
General chemoenzymatic route to two-stereocenter triketides employing assembly line ketoreductases†
Zhicheng
Zhang
a,
Alexis J.
Cepeda
a,
Mireya L.
Robles
 a,
Melissa
Hirsch
a,
Kaan
Kumru
b,
Jina A.
Zhou
a and
Adrian T.
Keatinge-Clay
*b
a,
Melissa
Hirsch
a,
Kaan
Kumru
b,
Jina A.
Zhou
a and
Adrian T.
Keatinge-Clay
*b
aDepartment of Chemistry, The University of Texas at Austin, 100 E. 24th St., Austin, TX 78712, USA
bDepartment of Molecular Biosciences, The University of Texas at Austin, 100 E. 24th St., Austin, TX 78712, USA. E-mail: adriankc@utexas.edu
First published on 28th November 2019
Abstract
Modular polyketide synthases (PKSs) are enzymatic assembly lines that fuse carbon fragments into complex chiral products. Here, their synthetic logic is employed to chemoenzymatically generate two-stereocenter triketides. Each of the four stereoisomers was constructed in a stereocontrolled manner using C-acylation and two PKS ketoreductases possessing opposite stereoselectivities.
Natural products are often lead compounds in the discovery and development of new medicines, with polyketides more than any other class of natural product giving rise to clinically-valuable pharmaceuticals (e.g., the antibacterial erythromycin, the immunosuppressant rapamycin, and the anticancer agent epothilone).1 Unfortunately, the low natural abundance of polyketides that possess desired bioactivities coupled with their complex structures hamper their development as medicines. Advances have been made in obtaining complex polyketides such as discodermolide2 and bryostatin3 through total synthesis, yet a general strategy to precisely set stereocenters during the union of carbon fragments has not yet been realized. While automated technologies have been utilized to rapidly synthesize biopolymers such as polypeptides4,5 and DNA6,7 for almost half a century, no similar process has been developed for polyketides.
Our group is inspired by polyketide synthase (PKS) assembly lines and how they construct complex polyketides from simple building blocks (Fig. 1).8–11 During polyketide biosynthesis, intermediates are covalently attached to acyl carrier protein (ACP) domains that dock with assembly line enzymes such as ketosynthases (KSs) that catalyze carbon–carbon bond formation and ketoreductases (KRs) that mediate stereocontrolled reductions that forge either one or two stereocenters.12 The extension of intermediates is accomplished through the decarboxylative Claisen-like condensation of malonyl thioesters in the active sites of KSs. While KRs have been utilized as robust biocatalysts,13 KSs have not.14 However, since a directly analogous C-acylation reaction is known,15,16 a chemoenzymatic synthesis of polyketides through cycles of C-acylation and KR biocatalysis should be achievable.17,18
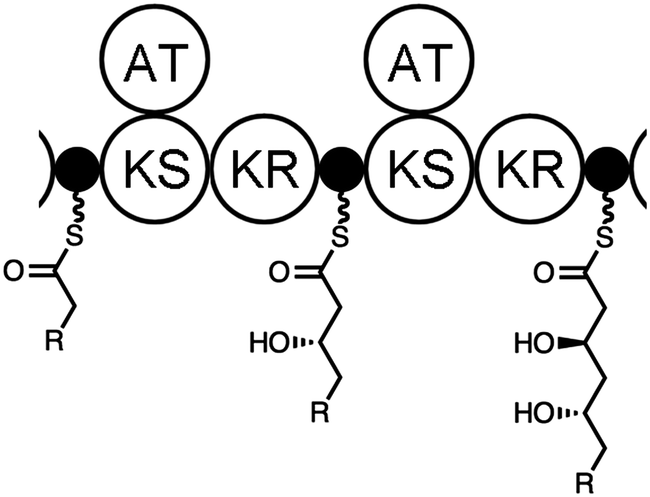 | ||
| Fig. 1 Polyketide assembly lines provide inspiration for the chemoenzymatic synthesis of polyketide fragments. If the power of the ketosynthase (KS) domains that make carbon–carbon bonds and ketoreductase (KR) domains that set stereocenters were accessed, the general synthesis of diverse, chiral fragments would be possible. | ||
Aiming at a general synthesis of polyketides, we sought to demonstrate the chemoenzymatic construction of chiral triketides and set our goal as the stereocontrolled synthesis of a library of four stereoisomers containing chiral centers at their β- and δ-carbons. Two KRs with opposite stereoselectivities were employed – the KR from the sixth module of the mycolactone PKS (MycKR6)19 and the KR from the second module of the tylosin PKS (TylKR2)20 (updated module definition used21,22). Both MycKR6, an A-type KR that generates L-β-hydroxy substituents, and TylKR2, a B-type KR that generates D-β-hydroxy substituents, have been shown to be stereoselectively-robust biocatalysts when excised from their assembly lines.23–26 β-Hydroxyl groups generated by KRs need to be protected before C-acylation due to the sensitivity of this reaction to labile protons (another consideration is the spontaneous cyclization of the δ-hydroxy thioester product).
We designed the route to begin with N-acetylcysteamine (NAC) opening propionyl-Meldrum's acid 1 to generate β-keto-diketide 2 (Scheme 1). Next, incubation of 2 with MycKR6 or TylKR2 and a nicotinamide adenine dinucleotide phosphate (NADPH) regeneration system [D-glucose and glucose dehydrogenase (GDH)] would yield either the L- or the D-β-hydroxy-diketide 3.13 The hydrolysis of 3 would afford the corresponding β-hydroxy acid 4, and protection would yield protected acid 5. C-Acylation of 5 with malonyl ethanethiol thioester would generate β-keto-triketide 6. In situ thiol–thioester exchange of 6 with NAC would give NAC-bound β-keto-triketide 7, which should then be reduced by MycKR6 or TylKR2 to yield a β-hydroxy triketide 8. Subsequent hydrolysis would provide a two-stereocenter triketide acid 9.
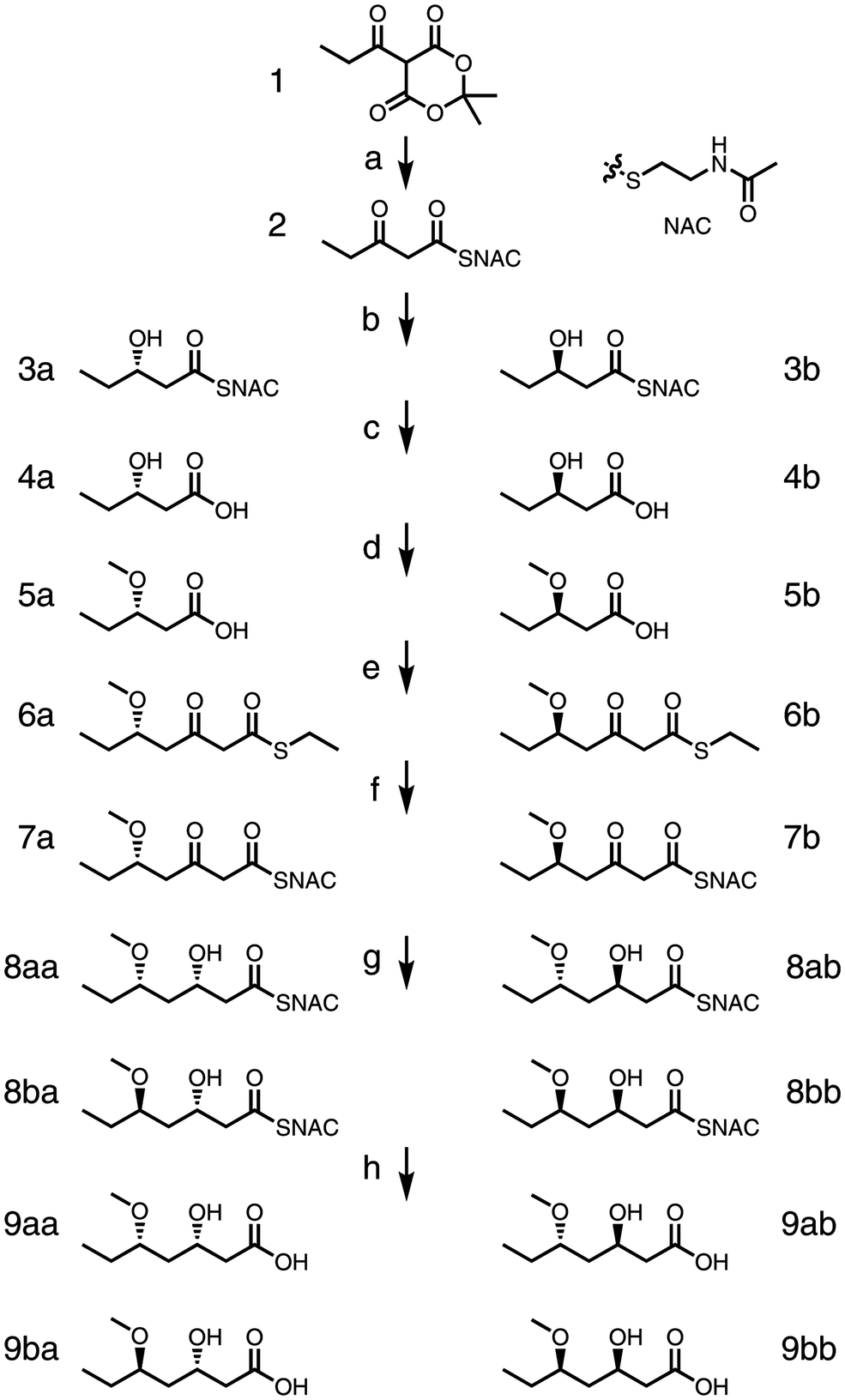 | ||
| Scheme 1 Chemoenzymatic route. (a) Pyridine (2.0 eq.), propionyl chloride (1.0 eq.), DCM, overnight, 0 to 22 °C (76% yield); NAC (0.95 eq.), toluene, 115 °C, 5 h (61%), (b) either MycKR6 (A-type KR) or TylKR2 (B-type KR), GDH, NADP+, pH 7.7, overnight, 22 °C (3a, 65%; 3b, 58%), (c) 5 M NaOH aq., 80 °C, overnight (4a, 51%; 4b, 59%), (d) 2.5 M n-BuLi (3.0 eq.), DMSO, MeI (2.4 eq.), under argon, 22 °C, overnight (5a, 73%; 5b, 78%), (e) 1,1′-carbonyldiimidazole (1.1 eq.), Mg(OEt)2 (0.55 eq.), malonyl ethanethiol thioester (1.1 eq.), anhydrous THF, 22 °C, overnight (6a, 35%; 6b, 34%), (f–h) NAC, pH 8.5, 22 °C, 2 h, then either MycKR6 or TylKR2, GDH, NADP+, pH 7.7, overnight, then 5 M NaOH, 70 °C, overnight (9aa, 57%; 9ab, 36%; 9ba, 44%; 9bb, 24%). | ||
The first two steps of this route are based on our previous work but were performed using a modified procedure to obtain higher purity.13 Propionyl chloride was added to Meldrum's acid in the presence of excess pyridine to afford propionyl-Meldrum's acid 1 without separation (70% crude yield).271 was refluxed at 115 °C with NAC to provide β-keto-diketide 2 (61% yield). Unreacted NAC could be removed with copper sulfate impregnated silica during flash chromatography. The NAC handle, compared with shorter handles such as ethanethiol, affords greater stereocontrol during KR biocatalysis.28 β-Keto-diketide 2 (soluble to ∼100 mM in water) was incubated at the gram scale overnight with either MycKR6 to provide 3a (65% yield) or TylKR2 to provide 3b (58% yield) as measured by chiral HPLC in comparison with standards.13 Through heating overnight in 5 M NaOH, crude 3a and 3b were hydrolyzed to L-β-hydroxypentanoic acid 4a and D-β-hydroxypentanoic acid 4b (51% and 59% yields, respectively); this enabled the removal of NAC and its dimer generated during the reduction reaction.
Several groups were considered to protect the β-hydroxyl substituent, including acetyl, methyl, methoxymethyl, and silyl. The acetyl group was attempted; however, as elimination reactions predominated, the more stable methyl protecting group was adopted. In the biosynthesis of polyketides, O-methyltransferase domains frequently methylate β-hydroxyl substituents, therefore the methyl group will not likely prevent entry into the active sites of downstream enzymes such as KRs, which is a concern with the larger silyl groups.29 The hydroxyl substituents of 4a and 4b were methylated using a solution of n-butyllithium (n-BuLi) and DMSO through treatment with MeI, generating β-methoxy acids 5a and 5b (73% and 78% yield, respectively).30 The reaction was monitored through the color change from the 4a/4b dianion (dark brown) to the monoanion (light orange); although the reaction had been developed to generate dimethylated alkoxy esters from hydroxy acids, it mainly provided the desired monomethylated alkoxy esters. The protected diketide acids 5a and 5b were C-acylated using the magnesium salt of malonyl ethanethiol thioester to give β-keto-triketides 6a and 6b (35% and 34% yields, respectively).15 This key, carbon–carbon bond-forming step mimicks the decarboxylative condensation catalyzed by KSs.
Thiol–thioester exchange in 0.33 M HEPES (pH 8.5) using 10 eq. of NAC converted 6a to 7a and 6b to 7b. After this handle swap, the pH was adjusted to 7.7 and the NADPH regeneration system as well as either MycKR6 or TylKR2 was added to generate a product with an L- or D-orientated β-hydroxyl group, respectively. Purifications of 8aa, 8ba, 8ab, and 8bb were challenging due to byproducts such as the NAC dimer. However, heating at 70 °C in aqueous 5 M NaOH converted the thioesters into carboxylic acids and enabled the removal of byproducts to facilitate the purification of triketide products 9aa, 9ba, 9ab, and 9bb (57%, 44%, 36%, and 24% yield, respectively, through three steps).
Chemical standards for triketide thioesters 8aa, 8ab, 8ba, and 8bb as well as the corresponding carboxylic acids 9aa, 9ab, 9ba, and 9bb were prepared (Scheme 2). Commercially-available 10 was hydrogenated at 100 atm using (Sa)-Ir-spiroSAP or (Ra)-Ir-spiroSAP as the catalyst to give enantiomers 11a or 11b, respectively.31,3211a and 11b were hydrolyzed by heating overnight in a solution of 5 M NaOH to generate 4a and 4b. Protection and extension of 4a and 4b into 6a and 6b were performed as in the chemoenzymatic route. Alcoholysis converted thioesters 6a and 6b to oxoesters 12a and 12b (27% and 16% yields, respectively; 30% of starting material was unreacted and could be reused). 12a was reduced using (Sa)-Ir-spiroSAP or (Ra)-Ir-spiroSAP and hydrolyzed to provide 9aa or 9ab, (77% and 97% yields, respectively, through two steps).31,32 Likewise, 12b was reduced using (Sa)-Ir-spiroSAP or (Ra)-Ir-spiroSAP and hydrolyzed to provide 9ba or 9bb (83% and 86% yields, respectively, through two steps). Coupling with NAC yielded 8aa, 8ab, 8ba, and 8bb.
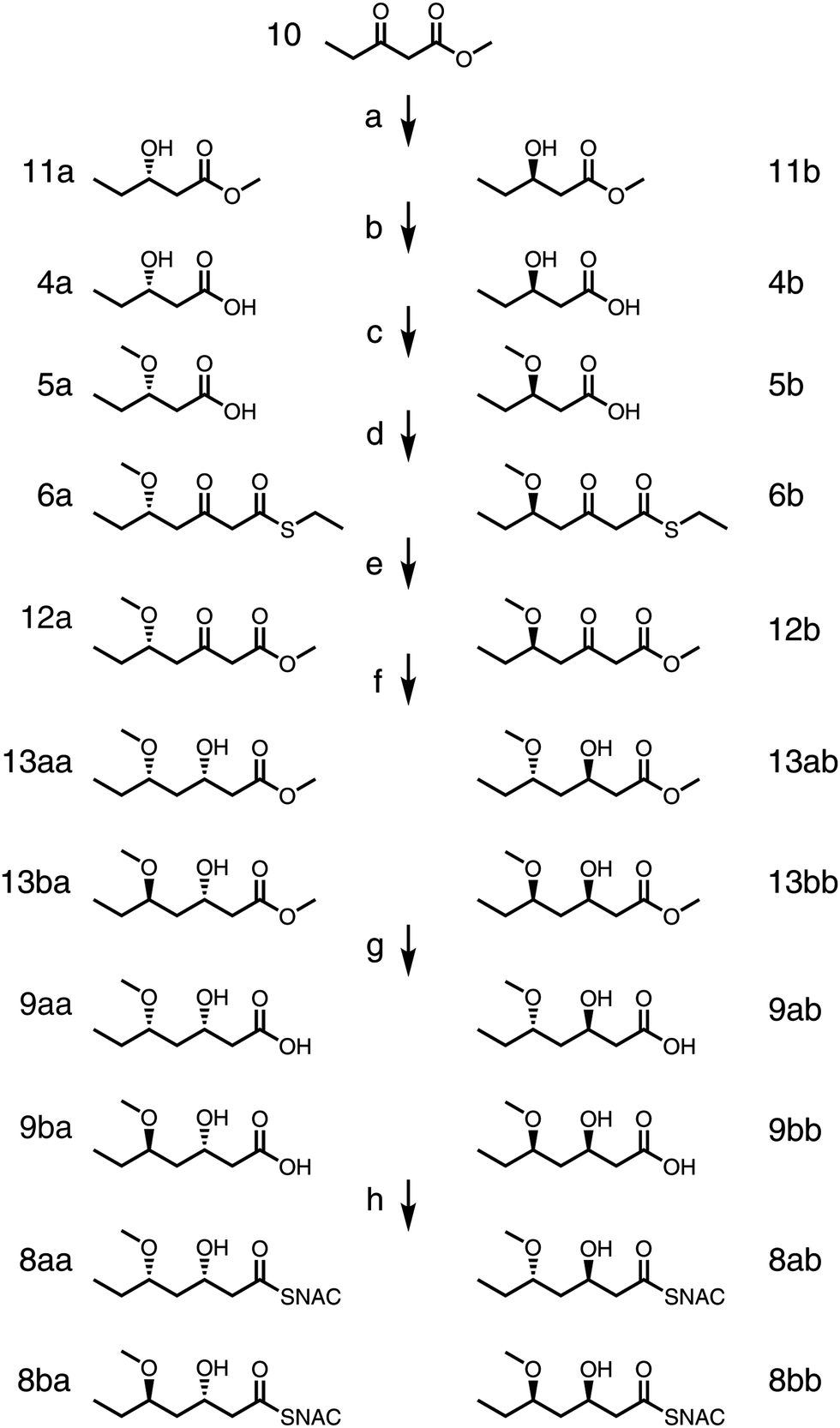 | ||
| Scheme 2 Synthesis of standards. (a) Either (Sa)-Ir-spiroSAP, 100 atm H2, MeOH, 22 °C, 48 h for 11a or (Ra)-Ir-spiro-SAP, 100 atm H2, MeOH, 22 °C, 48 h for 11b (both yields >99%), (b) 5 M NaOH, 80 °C, overnight (4a, 51% and 4b, 59%), (c) 2.5 M n-BuLi (3.0 eq.), DMSO, MeI (2.4 eq.), under argon, 22 °C, overnight (5a, 73%; 5b, 78%), (d) 1,1′-carbonyldiimidazole (1.1 eq.), Mg(OEt)2 (0.55 eq.), malonyl ethanethiol thioester (1.1 eq.), anhydrous THF, 22 °C, overnight (6a, 35%; 6b, 34%), (e) sodium methoxide (10.0 eq.), anhydrous MeOH, 22 °C, 6–9 h, (12a, 27%; 12b, 16%), (f–g) either (Sa)-Ir-spiroSAP, 100 atm H2, MeOH, 22 °C, 48 h then 5 M NaOH, 60 °C, overnight for 9aa and 9ba or (Ra)-Ir-spiroSAP, 100 atm H2, MeOH, 22 °C, 48 h then 5 M NaOH, 60 °C, overnight for 9ab and 9bb (9aa, 77%; 9ab, 97%; 9ba, 83%; 9bb, 86%). (h) THF, 4-dimethylaminopyridine (0.5 eq.), 1-ethyl-3-(3-dimethylaminopropyl)carbo-diimide-HCl (2.3 eq.), NAC (5.0 eq.), 22 °C, overnight. | ||
The syn-isomers 9aa and 9bb can be distinguished from the anti-isomers 9ab and 9ba through 1H NMR most apparently through the signal from the C5 hydrogen geminal to the methoxy group (4.2–4.3 ppm); however, impurities from the enzymatic reductions often gave rise to overlapping signals. The relative configurations of the isomers could be assessed through 13C NMR using Hoffmann's method of distinguishing between β-alkoxy alcohol diastereomers.33 The sum of the signals for the hydroxy- and alkoxy-bearing carbons should be lower for anti-isomers (in the chair made by the hydrogen bond between the hydroxy and alkoxy groups either the substituent on the hydroxy- or the alkoxy-bearing carbon is axial for the anti-isomers, resulting in steric hindrance and relatively upfield signals compared to the signals from the hydroxy- and alkoxy-bearing carbons in the syn-isomers that possess equatorial substituents) (Fig. 2A). For the chemoenzymatically- or chemically-generated syn-isomers 9aa and 9bb, the signals from the hydroxy-bearing C3 and the alkoxy-bearing C5 were 82.0 ppm and 67.9 ppm, respectively, and for the chemoenzymatically- or chemically-generated anti-isomers 9ab and 9ba, the signals from the hydroxy-bearing C3 and the alkoxy-bearing C5 were 79.7 ppm and 65.3 ppm, respectively. In agreement with Hoffmann's method, the sum of the signals for the syn-isomers was greater than the anti-isomers, by 4.9 ppm.
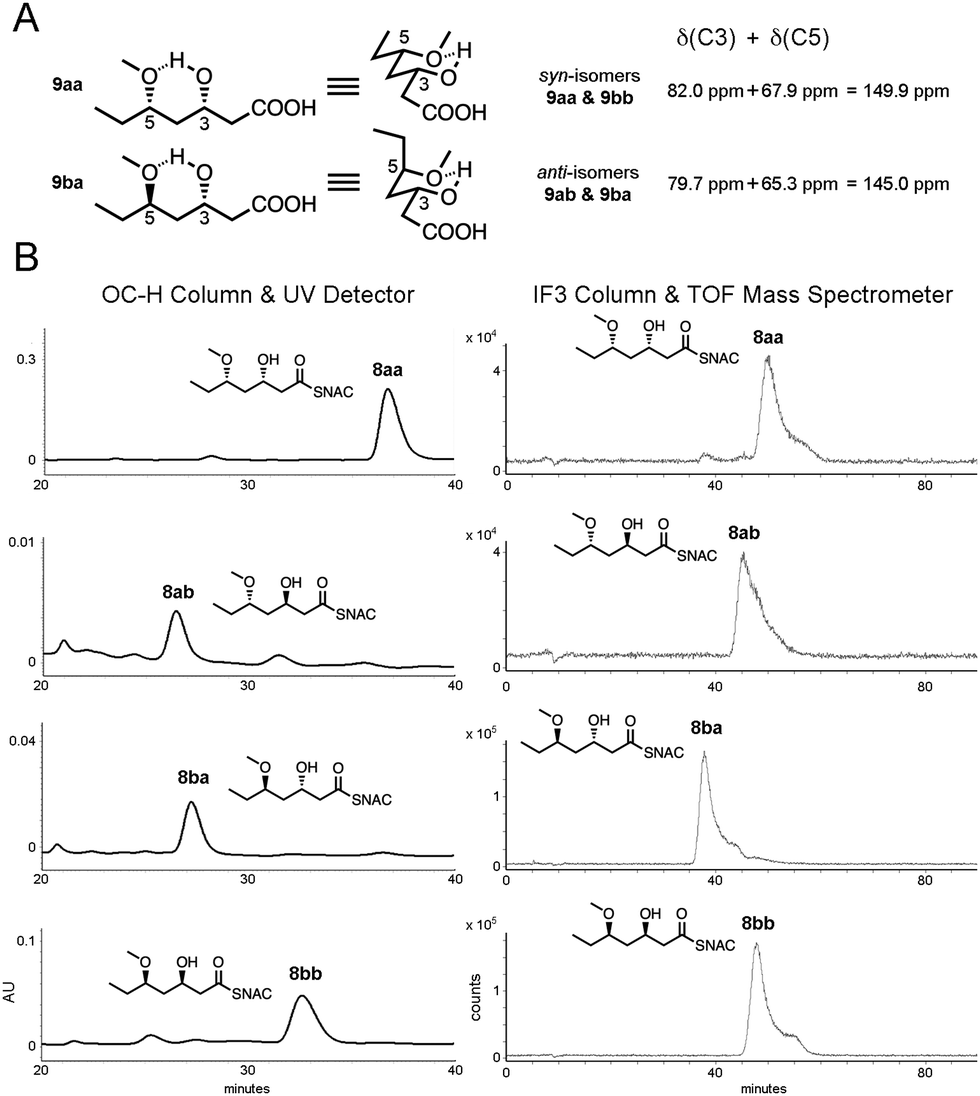 | ||
| Fig. 2 (A) The relative configurations of the triketide stereoisomers can be readily assigned using Hoffmann's method of summing the 13C NMR shifts for the C3 and C5 signals: the anti-isomers (e.g., 9ba) give a smaller δ(C3) + δ(C5) value than syn-isomers (e.g., 9aa) due to axial substituents on the chair formed by the hydroxyl and the methoxy groups. (B) Chiral chromatography of chemoenzymatically-generated triketides 8aa, 8ab, 8ba, and 8bb using two systems: an OC-H column coupled with a UV detector (left) and an IF3 column coupled with a time-of-flight mass spectrometer (right). The OC-H column was able to resolve the syn-products, while the IF3 column was able to resolve the anti-products. Retention times matched those of standards (Tables S1 and S2, ESI†). | ||
Chemoenzymatically- and chemically-generated triketide stereoisomers 8aa, 8ab, 8ba, and 8bb were also compared using complementary, chiral chromatography systems – an OC-H column coupled with a UV detector and an IF3 column coupled with a time-of-flight mass spectrometer (Fig. 2B and Fig. S1, Table S1, ESI†). The anti-isomers, 8ab and 8ba, eluted before the syn-isomers, 8aa and 8bb, in both chromatographies. While the anti-isomers could not be resolved from one another on the OC-H column, they could be on the IF3 column, and while the syn-isomers could not be resolved from one another on the IF3 column, they could be on the OC-H column. In all four cases, the targeted stereoisomer of the chemoenzymatic synthesis matched the chemically-generated stereoisomer. The A-type MycKR6 and the B-type TylKR2, known to be stereoselectively robust towards β-ketodiketide NAC thioesters,13 proved to be stereoselectively robust towards triketide NAC thioesters. KRs thus have an advantage over most asymmetric catalysts in the reduction of 1,3 polyol substrates in that they override the influence of neighboring stereocenters which commonly affect stereocontrol.32,34
The chemoenzymatic synthesis of a library of two-stereocenter triketide fragments was accomplished with overall yields of 2.0% for 9aa, 1.3% for 9ab, 1.7% for 9ba, and 0.9% for 9bb (in each case the targeted stereoisomer comprises at least 90% of all possible stereoisomers; stereoisomeric compositions are reported in Table S1, ESI†). This marks an initial success for the described general polyketide synthesis and a starting point for optimization. The yields should be improved, and a protecting group that can be easily removed at the end of the synthesis but does not affect KR stereocontrol should be identified. As many KRs are able to set two stereocenters when reducing α-substituted, β-ketoacyl substrates,12 the described route may be utilized to generate libraries of three- or four-stereocenter triketides, for example through the use of methylmalonyl ethanethiol thioester in C-acylation reactions. The biocatalytic employment of other PKS enzymes such as dehydratases and enoylreductases could further diversify the functionality of synthesized fragments. Reaction optimization should allow another round of C-acylation and reduction to generate tetraketides at milligram levels. If the described, stereoselective synthesis could be generally employed in the construction of stereocomplex fragments, it would be a boon to synthetic methodology as well as the development of new medicines.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- K. J. Weissman and P. F. Leadlay, Nat. Rev. Microbiol., 2005, 3, 925–936 CrossRef CAS PubMed.
- S. J. Mickel, et al. , Org. Process Res. Dev., 2004, 8, 122–130 CrossRef CAS.
- Y. Lu, S. K. Woo and M. J. Krische, J. Am. Chem. Soc., 2011, 133, 13876–13879 CrossRef CAS.
- A. R. Mitchell, Biopolymers, 2008, 90, 175–184 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154 CrossRef CAS.
- A. M. Michelson and A. R. Todd, J. Chem. Soc., 1955, 2632–2638 RSC.
- R. H. Hall, A. Todd and R. F. Webb, J. Chem. Soc., 1957, 3291–3296 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2012, 29, 1050–1073 RSC.
- A. T. Keatinge-Clay, Chem. Rev., 2017, 117, 5334–5366 CrossRef CAS.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- E. J. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- S. K. Piasecki, C. A. Taylor, J. F. Detelich, J. N. Liu, J. T. Zheng, A. Komsoukaniants, D. R. Siegel and A. T. Keatinge-Clay, Chem. Biol., 2011, 18, 1331–1340 CrossRef CAS.
- D. C. Gay, G. Gay, A. J. Axelrod, M. Jenner, C. Kohlhaas, A. Kampa, N. J. Oldham, J. Piel and A. T. Keatinge-Clay, Structure, 2014, 22, 444–451 CrossRef CAS.
- D. W. Brooks, L. D. L. Lu and S. Masamune, Angew. Chem., Int. Ed. Engl., 1979, 18, 72–73 CrossRef.
- Y. H. Pan and C. H. Tan, Synthesis, 2011, 2044–2053 CAS.
- C. Khosla, R. S. Gokhale, J. R. Jacobsen and D. E. Cane, Annu. Rev. Biochem., 1999, 68, 219–253 CrossRef CAS.
- A. D. Harper, C. B. Bailey, A. D. Edwards, J. F. Detelich and A. T. Keatinge-Clay, ChemBioChem, 2012, 13, 2200–2203 CrossRef CAS PubMed.
- T. P. Stinear, et al. , Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1345–1349 CrossRef CAS.
- E. Cundliffe, N. Bate, A. Butler, S. Fish, A. Gandecha and L. Merson-Davies, Antonie van Leeuwenhoek, 2001, 79, 229–234 CrossRef CAS.
- L. H. Zhang, et al. , Angew. Chem., Int. Ed., 2017, 56, 1740–1745 CrossRef CAS.
- A. T. Keatinge-Clay, Angew. Chem., Int. Ed., 2017, 56, 4658–4660 CrossRef CAS.
- R. Reid, M. Piagentini, E. Rodriguez, G. Ashley, N. Viswanathan, J. Carney, D. V. Santi, C. R. Hutchinson and R. McDaniel, Biochemistry, 2003, 42, 72–79 CrossRef CAS.
- P. Caffrey, ChemBioChem, 2003, 4, 654–657 CrossRef CAS.
- A. T. Keatinge-Clay, Chem. Biol., 2007, 14, 898–908 CrossRef CAS.
- J. T. Zheng and A. T. Keatinge-Clay, MedChemComm, 2013, 4, 34–40 RSC.
- Y. Oikawa, K. Sugano and O. Yonemitsu, J. Org. Chem., 1978, 43, 2087–2088 CrossRef CAS.
- C. B. Bailey, M. E. Pasman and A. T. Keatinge-Clay, Chem. Commun., 2016, 52, 792–795 RSC.
- M. A. Skiba, M. M. Bivins, J. R. Schultz, S. M. Bernard, W. D. Fiers, Q. Dan, S. Kulkarni, P. Wipf, W. H. Gerwick, D. H. Sherman, C. C. Aldrich and J. L. Smith, ACS Chem. Biol., 2018, 13, 3221–3228 CrossRef CAS.
- P. C. B. Page, Y. H. Chan, H. Heaney, M. J. McGrath and E. Moreno, Synlett, 2004, 2606–2608 CAS.
- W. Che, D. Y. C. Wen, S. F. Zhu and Q. L. Zhou, Helv. Chim. Acta, 2019, 102, e1900023 CrossRef.
- W. Che, Y. Z. Li, J. C. Liu, S. F. Zhu, J. H. Xie and Q. L. Zhou, Org. Lett., 2019, 21, 2369–2373 CrossRef CAS.
- R. W. Hoffmann and U. Weidmann, Chem. Ber., 1985, 118, 3980–3992 CrossRef CAS.
- S. Masamune, W. Choy, J. S. Petersen and L. R. Sita, Angew. Chem., Int. Ed. Engl., 1985, 24, 1–30 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Methods and characterization. See DOI: 10.1039/c9cc07966a |
| This journal is © The Royal Society of Chemistry 2020 |