
Supramolecular enantiomeric and structural differentiation of amino acid derivatives with achiral pillar[5]arene homologs†
Jiecheng
Ji‡
a,
Yizhou
Li‡
a,
Chao
Xiao
a,
Guo
Cheng
a,
Kui
Luo
 a,
Qiyong
Gong
a,
Dayang
Zhou
b,
Jason J.
Chruma
a,
Wanhua
Wu
*a and
Cheng
Yang
*a
a,
Qiyong
Gong
a,
Dayang
Zhou
b,
Jason J.
Chruma
a,
Wanhua
Wu
*a and
Cheng
Yang
*a
aHuaxi MR Research Center (HMRRC), Department of Radiology, West China Hospital, Key Laboratory of Green Chemistry & Technology, College of Chemistry, and Healthy Food Evaluation Research Center, Sichuan University, 29 Wangjiang Road, Chengdu, 610064, China. E-mail: wuwanhua@scu.edu.cn; yangchengyc@scu.edu.cn
bComprehensive Analysis Center, ISIR, Osaka University, Japan
First published on 29th November 2019
Abstract
Complexation of achiral pillar[5]arenes with chiral amines induced strong circular dichroism (CD) signals. The CD responses differed drastically depending on the nature of the amino acid guest, and they significantly varied and part of them even inverted, upon increasing the length of the alkyl chains of the pillar[5]arenes guests. Accordingly, this tactic allowed for the unprecedented simultaneous enantiomeric and structural differentiation of α-amino esters with homologous molecular hosts.
Sensing and differentiating molecular chirality is of great significance for both fundamental research and practical applications.1 Supramolecular optical chirality sensing is advantageous over conventional chromatographic analyses due to the rapid, cost-effective and high-throughput features of the technique.1a–d Several successful supramolecular optical systems have been explored,2 and such systems employ a sophisticated combination of the complexation component(s) and reporting group(s), such as bisporphyrins,2a,b coordinating complexes,2c supramolecular assemblies,2d,e aromatic boronic acids,2f binaphthyls,2g polymers2h,i and interlocked structures.2j,k However, this strategy is not always effective for differentiating the chemical structures of analysts, especially those having similar functional groups.2a,e,h Similarly, the spectral responses of homologous supramolecular sensing hosts toward similar analytes are often difficult to discriminate. In this regard, combining a supramolecular sensor array with statistical analysis could be a promising strategy,3 but it raises the challenge of constructing appropriate libraries of molecular sensors. An ideal sensor library should be composed of members that can specifically respond to different analytes so as to facilitate structural differentiation and reduce the number of sensors required. We have demonstrated that the supramolecular assembly strategy is powerful for photochirogenesis and chirality sensing.4 Herein, we report that the specific chiroptical responses of various alkylated pillar[5]arene (P[5]) derivatives when complexing with L-α-amino esters are highly dependent upon the length of the P[5] alkyl chain substituents, thus allowing for an unprecedented chirality and structure differentiation of the analytes by the supramolecular homologs.
P[5] derivatives have attracted extensive attention in the past decade due to their unique structures and excellent host–guest complexing properties.5 P[5]s lack chirality centers, but they can populate two enantiomeric conformations, PR or PS,6a which interconvert rapidly in solution via “oxygen-through-the-annulus” flipping (Fig. 1). Accordingly, P[5] derivatives can be rendered chiral by inhibiting this conformational flipping or by fusing the side ring(s) into one hydroquinone unit.6b–i The resulting chiral P[5] derivative demonstrate extremely strong circular dichroism (CD) signals. We have demonstrated that the PR and PS configurations have positive and negative CD extreme (CDex) at ca. 310 nm, respectively.6g It occurred to us that P[5]s could sever as convenient chiral sensors: complexation of a chiral guest should lead to a unequal population of the enantiomeric conformers (Scheme 1), and the induced CD signals of the preferred conformer can report the handedness of the guest.
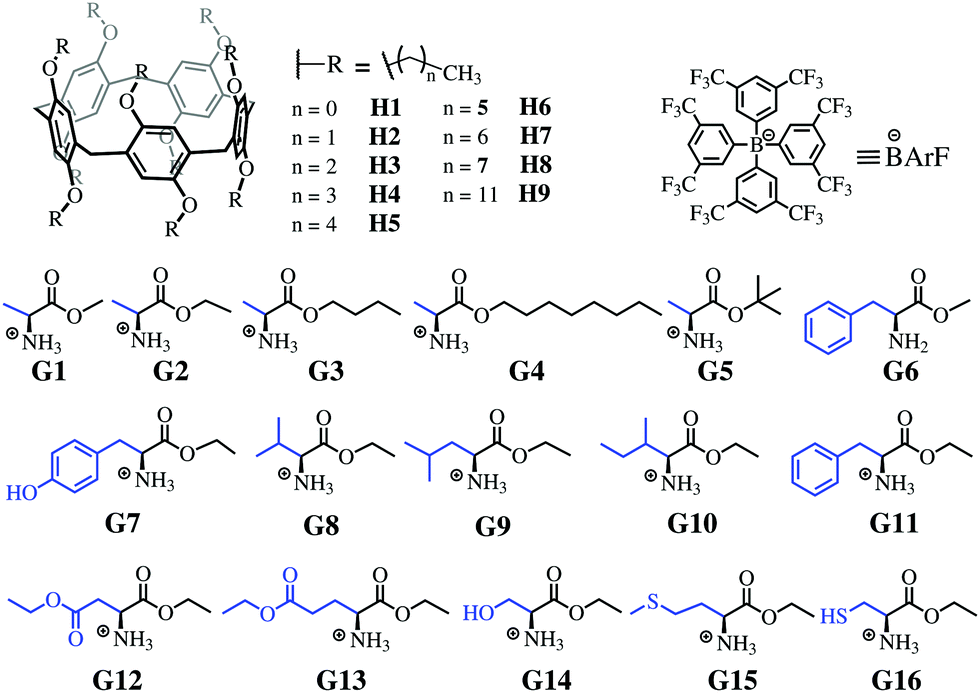 | ||
| Fig. 1 Chemical structure of P[5] derivatives and amino acid derivatives. | ||
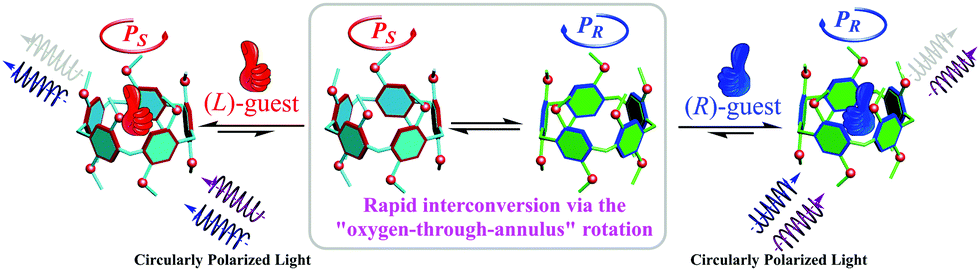 | ||
| Scheme 1 Schematic representation of the chiroptical induction of P[5] through complexation with chiral guests. | ||
To test this hypothesis, a library of P[5]s bearing alkyl chains of increasing length (H1–H9, Fig. 1) were synthesized and characterized by NMR and HRMS spectral studies (Fig. S1–S81, ESI†).7 The polyaromatic rings of P[5]s are known to strongly complex with organic ammoniums cations;5m our own spectroscopic studies unequivocally demonstrated such a complexation between P[5]s H1–H8 with the chiral α-ammonium ester BArF salts G2–G17 (Fig. 1 and Fig. S84–S93, ESI†).7 For example, increasing the concentration of L-G2 led to a clear hypochromic shift of the UV-vis spectra of H1 (Fig. 2a). The continuous variation Job's plot (Fig. S83, ESI†)7 showed a maximum at the fraction of 0.5, indicating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexation stoichiometry. H1 exhibited a weak fluorescence peak at 327 nm in CHCl3, and this fluorescence maximum experienced a slight bathochromic shift and a significant enhancement (up to 7 times) upon addition of G2 (Fig. 2b). The fluorescence lifetime decreased from 1.0 ns for H1 alone to 0.26 ns for the G2@H1 complex (Fig. S103, ESI†), suggesting that the complexation facilitated the radiation decay of H1.
:1 complexation stoichiometry. H1 exhibited a weak fluorescence peak at 327 nm in CHCl3, and this fluorescence maximum experienced a slight bathochromic shift and a significant enhancement (up to 7 times) upon addition of G2 (Fig. 2b). The fluorescence lifetime decreased from 1.0 ns for H1 alone to 0.26 ns for the G2@H1 complex (Fig. S103, ESI†), suggesting that the complexation facilitated the radiation decay of H1.
 | ||
| Fig. 2 (a) UV-vis, (b) fluorescence (λex = 302 nm) and (c) CD spectral changes of 0.05 mM H1 upon increasing the concentration of (L)-G2. Inset of (b): Normalized fluorescence spectra. (d) CD spectra of H1 (50 μM) in the presence of G2 of different ee's ([L-G2] + [D-G2] = 0.1 mM). All measured in CHCl3 at 25 °C. | ||
The CD spectra of H1 and the other P[5] homologs changed proportionally to the addition of the α-ammonium esters. For example, adding L-G2 to H1 resulted in the growth of a positive CD maximum at 310 nm and a negative CD signal at 283 nm (Fig. 2c). It is worth noting that both H1 and L-G2 alone did not show a CD signal in the wavelength range of 250–350 nm. The CD spectra of the complexes are consistent with that of PR P[5]s,6g indicating that the PR conformer is preferred when complexing with L-G2. Complexation of H1 with D-G2 led to an opposite CDex,7 and the CD intensities are proportional to the enantiomeric response was seen for all other ammonium esters G (Fig. S84–S93, ESI†).7 These results reveal for the first time, the promising perspective of P[5] supramolecular hosts for assaying the absolute confirmation and enantiopurity of chiral amines. Furthermore, the optical purity of chiral ammonium esters could be determined. As shown in Fig. 2d, the CD intensity of complexes formed by H1 and G2 was strongly dependent on enantiomeric compositions, the calibration curve built from the CD ellipticity at 301 nm vs. the enantiomeric excess revealed a perfect linear relationship (Fig. S105b (ESI†), R2 = 0.998), and the average errors were determined to be 2.2% (Table S3, ESI†).
We were intrigued to discover that the nature of the induced CD spectra upon addition of chiral α-ammonium esters to our P[5] homologs, was critically dependent on the length of the P[5] alkyl chain substituents (Fig. 1). The strong positive CDex observed upon addition of L-G2 to H1 (R = Me) became weaker with H2 (R = Et), inverted to negative with H3 (R = n-Pr), and went to strongly negative with further increases in the alkyl length (Fig. 3a). The maximum negative CDex was observed for the complex between L-G2 and hexyl ether H6; as the length of the P5 alkyl chains increased beyond hexyl, the corresponding CDex spectra for the complexes with L-G2 became proportionally less negative. Guests G1, G3, G4, G8 and G9 showed similar inverted CD signal upon increasing the length of the alkyl chain of pillararene. Overall, such spectral changes indicate that the enantio-preference of P[5]s is dependent upon the alkyl ether chain length. This observation is exceptional considering that the alkyl chain substituents are achiral and the central binding site of the P[5] hosts should be identical throughout the series of homologs. When the library of P[5] homologs H1–H9 were complexed individually with other investigated α-ammonium esters, such as G5 (Fig. 3b), G11 (Fig. 3c), or G12 (Fig. 3d), the resulting CDex spectra did not invert, but the intensity of the signals still were highly dependent on the length of the P[5] alkyl chains.
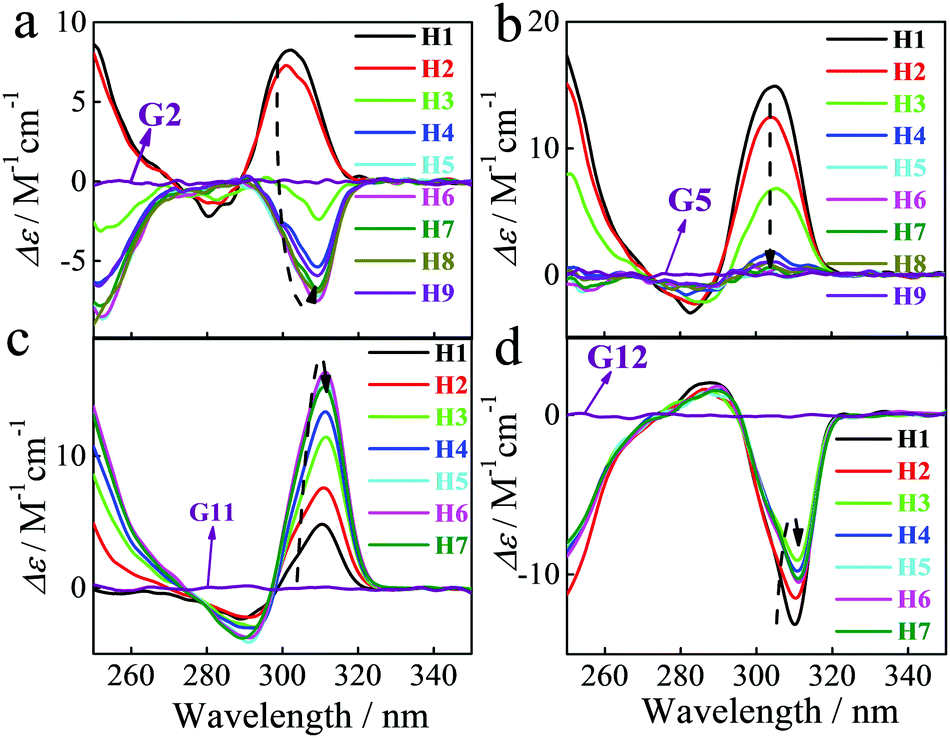 | ||
| Fig. 3 CD spectra of different P[5] derivatives (5.0 × 10−5 M) in the presence of compound (9.1 × 10−5 M) G2 (a), G5 (b), G11 (c) and G12 (d) in CHCl3 at 25 °C. | ||
The above investigation inspired us to further explore the chain length-dependent complexing behavior. Isothermal titration calorimetry studies indicated that complexation of G2 with all of the P[5] homologs are highly entropy-favored (Fig. S94–S102, ESI†). The TΔS values decreased from 19.1 kJ M−1 for H1 to 14.7 kJ M−1 for H2, and then increasing to 22.6 kJ M−1 for H3 and 27.8 kJ M−1H4 (Fig. 4a and Table S2, ESI,†7). Further elongation of the alkyl chain beyond four carbons (H5–H8) did not result in any substantial changes in the TΔS values. The enthalpy changes showed an opposite tendency, increasing from H1 to H2 and then decreasing with the chain length. By virtue of the large entropy gains, strong association constants on the order of 105 M−1 were observed for all of the P[5] homologs with G2. These values are higher by one order of magnitude than the constants that have been reported for the strong P[5] binder hexanedinitrile (HAN).5n
 | ||
| Fig. 4 (a) Entropy–enthalpy diagram of HAN (black) and G2 (red) complexing with P[5]s H1–H8 at 25 °C in CHCl3. The n value for each point, e.g. “n = 1”, indicates the number of carbons in the specific P5 alkyl chain; (b) single crystal X-ray structure of P[5] homologs H4–H7 complexed with HAN. Top: Front view. Below: Top view. C-gray, O-red, HAN-blue Hydrogen atoms were hidden for clarity. | ||
All efforts to grow X-ray quality single crystals of the P[5]/amino acids complexes failed due to the better crystallinity of P[5]s themselves. Fortunately, several HAN@P[5] single crystals were successfully obtained. As expected, the length of the alkyl chains did not result in any apparent changes to the structure of the central P[5] host domain, but the space bordering both rims of the binding site does become increasingly crowded (Fig. 4b and Fig. S123, ESI†). The dinitrile host HAN oriented only at the center P[5] unit and caused negligible steric interactions with the alkyl chains. As noted above (Fig. 4a), both HAN and G2 demonstrate similar variation tendencies in the thermodynamic parameters. That is to say, both G2 and HAN showed excellent entropy–enthalpy compensation relationships, suggesting that the main driving force for complexation is not changed with the difference in the P[5] alkyl ether chain length.
G2 should interact with P[5] mainly through the ammonium–oxygen hydrogen bonding.5m Molecular simulations (Fig. S124, ESI†) revealed that the ammonium group will locate at the opening of P[5] ring, while the rest of the guest is situated outside the host cavity. This is supported by the significant upfield shifts of the α and β protons of G2 upon complexing with H1 (Fig. 5a and b) and the correlations between protons on G2 with the alkyl chain in the ROESY spectrum (Fig. S126 and S127, ESI†).7 The signal for the β protons showed larger and larger upfield shifts upon increases in the P[5] chain lengths, affording a chemical shift up to −0.7 ppm with H7 (Fig. 5). This suggests that increasing the P[5] alkyl chain length forces the α-methyl group to insert deeper into the cavity, presumably due to stronger steric interactions. This chain-dependent difference in complexation might be responsible for the switching of enantio-preference with G2. Moreover, the sharp signal for the α-methyl protons in pure G2 became broad and separated into two peaks upon complexation with the P[5] homologs and this effect increased with the host alkyl chain length. A similar trend was observed with the methylene protons in the ethyl ester of G2 (protons d), indicating that the complexation/flipping kinetic become slower with the chain length. The separated proton signals can be ascribed to the diastereomeric complexes formed by the R- and S-conformer, respectively. The more upfield shifted α-methyl showed less integration, and ratios of 1.67/1, 1.78/1, 1.82/1 and 1.81/1 were calculated for H4, H5, H6 and H7, respectively, suggesting the diastereomeric complexes having relatively deeper entered G2 are less stable.
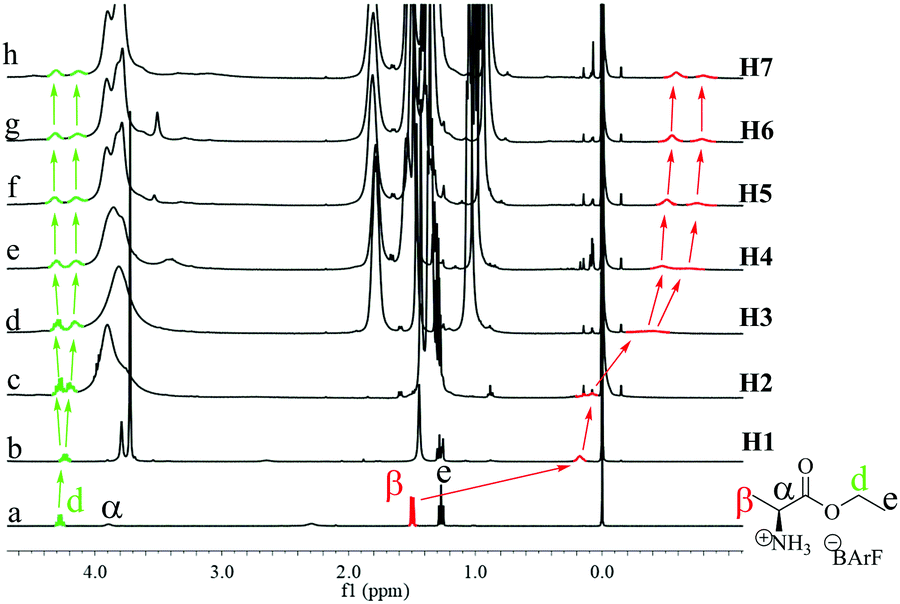 | ||
| Fig. 5 1H NMR spectra of G2 (a) in the absence of and (b–h) presence of a P[5] ([G2] = [P5] = 3.4 mM, 400 MHz, CDCl3). | ||
As mentioned above, the chiroptical response of the different P[5] derivatives varied drastically depending on the specific α-ammonium ester investigated, with changes in either the ester moiety or the α-substituent being readily detectable. For example, a rapid decrease of the positive CDex induced by tert-butyl alanate G5 was observed going from H1 to H4, with only a small positive CDex resulting upon further increases in the P[5] alkyl chain length (Fig. 3b). Ethyl phenylalanate G11 induced an increasingly positive CDex with longer P[5] chain lengths (Fig. 3c), whereas the negative CDex induced by diethyl aspartate G12 decreased with increasing P[5] alkyl chain length. The unique relationship between both the configuration and specific structure of the α-ammonium ester guests on the sign (Fig. 2d) and intensities (Fig. 3) of the induced CD spectra for the library of P[5] hosts provides a powerful optical tool for simultaneously determining the chirality and structure of the chiral analytes. Indeed, the g factor changing model vs. chain length showed a clear distinction for the different α-ammonium esters (Fig. 6, top). The changing models are similar groups of analytes. For examples, different esters of the same amino acids (G1–G5) form one group, ethyl esters of different α-alkyl-substituted ammonium esters (G2, G8–G10, G14) form another similarity group, and analytes that possess aromatic residues (G6–G7, G11) form a third cluster. These trends allowed for analyte classification via hierarchical clustering analysis (Fig. 6, bottom).
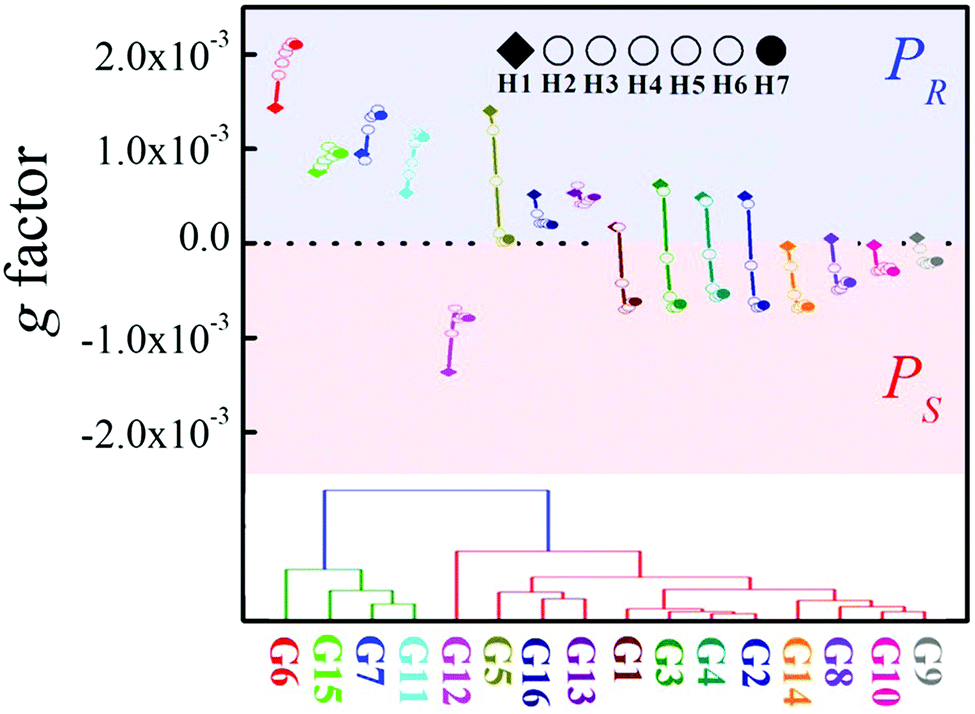 | ||
| Fig. 6 Top: g factor at 310 nm as a function of chain length. Bottom: The hierarchical clustering dendrogram analysis based on the CDex changes. | ||
In conclusion, we demonstrated that a library of P[5] derivatives, which differ only in the length of the alkyl ether chains, showed strong and diverse CD spectral responses by complexing with amino acid derivatives, revealing a potential tool for sensing the handedness and enantiopurity of chiral amines. The chain length on the P[5] derivatives significantly changed the complexation behavior as well as the resulting CD response. The diversity of CD responses and the P[5] alkyl chain length-dependent trends with different chiral amines provide a powerful library for the quantitative assignment of the α-amino esters, thus opening a new venture for chiral and structural discrimination with P[5] homologs.
We acknowledge the support of this work by the National Natural Science Foundation of China (No. 21871194, 21971169, 21572142), National Key Research and Development Program of China (No. 2017YFA0505903), Science & Technology Department of Sichuan Province (2019YJ0160, 2019YJ0090, 2017SZ0021), Comprehensive Training Platform of Specialized Laboratory, College of Chemistry and Prof. Peng Wu of Analytical & Testing Center, Sichuan University.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) E. V. Anslyn, J. Org. Chem., 2007, 72, 687–699 CrossRef CAS
; (b) G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS
-
(a) V. V. Borovkov, I. Fujii, A. Muranaka, G. A. Hembury, T. Tanaka, A. Ceulemans, N. Kobayashi and Y. Inoue, Angew. Chem., Int. Ed., 2004, 43, 5481–5485 CrossRef CAS
-
(a) K. L. Diehl and E. V. Anslyn, Chem. Soc. Rev., 2013, 42, 8596–8611 RSC
-
(a) X. Wei, W. Wu, R. Matsushita, Z. Yan, D. Zhou, J. J. Chruma, M. Nishijima, G. Fukuhara, T. Mori, Y. Inoue and C. Yang, J. Am. Chem. Soc., 2018, 140, 3959–3974 CrossRef CAS PubMed
-
(a) T. Ogoshi, S. Kanai, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed
-
(a) T. Ogoshi, K. Kitajima, T. Aoki, S. Fujinami, T.-A. Yamagishi and Y. Nakamoto, J. Org. Chem., 2010, 75, 3268–3273 CrossRef CAS PubMed
- For the details see ESI†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC H4@HAN: 1880191, H5@HAN: 1880195, H6@HAN: 1880192 and H7@HAN: 1880193. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc08541f |
| ‡ J. Ji and Y. Zhou contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |