdO11S |
C31.65H43Br1.35F4.94N3NdO19.29S1.65 |
C33H46F9N3NdO25S3 |
C32H56Br2N3NdO21 |
C32H44Br2N3NdO15 |
| Formula weight |
1071.66 |
1191.27 |
1014.71 |
1173.14 |
1296.15 |
1122.85 |
1014.76 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P (# 2) |
P (# 2) |
P (# 2) |
P21/c (# 14) |
P (# 2) |
P (# 2) |
P (# 2) |
|
a/Å |
9.4378(3) |
9.5721(4) |
9.4141(3) |
18.7643(2) |
10.1157(3) |
9.1533(9) |
8.7309(9) |
|
b/Å |
12.3546(3) |
14.4092(6) |
12.0776(3) |
25.6962(3) |
14.2420(4) |
14.3573(13) |
12.2707(11) |
|
c/Å |
18.8810(4) |
18.7321(7) |
18.7454(5) |
9.85675(12) |
19.1578(7) |
18.8779(16) |
19.3079(11) |
|
α/° |
104.683(2) |
106.854(4) |
105.103(2) |
90 |
110.861(3) |
107.505(8) |
106.661(7) |
|
β/° |
98.809(2) |
103.019(4) |
98.057(2) |
102.8557(12) |
101.670(3) |
103.919(8) |
100.429(7) |
|
γ/° |
99.428(2) |
99.792(4) |
99.489(2) |
90 |
91.706(3) |
96.822(8) |
91.716(8) |
| Volume/Å3 |
2056.71(10) |
2331.09(18) |
1991.69(9) |
4633.50(10) |
2510.09(16) |
2247.0(4) |
1941.6(3) |
|
Z
|
2 |
2 |
2 |
4 |
2 |
2 |
2 |
| Reflections collected |
12580 |
14235 |
12783 |
16990 |
15789 |
13462 |
12498 |
| Independent reflections |
7446, Rint = 0.0275 |
8450, Rint = 0.0310 |
7241, Rint = 0.0255 |
8412, Rint = 0.0245 |
9109, Rint = 0.0595 |
8126, Rint = 0.0427 |
7055, Rint = 0.0284 |
| Final R indices [I ≥ 2σ(I)] |
R
1 = 0.0449, wR2 = 0.1163 |
R
1 = 0.0612, wR2 = 0.1752 |
R
1 = 0.0367, wR2 = 0.0944 |
R
1 = 0.0464, wR2 = 0.1295 |
R
1 = 0.0526, wR2 = 0.1363 |
R
1 = 0.0430, wR2 = 0.1097 |
R
1 = 0.0251, wR2 = 0.0589 |
| Final R indices [all data] |
R
1 = 0.0463, wR2 = 0.1179 |
R
1 = 0.0642, wR2 = 0.1788 |
R
1 = 0.0378, wR2 = 0.0952 |
R
1 = 0.0552, wR2 = 0.1363 |
R
1 = 0.0556, wR2 = 0.1402 |
R
1 = 0.0498, wR2 = 0.1163 |
R
1 = 0.0272, wR2 = 0.0600 |
| GooF on F2 |
1.038 |
1.047 |
1.050 |
1.068 |
1.042 |
1.038 |
1.007 |
Powder diffraction patterns were collected from gently mortar ground bulk samples on a PANalytical X'Pert Pro alpha 1 diffractometer in the Bragg–Brentano geometry using a fixed-anode Cu tube with a Johansson monochromator (CuKα1λ = 1.5406 Å; 45 kV and 40 mA). Data were routinely collected from samples prepared on a spinning concave zero-background (ZBC) silicon disc with an X'Celerator detector over a 2θ-range of 5–90°. The step size and time per step were respectively 0.008° and 180 s. When analyzing the phase transition of fresh TTTPC NdBr2OAc, a wet crystalline sample was gently smeared between two Kapton plastic films in a proprietary stainless steel holder, and then data were immediately collected using the parameters above, followed by waiting for 24 h and measuring the same sample in the intact holder again. Real time ambient phase transition monitoring was conducted using again the ZBC disc, but with data collected repeatedly over a 2θ-range of only 5–50° and setting the step size and time to 0.017° and 16 s, respectively, so that one run took approximately six minutes. Diffraction data were analyzed on a computer using the PANalytical HighScore Plus program (v. 4.7).
2.5 Thermogravimetry
Dry crystalline samples of 5–10 mg in weight were analyzed with a PerkinElmer STA 6000 TG/DSC simultaneous thermal analyzer over a temperature range of 25–850 °C. Measurement was conducted in an open platinum crucible under an air or nitrogen atmosphere with a gas flow rate of 40 ml min−1 and a heating rate of 10 °C min−1. Device temperature calibration was made with the melting point onsets of indium and aluminum (standard values of 156.60 °C and 660.1 °C, respectively). Heat flow calibration was made using the standard enthalpy of fusion of indium (28.45 J g−1). Weight calibration was made at ambient temperature using a manufacturer provided steel ball bearing with a standardized reference weight of 55.98 mg. Samples were prepared before measurement at room temperature either by drying in a desiccator over a silica gel bed or by keeping them in a constant 1 × 10−6 bar vacuum for three days.
2.6 SEM imaging and EDX
Scanning electron microscopy (SEM) imaging and energy-dispersive X-ray fluorescence spectroscopy (EDX) analyses were conducted from vacuum dried gold coated crystalline bulk samples. Gold coating was applied using a JEOL fine coat ion sputter JFC-1100 device. The microscope equipment used was Zeiss EVO-50XVP with a Bruker Quantax 400 ED spectrometer installed.
2.7 Solid state UV-vis spectrophotometry
Absorption spectra of solid powder samples were measured from 800 to 200 nm under ambient conditions using a PerkinElmer Lambda 650 spectrophotometer equipped with a Spectralon-coated 150 mm integrating sphere assembly. Samples were center-mounted in the sphere between two vertical quartz plates in a clamp holder that aligned the sample approximately perpendicular to the incoming light beam, and only a thin layer of the sample was prepared to allow adequate pass-through of incident and scattered light.
2.8 BET surface area analysis
Brunauer, Emmett and Teller (BET) surface area30 analyses were performed using a Micromeritics Gemini VII 2390 t surface area analyzer using N2 as the adsorptive gas under liquid nitrogen isothermal conditions. Crystalline samples with amounts of 100–160 mg were freshly prepared and kept slightly moist before introducing them first into a Micromeritics VacPrep061 for a minimum of 4 h of degassing at 150 °C, followed by the appropriate analysis procedure.
3 Results and discussion
3.1 Structural description of the as-synthesized network solids
TTTPC LnBr
3
, TTTPC LnBr
2
OTf and TTTPC Ln(NO3)3 are all topologically identical, crystallize in the same triclinic crystal system and space group P, and mainly differ only by their anion distribution in the crystal lattice. These factors result in slight but noticeable differences e.g. in the unit cell parameters (Table 1), metal coordination environment (Fig. 1), and structure porosity and dimensions (e.g. Fig. S25†). In the cases of TTTPC LnBr3 and TTTPC LnBr2OTf, as well as the two following structure types, Nd structures will be discussed in detail as archetypes. However, a comparative emphasis on TTTPC YbBr3 as a representative of heavier lanthanoids is also given. The trinitrate structures are presented and discussed entirely in the ESI, to prevent exhaustive repetition. The evacuated single crystal structure of TTTPC NdBr2OTf (vac) is discussed in its own section (3.2). TTTPC LnBr(OTf)2 is the only type of structure in this paper exhibiting monoclinic symmetry in the space group P21/c (# 14) with Z = 4. TTTPC Ln(OTf)3 and TTTPC NdBr2OAc crystallize again in the P space group and they present their own traits in the metal coordination sphere. After drying under ambient conditions, the structure of TTTPC NdBr2OAc (dry) is also presented in its own section (3.3) with emphasis on well resolved H-bonding in the water channel. All structures exhibit similar ligand–anion interactions, which will be first discussed in detail to better account for the build-up of the structures.
 |
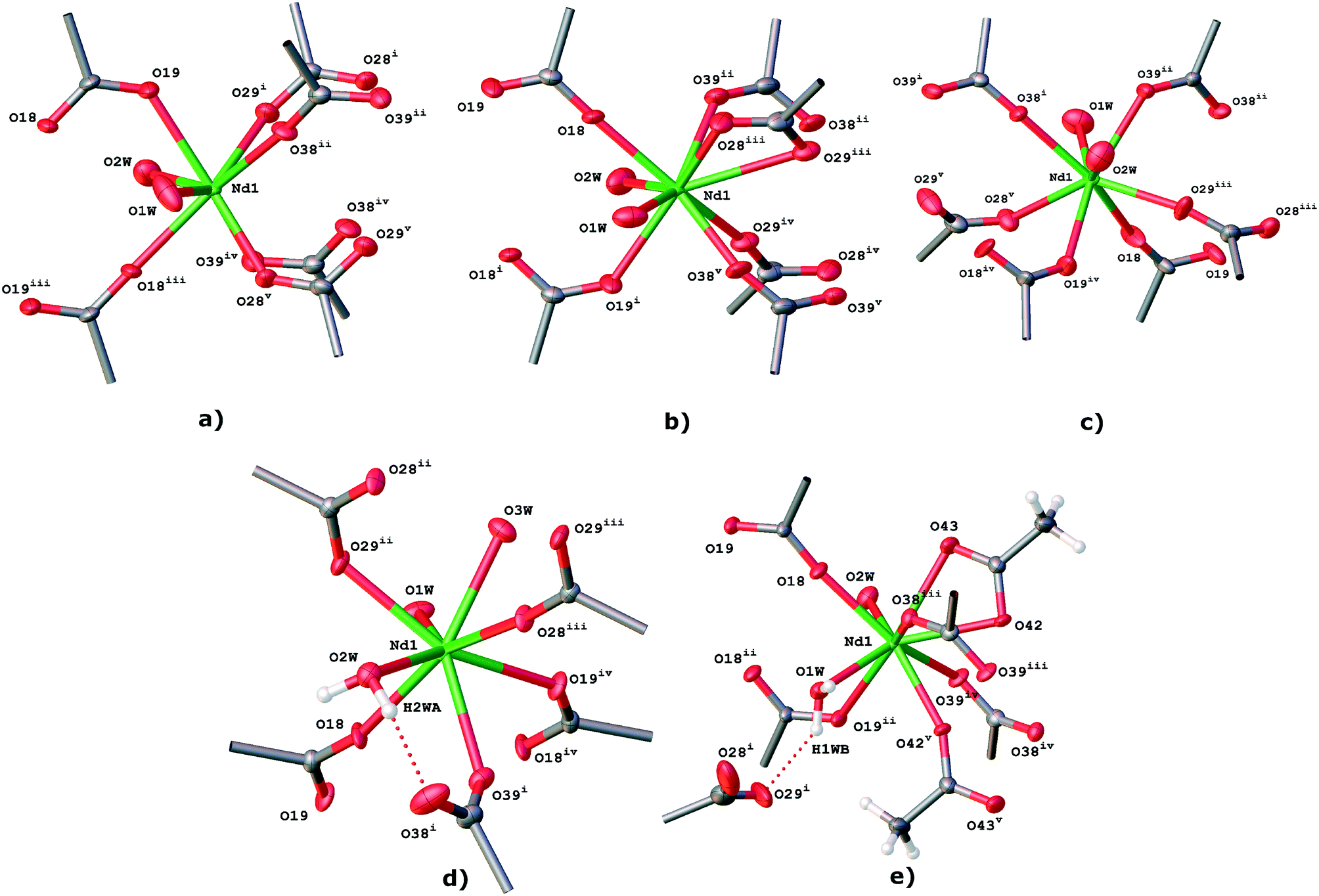
| | Fig. 1 Coordination environments of TTTPC a) NdBr3, b) NdBr2OTf, c) NdBr(OTf)2, d) Nd(OTf)3, and e) NdBr2OAc (for the legend of symmetry labels, see Table S1†). Thermal ellipsoids are depicted at the 50% probability level here, and in the following figures where applicable. Some hydrogen atoms are removed for clarity. | |
3.1.1 Asymmetric unit, disorder and ligand–anion interactions.
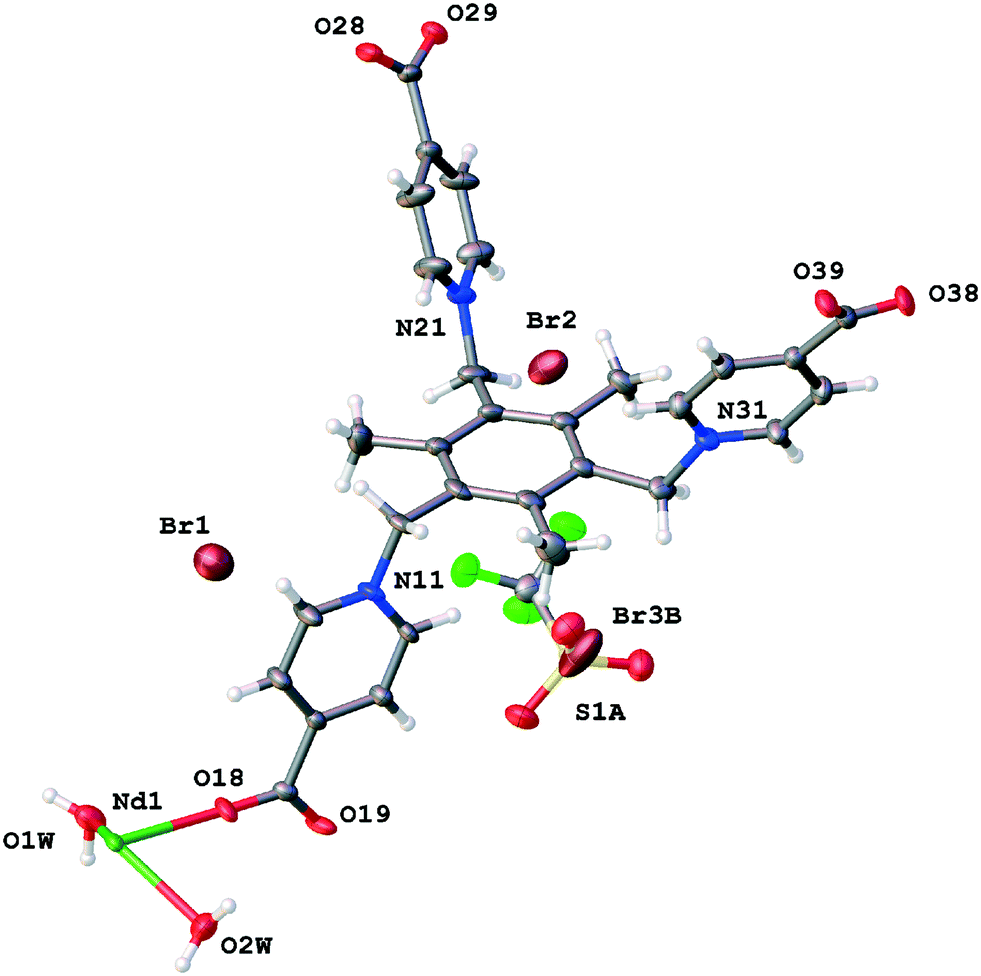
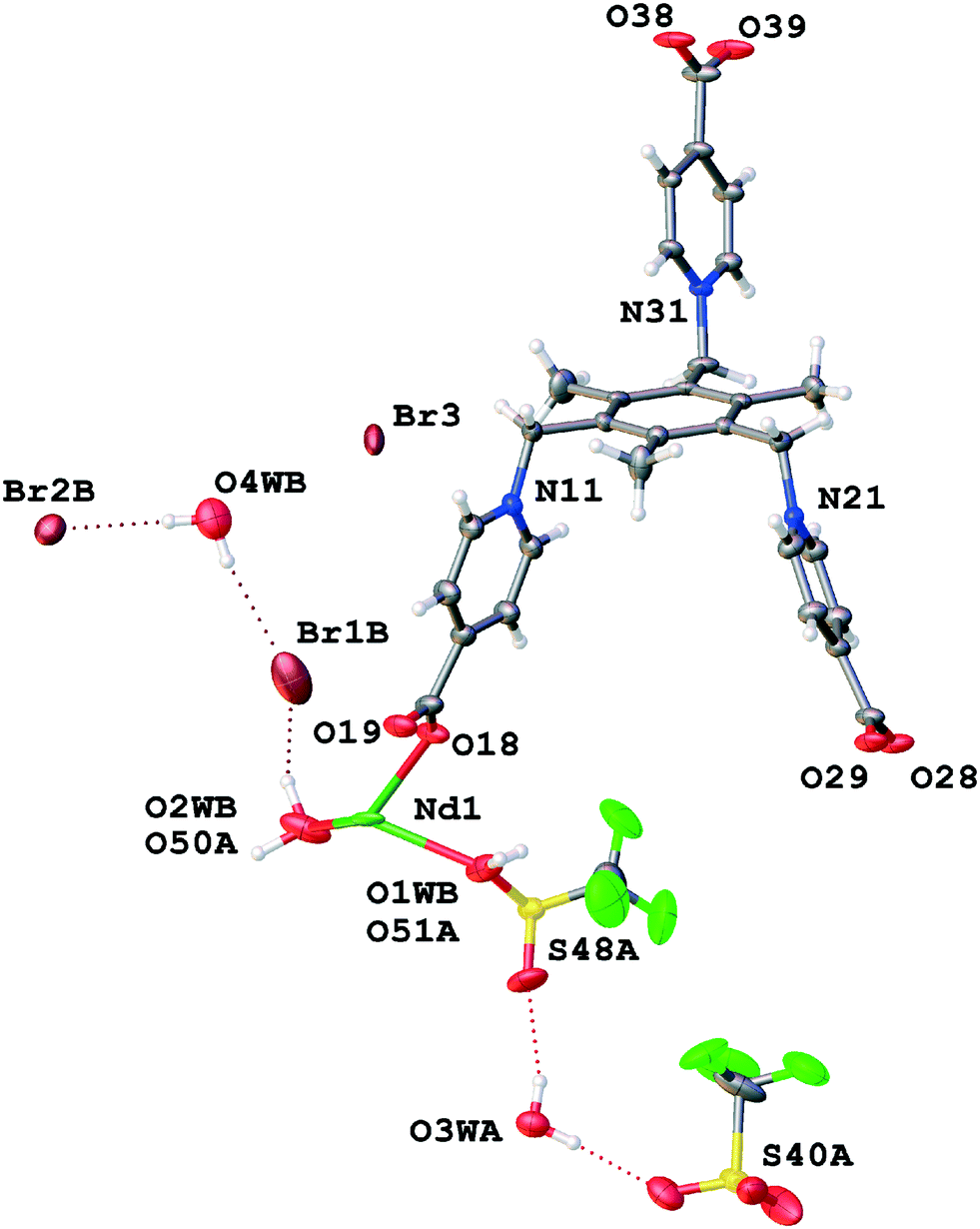
The asymmetric unit of TTTPC NdBr3 contains one crystallographically independent neodymium (III) cation (Nd1) with two coordinated water molecules (O1W and O2W) and one zwitterionic TTTPC ligand molecule. The ligand has an anti “chair” conformation and it coordinates to six metal centers, with each carboxylate acting as a bidentate and bridging two metals together. This coordination mode is one of the most commonly observed, and can be indicated with a notation of μ2-η1η1 ZZ.8 The coordination number of the Nd1 center is eight and its geometry follows a slightly distorted bicapped trigonal prismatic (or alternately distorted square antiprismatic) motif. Fig. 2 displays the contents of the asymmetric unit of TTTPC NdBr3 and Fig. 1a displays the coordination sphere geometry and relationships of the Nd1 center. A total of three bromides are situated between the ligand moieties and are not directly coordinated to the metal. Two of the three bromides (Br1 and Br2) have full occupancy, but one bromide is disordered over two positions (Br3A and Br3B, with occupancies of approximately 40% and 60%, respectively). This is probably because of specific C–H⋯Br− interactions between the ligand framework and the anions, and the lack thereof, in addition to conventional H-bonding with solvent water. Br1 is almost completely enclosed by the partially electron-deficient ligand framework (Fig. S1†), bound with distinctive pyridinium ring ortho-C–H⋯Br− HB direct contacts (o-pyr, C12–H12⋯Br1 and C16–H16⋯Br1). Br2 and Br3A are similar in such a sense that they are more exposed than Br1, and they both symmetrically occupy the water channel walls bound with pyridinium C–H⋯Br− interactions (C22–H22⋯Br2 (o-pyr), C26–H26⋯Br2 (o-pyr), C32–H32⋯Br3A (o-pyr) and C35–H35⋯Br3A (meta-pyridinium, m-pyr)). Br3A additionally has contacts to two mesitylene center methyl groups (C8–H8B⋯Br3A and C9–H9C⋯Br3A) and to the pyridinium ring next to it (N11, C12, etc.). o-Pyr, m-pyr and CH3 contact types are displayed with Br3A in Fig. 3. Br3B has only a single methyl group contact (C8–H8C⋯Br3B) with the ligand framework. Since it resides entirely in the water channel, it is conventionally hydrogen bonded with diffuse water instead of having prominent C–H⋯Br− interactions. As described in the experimental part, solvent water has been removed, and its electron density suggests approximately seven molecules to be present in the asymmetric unit in addition to those coordinated to the metal center. The Yb counterpart of this structure, TTTPC YbBr3 (Fig. S2†), bears resemblance to the Nd system in almost every way by having a similar topology, metal coordination geometry and ligand–anion interactions. The only difference is that bromide anions are distributed differently and have different contact points due to ligands being in a slightly different preferred conformation. Br1 is analogous to the Br2 in the previous Nd structure (and vice versa). Br1 has one CH3 contact (C7–H7A⋯Br1) and three pyridinium ring contacts (C12 (o-pyr), C15 (m-pyr) and C16 (o-pyr)). C15 and C16 thus make a bridging interaction (μ-pyr), forming a structure similar to a chelate ring. Br2 has one CH3 contact (C7–H7⋯Br2) and two o-pyr contacts (C22 and C26). Br3 is disordered over two positions (occupancies of Br3A: 75% and Br3B: 25% approximately). Br3A has two o-pyr contacts (C32 and C36) as well as one CH3 contact C8–H8A⋯Br3A. Br3B is not situated in the middle of the channel, but resides next to Br3A, offset towards the metal center. It only has a borderline m-pyr contact (C33–H33⋯Br3B, Fig. S3†) in addition to apparent H-bonding with the solvent water. Overall, the C-H⋯Br− interactions in TTTPC NdBr3 and TTTPC YbBr3 fall within reasonably similar distances (H-Br−, from 2.620 Å to 2.999 Å) and bond angles (C–H–Br−, from 131.30° to 175.07°), with the o-pyr type interactions being the most prominent. All identified C–H⋯Br− contacts in these structures are listed in Table S3.† Some of the listed o-pyr interactions with a sufficiently small bond angle may also be interpreted as being half of a μ-pyr system, such as C22/C23 in TTTPC NdBr3 and C32/C33 in TTTPC YbBr3. Lastly, the direct pyridinium ring contact of Br3A in TTTPC NdBr3 is a strong indication of electrostatic attraction between the anion and the framework, seen also with almost all other anions in every system presented in this paper, but not as clearly. These interactions can be identified as being most likely charge transfer type interactions, instead of anion–π, because the anions tend to be located clearly offset from the ideal position (not directly over the ring centroid).21 Most likely these interactions are complementary driving forces to the C–H⋯Br− H-bonding when packing and self-assembly during crystallization are considered, and they may also contribute to anion binding in the solution, although such studies are beyond the scope of this paper.
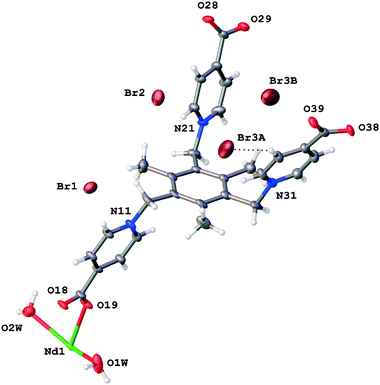 |
| | Fig. 2 Asymmetric unit contents of TTTPC NdBr3. | |
 |
| | Fig. 3 Br3A of TTTPC NdBr3 bound by o-pyr (C32) and m-pyr (C35) pyridinium ring C–H⋯Br− interactions (HBs) with a CH3 type interaction originating from C9–H9C. | |
The TTTPC NdBr2OTf asymmetric unit (Fig. 4) consists of the same framework components as the TTTPC NdBr3, namely a neodymium center (Nd1) with two water molecules (O1W and O2W) and the zwitterionic ligand in the anti conformation. The coordination number of Nd1 is formally nine and its geometry follows a slightly distorted tricapped trigonal prismatic motif, with one tridentate carboxylate chelating a metal with both oxygen atoms (O28 and O29) with an additional contact to the next center (Fig. 1b, coordination mode μ2-η2η1, distances O28–Nd1: 2.489 Å and O29–Nd1: 2.925 Å). Anionic components include two bromides (Br1 and Br2) with full occupancy and one disordered triflate and bromide (S1A etc. and Br3B) with respective occupancies of approximately 70% and 30%. In structure solution, the disordered bromide atom could be found from the difference electron density map as a strong peak next to the triflate sulfur atom, and it couldn't have been modelled with triflate sulfur disorder alone, as no other adjacent disordered triflate atoms could be found. Br1 and Br2 are both situated in the walls of the water channel (a total of eleven water molecules are removed), but their binding interactions are different. Br1 has an o-pyr contact and a contact from the adjacent methylene bridge hydrogen, namely C26–H26⋯Br1 and C20–H20A⋯Br1, making the overall binding effectively a bridging type interaction (μ-pyr–CH2). Br1 also has a single CH3 contact from C7–H7C, and a possible borderline contact may be found from the C23 meta-position (distance 3.061 Å, angle 132.94°). Br2 has conventional interactions of two o-pyr contacts from C32 and C36. There is also a possible μ-pyr contact involved with the C32/C33 pair, albeit with a long H33–Br2 bond distance of 3.075 Å and an angle of 121.80°. The disordered triflate and bromide occupy the deepest position inside the framework analogous to Br1 in the TTTPC NdBr3 structure, but with the triflate being slightly larger in volume, together they take up more space and push the ligand moieties further away. Br3B has conventional o-pyr contacts with C12 and C16, with elongated bond lengths of 3.108 Å and 3.017 Å and angles of 162.17° and 144.48°, respectively, and a borderline C9–H9C methyl contact (3.111 Å, 158.65°). The triflate has several analogous contacts to the bromide, with a μ-pyr-type chelate C15–H15⋯O2A–S1A–O3A⋯H16–C16 and a μ-pyr–CH2-type chelate C10–H10A(B)⋯O4A⋯H12–C12 (Fig. 5). It also has a methyl contact C9–H9C⋯O4A and three C–H⋯F–C contacts, namely C7–H7A⋯F6A, C8–H8C⋯F8A and C20–H20B⋯F7A. Bond lengths, angles and types are listed in Table S6.† There is also a rather clear indication of the presence of electrostatic charge transfer interactions (Fig. S4†), because of the rather precise positions of the anions within the ligand framework. Analogous structures, TTTPC LnBr2OTf (Ln = La, Sm, Eu, Gd and Tb) (Fig. S5–S9†), are somewhat identical to the neodymium structures with only small differences in the structural details. Their asymmetric units contain a metal center with two coordinated water molecules and the ligand in the anti conformation. The metal coordination environment follows the slightly distorted tricapped trigonal prismatic motif with La and reverts to the formally bicapped one with Sm, Eu, Gd and Tb. Additionally, two bromides and a triflate are included but their disorder varies between metals. With La, Sm and Eu the disorder is the same as the triflate–bromide type presented in the TTTPC NdBr2OTf structure (occupancies of Br: 15–25% and triflate: 75–85%), but with Gd and Tb, the disorder is of the bromide–bromide type analogous to the TTTPC YbBr3 (occupancies of Br2A 75% and Br2B 25%). In the latter structures, concordantly, the single triflate has been assigned full occupancy instead of partial, because no strong peak in its vicinity could be found. It is therefore possible to postulate that the heavier lanthanoids prefer the Br2A/Br2B disorder whereas the lighter metals prefer the OTf/Br type, probably due to minute differences in the ligand conformation derived from the change in the ionic radius of the metal. Finally, the diffuse water removed from the structures is also of relatively uniform proportions (La: 10×, Sm: 11×, Eu: 11×, Gd: 10× and Tb: 9× H2O).
 |
| | Fig. 4 Asymmetric unit contents of TTTPC NdBr2OTf. | |
 |
| | Fig. 5 Disordered triflate and bromide of TTTPC NdBr2OTf bound by μ-pyr (C15/C16) and μ-pyr–CH2 (C10/C12) bridging interactions. | |
Not taking into account the crystal system or symmetry, the asymmetric unit of TTTPC NdBr(OTf)2 (Fig. 6) consists of familiar components, which are an independent neodymium center (Nd1) with two coordinated water molecules (O1W and O2W) and one zwitterionic ligand in the anti conformation. The coordination environment of the metal (Fig. 1c) is of the bicapped trigonal prismatic type, albeit slightly more distorted than with the previously discussed systems, and the coordination number is consequentially eight. All carboxylates follow the μ2-η1η1 ZZ coordination mode. The trio of anions comprises a fully occupied bromide (Br1), a fully occupied triflate (S40 etc.) and a disordered triflate–bromide pair (S48A etc. and Br3B, respective occupancies of 65% and 35%). Additionally, next to the disordered bromide, a water molecule oxygen (O3B) with the same occupancy of 35% has been assigned due to the leftover residual electron density that the OLEX2 SM was not able to take into account. A total of six water molecules were calculated and removed in the asymmetric unit as diffuse solvent water. The distances and angle between the oxygen O3B and bromides Br3B and Br1 (O3B–Br3B: 3.860 Å and O3B–Br1: 3.297 Å, Br1–O3B–Br3B angle: 116.92°) suggest a possible HB, but hydrogen positions could not be resolved from the electron density map. A clear o-pyr contact (C36–H36⋯O3B) is existent, though. Br1 is positioned inside the wall of the water channel and it is directly coordinated to the water molecules next to the Nd center with distances of 3.367 Å (O1W–Br1) and 3.512 Å (O2W–Br1). Some C–H⋯Br− interactions with the Br1 and the ligand molecules may also be present, but they are not entirely obvious, with the closest hydrogen atoms being over 3.3 Å away (Fig. S10†). The fully occupied triflate resides within the framework cavity (Fig. S11†) and exhibits one μ-pyr–CH2 chelate contact (C10–H10A⋯O43⋯H16–C16), one o-pyr contact (C12–H12⋯O42), two methylene (CH2) contacts (C20–H20B⋯O42 and C30–H30A⋯O41) and two CH3 contacts (C7C–H7C⋯O43 and C9–H9C⋯F47). The disordered triflate and bromide reside in the water channel cavity opposite Br1, and of these Br2B exhibits two typical elongated o-pyr contacts (C22 and C26), whereas the triflate has more diverse interactions, namely a μ-pyr chelate (C22–H22⋯O50A–S48–O51A⋯H23-C23), a μ-pyr–CH2 chelate (C20–H20A⋯O49A⋯H26–C26), a CH2 contact (C10–H10B⋯O50A) and a CH3 contact (C7–H7B⋯O50A). Bond lengths, angles and types are listed in Table S8.† Structural analogues, TTTPC LnBr(OTf)2(Ln = Sm, Eu, and Tb) (Fig. S12–S14†), have similar asymmetric unit contents to the Nd structure, with the only exception being the Sm structure which does not need to assign an O3B placeholder atom for residual electron density from the SM procedure (Sm: 8×, Eu: 6×, Tb: 8× water removed). The anion distribution is the same for all structures with approximately 60% OTf (S48A etc.) and 40% Br2B occupancy, well in line with the Nd structure. The interactions and geometry between the asymmetric unit components also generally follow the same motifs as discussed above.
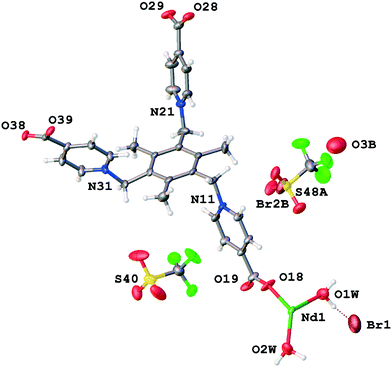 |
| | Fig. 6 Asymmetric unit contents of TTTPC NdBr(OTf)2. | |
The last structure in the TTTPC neodymium Br–OTf series is TTTPC Nd(OTf)3, which exhibits triclinic symmetry in P (# 2), with one crystallographically independent metal center, three triflates (one with 50%:50% disorder) and the zwitterionic ligand in the anti conformation (Fig. 7). What is different from the previous similar structures is the degree of metal hydration with three coordinated water molecules (O1W, O2W and O3W) and the ligand–metal connectivity. In this structure, the ligand is coordinated only to five different metal centers, with one carboxylate oxygen left uncoordinated (O38) and having a HB to one of the water molecules (O2W) instead (Fig. 1d). The coordination modes for the bidentate carboxylates are thus μ2-η1η1 ZZ (C17 and C27) and the simple η1 for the monodentate C37 carboxylate. The metal coordination geometry still follows the familiar slightly distorted bicapped trigonal prismatic motif, with the coordination number being eight. One non-disordered secondary coordination sphere water molecule (O4W) could be found from the electron density map with H-bonding to a triflate oxygen (O2W–H2WB⋯O4W and O4W–H4WA⋯O41). Additionally, a residual peak next to the disordered triflate sulfur atom (S56A) was assigned to a disordered secondary sphere water molecule (O5WB with 50% occupancy), after SM had been applied with approximately 5.5 water molecules worth of electron density removed. It is likely that this water molecule is H-bonded to the farther disordered triflate oxygen (O57B), but hydrogen positions could not be resolved (O5WB–O57B distance: 2.819 Å). The disordered triflate pair is flanked by one regular bridging and two monodentate ligand moieties in the water channel wall cavity, without apparent pyridinium ring H-bonding (Fig. S15†). The staggered ligands most likely exhibit only charge transfer interactions towards these triflates. A single CH3 type interaction for both triflate parts can be found (C9–H9B⋯O59A and C8–H8A⋯O58B) in addition to the regular H-bonding with the first and second coordination sphere water molecules, though. The two fully occupied triflates share similar environments and ligand–anion interactions between parallel pyridinium rings (N11 and N21). All regular C–H⋯OTf− types can be found (with possible charge transfer from nearby perpendicular rings), ranging from CH2 and CH3 to more prevalent o-pyr, μ-pyr and μ-pyr–CH2 interactions. These are conventionally listed in Table S10.† The TTTPC Yb(OTf)3 structure was determined as a counterpart system, and compared to the Nd structure, it had some distinctive features most likely attributed to the change of metal to Yb. The asymmetric unit (Fig. S16†) comprises the metal, the ligand in a similar conformation and connectivity to that of the Nd structure, three anions (two with disorder: OTf/Br and OTf/OTf) and only two metal coordinated water molecules (5 diffuse water molecules removed). Due to the metal coordination number being only seven, the metal coordination geometry follows a slightly distorted pentagonal bipyramidal motif. Closest to the metal, a triflate is coordinated to the water molecules between perpendicular pyridinium rings and disordered with an adjacent bromide, which is assigned to similar reasoning to that in TTTPC NdBr2OTf, although the OTf:Br disorder here is only 90%:10%, respectively. Another disordered triflate resides in the water channel wall between parallel pyridinium rings (OTf:OTf, 60%:40%) with familiar C–H⋯OTf− interactions, but apparently with some freedom to tilt on its foothold. Finally, the fully occupied triflate is once again buried deepest inside the framework cavity. A rather peculiar intermediate between the systematic structures presented thus far was also found and its crystal structure was determined, namely TTTPC2Sm2(OTf)6, with double the conventional unit cell size and contents (Fig. S17†), but half of the metal centers with two (and half with three) coordinated water molecules. It is likely that this structure is not a systematic representative of the middle lanthanoids, but rather just an obscure crystallization product (see more details in the ESI†). However, it is notable that while screening for crystals, similar aberrations based on unit cell proportions could occasionally be found in minute quantities with other samples as well, although they were of such poor quality that additional complete data could not be obtained.
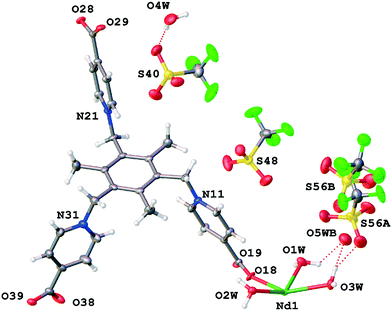 |
| | Fig. 7 Asymmetric unit contents of TTTPC Nd(OTf)3. | |
Finally, the asymmetric unit of fresh TTTPC NdBr2OAc consists of the otherwise familiar components (Nd1 center with two water molecules, O1W and O2W, two bromide anions, ligand in the anti conformation, no disorder), but an entirely different coordination network is obtained by having a single directly metal coordinated acetate displacing one of the zwitterionic ligand arms (Fig. 8). Two regular bidentate carboxylates (C17 and C37) follow the μ2-η1η1 ZZ coordination mode and the acetate (C41) has assumed the chelating tridentate μ2-η2η1 mode, making the overall metal coordination number nine (tricapped trigonal prismatic geometry). All Nd–O distances fall below 2.6 Å, so the coordination number is rather unambiguous. The uncoordinated carboxylate (C27) is drawn towards a metal center by forming a HB between O29 and O1W. O28 points in the water channel, but with the diffuse solvent removed (11 molecules in total), no interactions can be determined. The two bromides have typical ligand–anion interactions as discussed before (Br1: o-pyr × 2, CH3 and Br2: o-pyr × 3, CH3), listed in Table S13,† and they occupy cavities in the water channel walls symmetrically. The third o-pyr contact with Br2 (C22–H22⋯Br2) is formed with the displaced ligand arm, indicating a possible synergy with the aforementioned HB.
 |
| | Fig. 8 Asymmetric unit contents of TTTPC NdBr2OAc. | |
3.1.2 Packing and other supramolecular features.
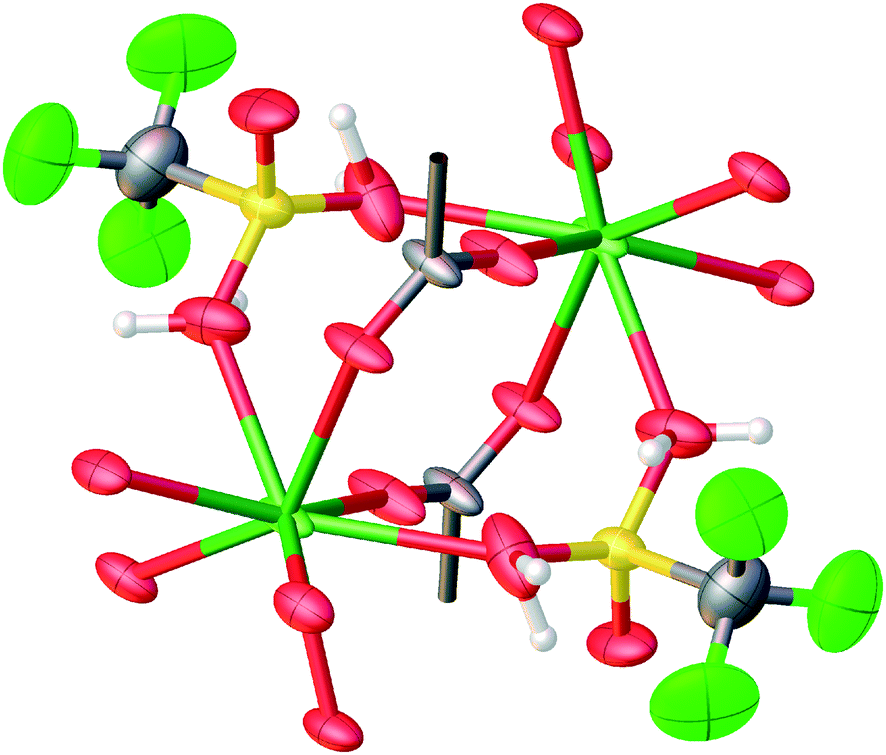
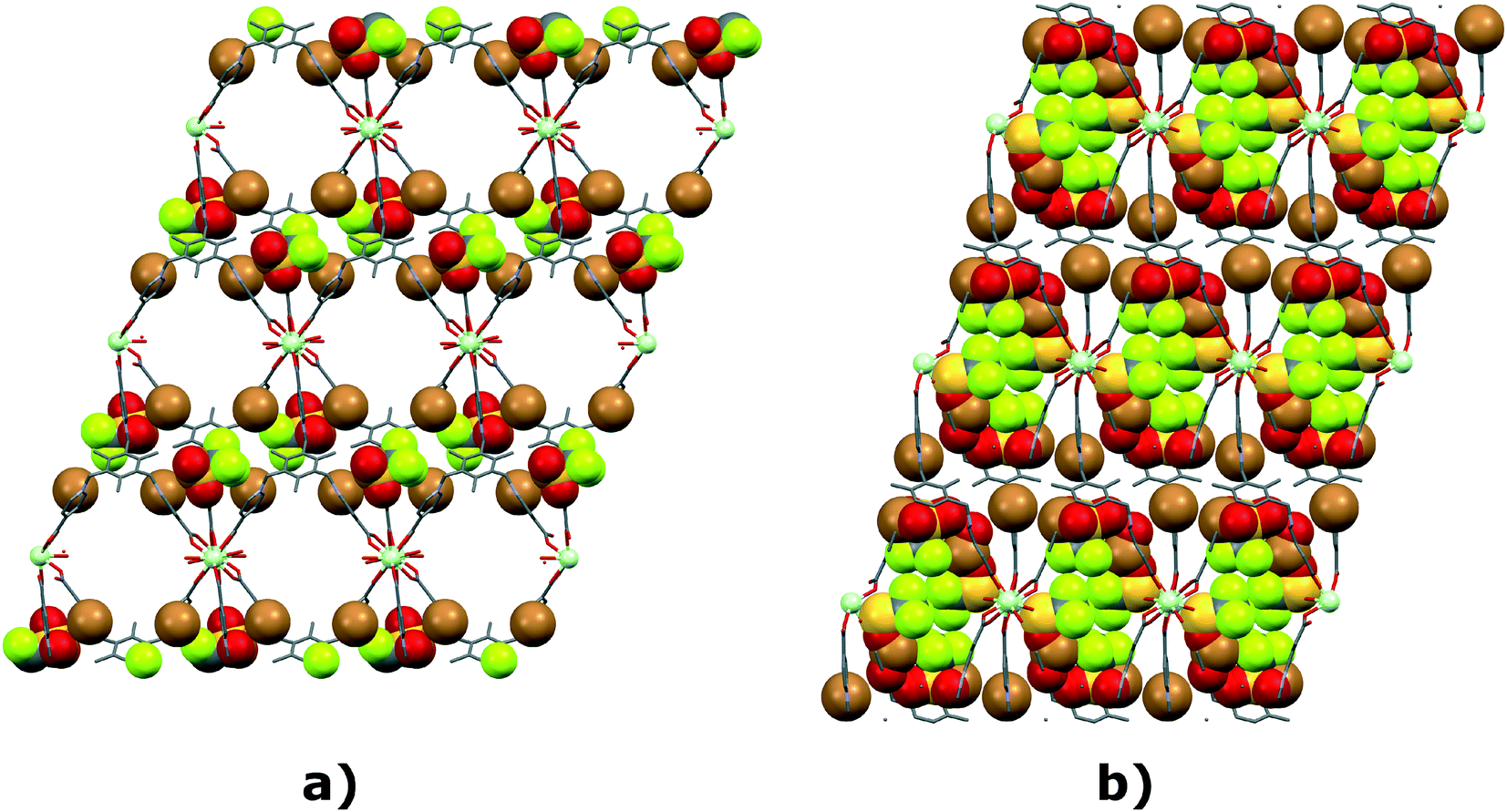
Features in TTTPC LnBr3, TTTPC LnBr2OTf and TTTPC Ln(NO3)3 are very similar as reported by Wen et al. and Bag et al. in their reported structures with TTTPC.16,17 Here, TTTPC NdBr3 is discussed as a representative of all the aforementioned structures, since they can be described in the same manner. In this structure, the metal centers are aligned along the crystallographic a-axis in an infinite 1D chain, and ligands take turns in bridging the centers in groups of two and four. Four water molecules are aligned two by two on opposite sides of two consecutive metal centers, and overall this repetition can be described as a rectangular paddlewheel type secondary building unit (SBU)31 of [Nd2(COO)4(H2O)4] (Fig. 9a) bridged by two additional carboxylate groups. More accurately, the repeating unit can be described in a linear fashion by running along the metal chain as [Nd(COO)2(H2O)4Nd(COO)4]n (Fig. S26a†). The ligands can be seen forming a “three-up-three-down” motif (Fig. 10a and S27a†) with respect to the row of metals, and the ligand–metal overall 3D framework defines an anisotropic open structure with solvent accessible pores running in the same direction as the bridged metal centers (Fig. 11a, S25, and S30a†).
 |
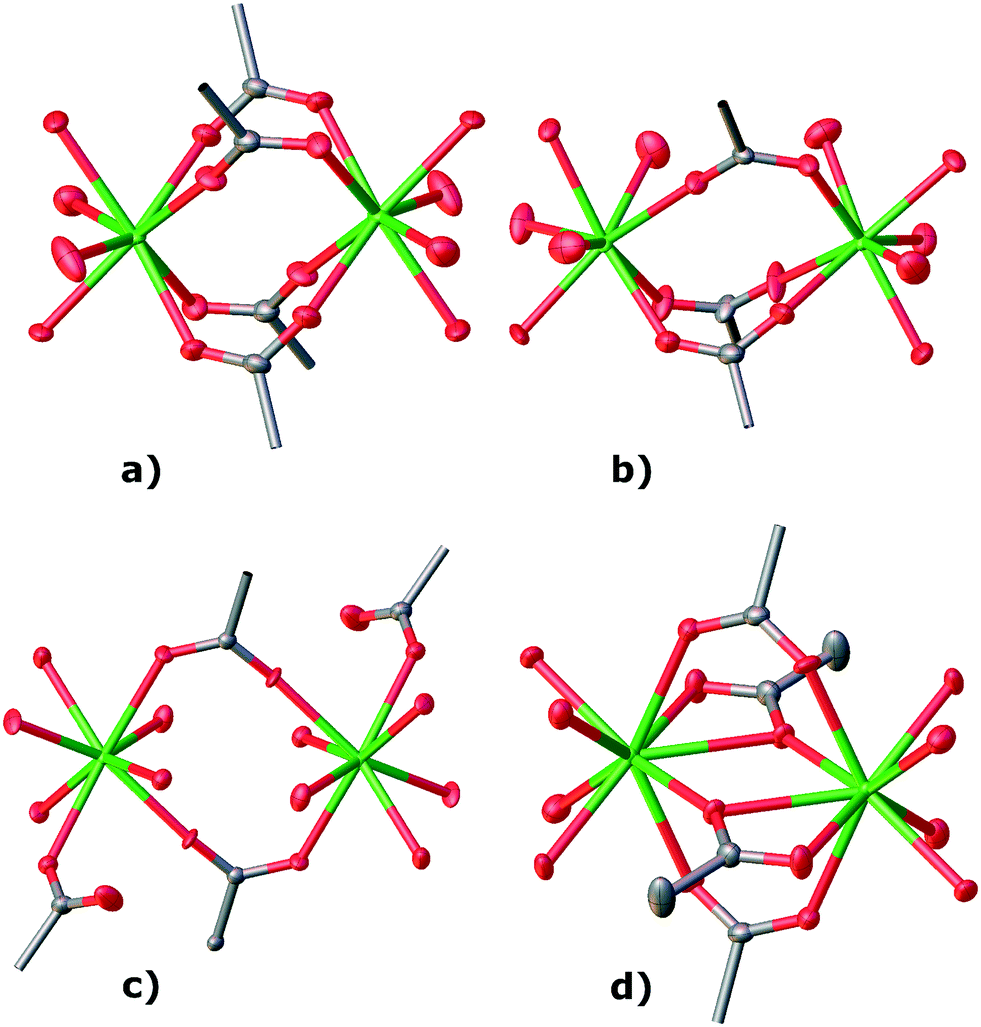
| | Fig. 9 Secondary building units (SBUs) of a) TTTPC NdBr3, b) TTTPC NdBr(OTf)2, c) TTTPC Nd(OTf)3 and d) TTTPC NdBr2OAc (hydrogen atoms omitted for clarity). | |
 |
| | Fig. 10 Ligand orientations of a) TTTPC NdBr3, b) TTTPC NdBr(OTf)2, c) TTTPC Nd(OTf)3 and d) TTTPC NdBr2OAc along the water channel direction (hydrogen atoms omitted for clarity). | |
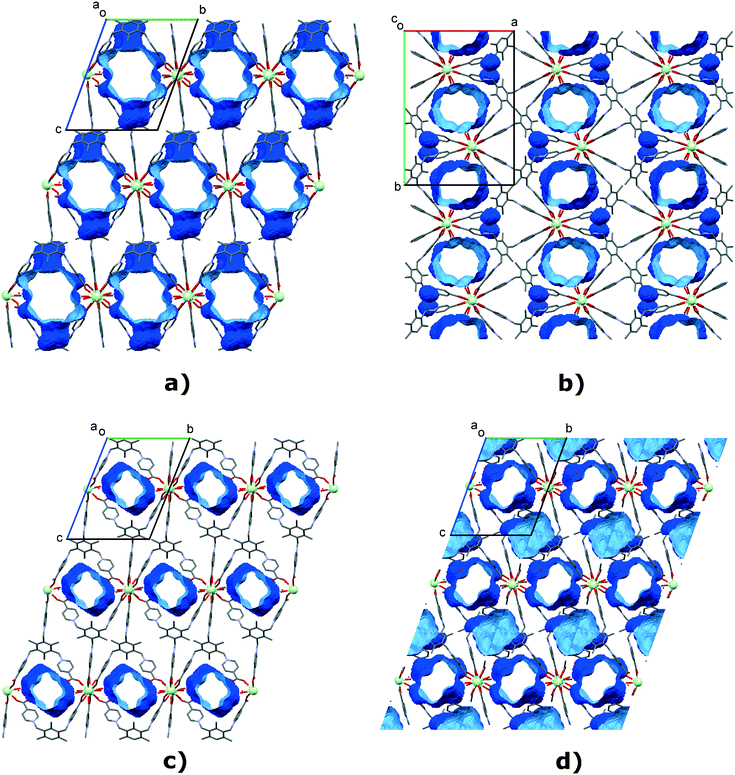 |
| | Fig. 11 Voids (water channels) of a) TTTPC NdBr2OTf, b) TTTPC NdBr(OTf)2, c) TTTPC Nd(OTf)3 and d) TTTPC NdBr2OAc (hydrogens, bromides and triflates omitted for clarity, Mercury contact surface mapping with a 1.2 Å probe and 0.2 Å grid). For TTTPC NdBr3 water channels, see Fig. S28.† | |
TTTPC LnBr(OTf)
2
structures are based on a similar rationale, but the SBU and ligand orientations differ. TTTPC NdBr(OTf)2 is discussed here as an archetype. The ligands do not take turns in groups of two and four, but instead in staggered pairs of three. This results in an SBU of [Nd2(COO)3(H2O)4] that can be described as a paddlewheel, with one “paddle” formed by a pair of water molecules (Fig. 9b). Respectively, these SBUs are bridged by three carboxylates. In the previous linear notation, the repetition along the metal chain would be simply [Nd(COO)3(H2O)2]n (Fig. S26b†). When viewed along the c-axis, the ligands follow a “two-up-four-down” motif (Fig. 10b and S27b†), and the complete 3D network structure also has solvent accessible channels in the same direction (Fig. 11b and S30b†), as well as smaller isolated pockets.
Moving on to TTTPC Nd(OTf)3 the “loose leg” of the ligand causes no significant changes in the SBU compared to TTTPC NdBr3 and the like, except for the addition of a single water molecule onto both metals. The rectangular paddlewheel SBU is thus [Nd2(COO)2(COO′)2(H2O)6] (Fig. 9c), with COO′ denoting the monodentate carboxylate. Again, the SBUs are bridged by two additional carboxylates. According to the linear notation, the repeating unit can thus be described as [Nd(COO)2(H2O)4Nd(COO)2(COO′)2(H2O)2]n (Fig. S26c†). Consequentially, the ligand orientation is similar to the “three-up-three-down” system, only with the anomalous carboxylate taken into account (Fig. 10c and S27c†). TTTPC Yb(OTf)3 is otherwise similar to its Nd counterpart, but due to a missing water molecule for each metal, its SBU is [Yb2(COO)2(COO′)2(H2O)4] with two bridging carboxylates, and its description by linear notation is respectively [Yb(COO)2(H2O)4Yb(COO)2(COO′)2]n. Both the Nd and Yb structures have water channels running along the crystallographic a-axis (Fig. 11c and S30c†).
Finally, the fresh TTTPC NdBr2OAc structure presents yet another variation to the structural motifs as already discussed, by having a ligand arm displaced by an acetate, but otherwise following the “three-up-three-down” trend (Fig. 10d and S27d†). The SBU is [Nd2(COO)2(COO′′)2(H2O)4] (Fig. 9d), with COO′′ denoting the coordinated acetate. As previously discussed, two carboxylates act as SBU bridging groups. The description with linear notation is, analogously, [Nd(COO)2(H2O)4Nd(COO)2(COO′′)2]n (Fig. S26d†). The water channels in TTTPC NdBr2OAc run in the direction of the crystallographic a-axis, but there are separate larger pockets as well (Fig. 11d and S30d†). Notable in this last case, though, is the dimensionality of the structure, since it is only a 2D coordination network, by definition.1TTTPC NdBr2OAc is composed of 2D layers bound together by the previously described HBs between the displaced carboxylate and coordinated water (Fig. 12).
 |
| | Fig. 12
TTTPC NdBr
2
OAc 2D layers connecting each other with HBs from the displaced ligand “arms” (bromides and hydrogen atoms omitted for clarity). | |
3.2 Structural description of the evacuated single crystal, TTTPC NdBr2OTf (vac)
The single crystal selected from an evacuated sample mixture of bromide–triflates (see section 2.3.3) turned out to correspond to the TTTPC NdBr2OTf structure type, crystal system and stoichiometry, but with some notable differences apparently caused by the evacuation process. The asymmetric unit of TTTPC NdBr2OTf (vac) (Fig. 13) has a ligand–metal framework completely intact with the ligand in the familiar anti conformation, binding six Nd centers, one of which is crystallographically independent. There is a one total triflate (S40A etc. and S48A etc.) and a one total bromide (Br1B and Br2B) disordered over two positions, with both of their parts' occupancies being 55% and 45%. One bromide enclosed in the framework cavity by typical C–H⋯Br− interactions is at full occupancy. H-bonding water molecules could be identified from the electron density map between triflates (O3WA) and between bromides (O4WB), and their occupancy was determined to be close to 55% with free variable refinement. The remarkable part of this structure turned out to be the metal coordination environment, as the triflate (S48A etc.) has been coordinated to two adjacent metal centers in a familiar μ2-η1η1 ZZ manner through O50A and O51A. Furthermore, since the triflate has substitutional disorder, there are also two water molecules (O1WB and O2WB, both with 45% occupancy) complementing the oxygen species in the coordination sphere (an EXYZ constraint was needed to model the atomic positions, Fig. 14). The coordination geometry is practically unchanged and follows a typical bicapped trigonal prismatic motif with a coordination number of eight. Technically, though, a change from a nine- to eight-coordinate system with Nd is observed, if compared to the un-evacuated structure. The two disordered H-bonded anion systems coexist in the structure, but they are mutually exclusive, as they inhabit the same space (Fig. S18†). It is therefore a reasonable assumption that when on one side of the metal there is a bromide system, simultaneously on the other side there is a triflate system (as depicted in Fig. 13). The anionic parts of the structure interact with the pyridinium rings as expected with the o-pyr contacts being the most prevalent. A list of these contacts is presented in the ESI† (Table S15). Packing and supramolecular features are highly similar to those of the un-evacuated sample, with the only exception being of course the difference in the coordination environment, inherently leading to a change only in the linear description of the repeating unit ([Nd(COO)2(OTf)1.1(H2O)1.7Nd(COO)4]n). The SBU and ligand orientations around the row of metals are essentially the same. Since in this structure there was no diffuse solvent present as in the previously illustrated water channels, no solvent mask was needed, and consequentially some space in the crystal structure was truly left devoid of solvent (Fig. S29†). Also in comparison with the un-evacuated structure of TTTPC NdBr2OTf, the previously open pore of the MOF structure can be seen here partially filled with the displaced anionic species (Fig. 15). Notably the crystal dimensions have also contracted (refer to Table 1), made possible by the properties of a flexible ligand used with a similarly flexible metal coordination.
 |
| | Fig. 13 Asymmetric unit contents of TTTPC NdBr2OTf (vac). | |
 |
| | Fig. 14 Coordination environment of TTTPC NdBr2OTf (vac) with coordinated partly disordered bridging triflates in the same positions as conventional water molecules. | |
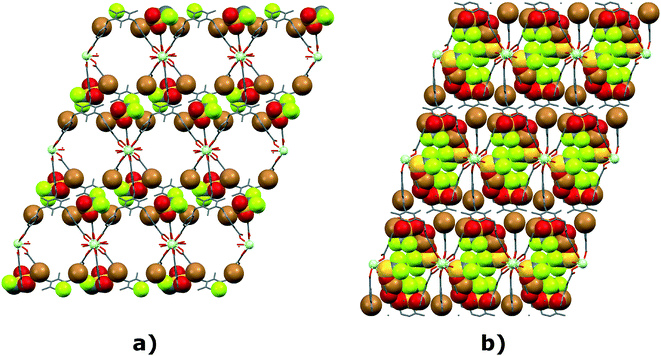 |
| | Fig. 15 View down the crystallographic a-axis of a) TTTPC NdBr2OTf and b) TTTPC NdBr2OTf (vac) with an emphasis on differences between a non-evacuated and an evacuated structure. | |
3.3 Structural description of the dry single crystal, TTTPC NdBr2OAc (dry)
The initial measured bulk powder diffraction patterns of TTTPC NdBr2OAc were clearly different from the simulated ones of the single crystal structure of the fresh sample, which led to suspicions about a possible different crystal structure of the dry material, but no sufficient quality single crystal could be obtained for structure determination. After making a new, slowly dried batch of TTTPC NdBr2OAc, indeed a different crystal structure of TTTPC NdBr2OAc (dry) could be obtained. Compared to the fresh structure, the most striking differences were the unambiguous positions of water molecules that were not diffuse (solvent mask was not needed), followed by the apparent dehydration and reorganization of the metal coordination sphere, consequently leading to the already well established 3D coordination network structure type discussed previously. The asymmetric unit of TTTPC NdBr2OAc (dry) (Fig. 16) consists of one ligand, two bromides, one Nd coordinated acetate and seven water molecules. The water molecules form an elaborate, highly symmetrical and infinite H-bonded network between the anion and ligand moieties in the water channels running along the a-axis. The main network (Fig. 17) consists of repeating units of square and hexagonal arrays of water molecules that are formed from five crystallographically independent water molecules (O1W–O5W) through symmetry. The remaining two independent water molecules (O6W and O7W) extend the network toward the two bromides (Fig. S19†) that reside within the ligand framework held in place by o-pyr, CH2 and CH3 contacts. The ligand orientation is analogous to the one presented in Fig. 10a, but the water molecules have been replaced by the μ2-η1η1 ZZ bridging acetate oxygens so that the overall notations of the SBU and linear metal chain can be expressed as [Nd2(COO)2(COO′′)2] (the same as Fig. 9d) and [Nd(COO)4Nd(COO)2(COO′′)2]n, respectively (COO′′ denoting the acetate, as previously discussed) (Fig. S20†).
 |
| | Fig. 16 Asymmetric unit contents of TTTPC NdBr2OAc (dry). | |
 |
| | Fig. 17 Main repeating part of an infinite H-bonded water network in TTTPC NdBr2OAc (dry). | |
4 Conclusions
In an in-depth survey of several lanthanoid network solids based on a flexible zwitterionic ligand TTTPC, the underlying ligand–metal and ligand–anion interactions have been elucidated in the diverse yet similar crystal structures, and supporting information has been acquired as well. Most network solids are found to be based on metal centers that adopt a slightly distorted bi- or tricapped trigonal prismatic symmetry with a coordination number of eight or nine, which is very common for lanthanoids.6 The choice of metal from fifteen lanthanoids appears to be arbitrary with regard to the resulting network topology, but in detail, differences can be found between the heavy and light metals, such as the preferred anion disorder and the degree of metal hydration. This design through lanthanoid contraction is viable, but not immensely rewarding. The choice of anions in a bianionic system, however, is found to promote new structures by first affecting the outcome through initial crystallization conditions, and afterwards by affecting the resulting products through gradual change of amounts in the crystallization vessel. From a solution where triflate is dominant over the bromide, a tri-triflate TTTPC Ln(OTf)3 structure crystallizes first, diminishing the amount of triflate in the solution and gradually increasing the relative amount of bromide. The TTTPC LnBr(OTf)2and TTTPC LnBr2OTf structure types follow suit and crystallize, when the conditions are more bromide-rich. This results in bulk products of mixed species, but through optimization and control of conditions there could be a possibility to obtain pure fractions. If the two anions are chosen in a way, where one has significant affinity towards the ligand and one towards the metal, a more controlled crystallization can also be conducted. For example in our last case, the bromide can be described as ligandophilic and the acetate as metallophilic, resulting in TTTPC LnBr2OAc and TTTPC LnBr2OAc (dry) structures that have unique properties because of the metal coordinated acetate. In future studies, this classification of anions in systems, where the ligand can interact with anionic species through attractive electrostatic forces or other weak interactions, can prove to be a helpful tool in designing new systems. Bromide, for instance is shown to be well suited for the ligandophilic part if the ligand is capable of interacting with it through H-bonding. However, the good bond acceptor character and water affinity of bromide may also be undesired properties, so further suitable anions could be experimented with. Alternatively, there is a possibility to conduct experiments in non-aqueous environments, if hydrophilicity proves to be a hindrance. Overall, TTTPC is a versatile ligand and will definitely see further use in coordination polymer designs either with or without auxiliary ligands. Due to its flexibility and the choice of similarly flexibly coordinating metals in this work, some unintentional, if not even serendipitous, structures could be obtained, as predicted.4 It is therefore of utmost importance to either set tighter conditions, or alternatively even further expand the existing thought models when working with this ligand, to obtain new and exciting results.
Conflicts of interest
We declare no conflicts of interest regarding the work presented in this paper.
Acknowledgements
We are grateful to the University of Jyväskylä (JYU), Department of Chemistry for providing funding and infrastructure throughout this work. M. L. kindly acknowledges the financial support from the Academy of Finland (project no. 277250). We also want to acknowledge MSc Juuso Järvinen (JYU) for his contribution during his BSc thesis, laboratory technicians Elina Hautakangas and Hannu Salo (JYU) for assistance in the CHN and SEM analyses, respectively, laboratory engineer Pasi Myllyperkiö (JYU) similarly for the solid state UV-vis measurements and finally laboratory manager Minna Pakkanen (VAPO Ventures) for providing the BET measurement results.
Notes and references
- S. Batten, N. Champness, X. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715 CAS.
- K. Gangu, S. Maddila, S. Mukkamala and S. Jonnalagadda, Inorg. Chim. Acta, 2016, 446, 61 CrossRef CAS.
- B. Li, M. Chrzanowski, Y. Zhang and S. Ma, Coord. Chem. Rev., 2016, 307, 106 CrossRef CAS.
- Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867 RSC.
- B. Li and B. Chen, Struct. Bonding, 2015, 163, 75 CrossRef CAS.
- J.-C. Bünzli, J. Coord. Chem., 2014, 67, 3706 CrossRef.
- R. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
-
R. Wang and Z. Zheng, in Rare Earth Coordination Chemistry: fundamentals and applications, ed. C. Huang, John Wiley & Sons (Asia), Singapore, 1st edn, 2010, ch. 3, pp. 91–136 Search PubMed.
- G.-Q. Kong and C.-D. Wu, Cryst. Growth Des., 2010, 10, 4590 CrossRef CAS.
- G.-M. Zhuang, X.-B. Li, Y.-Q. Wen, C.-Y. Tian and E.-Q. Gao, Eur. J. Inorg. Chem., 2014, 22, 3488 CrossRef.
- M. Zhao, J. Su, S. Wang, J. Zhang, S. Zhang, J. Wu and Y. Tian, J. Coord. Chem., 2016, 69, 879 CrossRef CAS.
- J. Su, L. Yao, J. Zhang, S. Yuan, F. Xie, Y. Ding, M. Zhao, S. Wang, H. Li, S. Shang, J. Wu and Y. Tian, New J. Chem., 2016, 40, 97 RSC.
- W. An, D. Aulakh, X. Xhang, W. Verdegaal, K. Dunbar and M. Wriedt, Chem. Mater., 2016, 28, 7825 CrossRef CAS.
- J. Zhang, M. Zhao, W. Xie, J. Jin, F. Xie, X. Song, S. Zhang, J. Wu and Y. Tian, New J. Chem., 2017, 41, 9152 RSC.
- J. Zhou, J.-S. Zhao, J. Feng, X.-F. Zhang, J. Xu, L. Du, M.-J. Xie and Q.-H. Zhao, J. Mol. Struct., 2018, 1155, 303 CrossRef CAS.
- R.-M. Wen, S.-D. Han, G.-J. Ren, Z. Chang, Y.-W. Li and X.-H. Bu, Dalton Trans., 2015, 44, 10914 RSC.
- P. Bag, X.-S. Wang and R. Cao, Dalton Trans., 2015, 44, 11954 RSC.
- Z. Bai, Y. Wang, Y. Li, W. Liu, L. Chen, D. Sheng, J. Diwu, Z. Chai, T. Albrecht-Schmitt and S. Wang, Inorg. Chem., 2016, 55, 6358 CrossRef CAS PubMed.
- L. Liang, R. Zhang, N. Weng, J. Zhao and C. Liu, Inorg. Chem. Commun., 2016, 64, 56 CrossRef CAS.
- C. Lian, Y. Chen, S. Li, M.-Y. Hao, F. Gao and L.-R. Yang, J. Alloys Compd., 2017, 702, 303 CrossRef CAS.
- O. Berryman, V. Bryantsev, D. Stay, D. Johnson and B. Hay, J. Am. Chem. Soc., 2007, 129, 48 CrossRef CAS PubMed.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- L. Palatinus and A. van der Lee, J. Appl. Crystallogr., 2008, 41, 975 CrossRef CAS.
- L. Palatinus, S. J. Prathapa and S. van Smaalen, J. Appl. Crystallogr., 2012, 45, 575 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- C. Groom, I. Bruno, M. Lightfoot and S. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- C. Macrae, I. Bruno, J. Chisholm, P. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- D. Tranchemontagne, J. Mendoza-Cortés, M. O'Keeffe and O. Yaghi, Chem. Soc. Rev., 2009, 38, 1257 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Manu
Lahtinen
and
Manu
Lahtinen