
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Use of pyridazinediones as extracellular cleavable linkers through reversible cysteine conjugation†
Calise
Bahou‡
 a,
Richard J.
Spears‡
a,
Abil E.
Aliev
a,
Antoine
Maruani
a,
Marcos
Fernandez
a,
Faiza
Javaid
a,
Peter A.
Szijj
a,
James R.
Baker
*a and
Vijay
Chudasama
*ab
a,
Richard J.
Spears‡
a,
Abil E.
Aliev
a,
Antoine
Maruani
a,
Marcos
Fernandez
a,
Faiza
Javaid
a,
Peter A.
Szijj
a,
James R.
Baker
*a and
Vijay
Chudasama
*ab
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk; j.r.baker@ucl.ac.uk
bResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, 1649-004 Lisbon, Portugal
First published on 18th November 2019
Abstract
Herein we report a retro-Michael deconjugation pathway of thiol–pyridazinedione linked protein bioconjugates to provide a novel cleavable linker technology. We demonstrate that the novel pyridazinedione linker does not suffer from off-target modification with blood thiols (e.g., glutathione, human serum albumin (HSA)), which is in sharp contrast to an analogous maleimide linker.
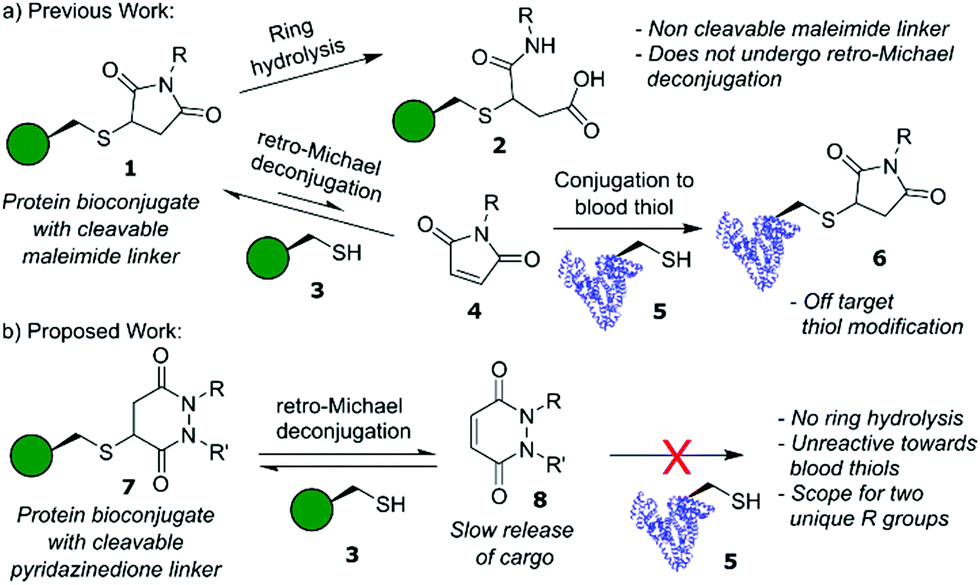
Site-selective modification of proteins lies at the forefront of contemporary approaches to problem solving in chemical biology.1,2 Construction of these protein bioconjugates frequently uses cysteine as a handle for bioconjugation,3,4 owing to its favourable properties (high nucleophilicity of the thiol side chain at neutral pH (pKa ≈ 8), low natural abundance (<2%) and ease of incorporation via site-directed mutagenesis).5 In particular, cysteine modification features heavily in the synthesis of antibody–drug conjugates (ADCs), a class of therapeutics that combines the selective targeting capabilities of an antibody with the potent toxicity of small molecule drugs.6 Currently, five ADCs are FDA-approved, including Adcetris™ and Polivy™ whose payloads are linked directly via cysteine bioconjugation.7,8 Mechanistically, ADCs operate via either internalising or non-internalising pathways depending on whether endocytosis is initiated following antigen binding,9 with toxic cargo release dependent on whether a “non-cleavable linker” or “cleavable linker” is used when constructing the ADC.10 For non-cleavable linkers, drug release occurs following lysosomal degradation of the internalised ADC, whereas for cleavable linkers drug release is facilitated through a stimulus pre- and/or post-internalisation, e.g., triggered by a change in cancer microenvironment conditions.
Historically, many ADCs relied on internalising targets to enable drug release, however, in recent years there has been increasing interest in non-internalising ADCs. These non-internalising ADCs require extracellular cleavable linkers in order to operate efficiently.11 The two most common types of extracellular cleavable linkers are disulfide based linkers12 and dipeptide valine–citrulline (Val–Cit) linkers.13,14 Disulfide linkers release cargo through disulfide exchange in environments containing high concentrations of thiols.12 The Val–Cit linker undergoes enzymatic cleavage by cathepsin B, a cysteine protease that is typically overexpressed in cancers;11,14 although this protease is usually found in the lysosome it has also displayed extracellular activity in cancers.11 These linkers are not without their disadvantages however; exposed disulfide linkers are susceptible to reduction before the ADC reaches its target.10 Whilst hindered disulfides have (to some extent) addressed the issue of stability of disulfide linkers in blood they are, to the best of our knowledge, not viable for application as extracellular cleavable linkers, i.e., they only cleave post-internalisation into an intracellular environment.15,16 Val–Cit linkers are also susceptible to other protease-related activity, which can hamper their stability in blood plasma.10 An alternative system explored within the context of cleavable linkers is that of maleimide–thiol linkages (Fig. 1a),17,18 which can cleave via a retro-Michael pathway and the released maleimide can be trapped by other abundant thiols in the tumour environment. However, the rapid kinetics of maleimide–thiol conjugation makes any released maleimide, before the maleimide–thiol conjugate reaches its desired destination, highly susceptible to off-target thiol modification by other biomolecules in vivo (e.g., HSA),19 thus limiting their use as cleavable systems in a biological context. Furthermore, maleimide–thiol linkages can undergo competitive hydrolysis; terminating the retro-Michael deconjugation pathway and hindering overall cleavage and ultimately cargo release.
 | ||
| Fig. 1 (a) Competing reactions of cysteine–maleimide conjugates including reversible retro-Michael deconjugation and irreversible hydrolysis of the succinimide motif. (b) Proposed reaction for cysteine–pyridazinedione conjugates involving retro-Michael deconjugation only. | ||
Over the past decade, we have reported on the use of bromopyridazinediones (BrPDs) as tools for efficient cysteine bioconjugation, particularly in the synthesis of dually functionalised conjugates, nanoparticle constructs, and therapeutically relevant biomolecules such as ADCs.20–22 Unlike maleimides, conjugates synthesised using BrPDs do not undergo ring hydrolysis, and the unsaturated ring scaffold is otherwise stable under aqueous conditions.23 We were intrigued, however, as to whether bioconjugates containing a saturated pyridazinedione scaffold (synthesised as previously described)23 could proceed down a similar reversible retro-Michael deconjugation pathway displayed by classical maleimides and other Michael acceptors, e.g., cyanoacrylates.24 In the context of a novel cleavable linker, we hypothesised that if Michael addition is observed with PDs the lack of ring hydrolysis would avoid termination of the retro-Michael pathway as seen in maleimide conjugates. Moreover, a different reactivity profile for conjugation and retro-Michael on PDs (compared to other acceptors) could also provide novel opportunities, e.g., a dynamic system with a slow release of PDs that do not react with blood thiols would be a useful addition to the extracellular non-internalising ADC linker toolbox.
Our initial assessment of PDs as a platform for reversible cysteine modification began with the synthesis of small molecule Boc-Cys(PD)-OMe 9 (from Boc-Cys-OMe 10 and PD 11) to act as a model system for our studies (Fig. 2a). We noted that, compared to the 5-membered ring of the thioether succinimide motif obtained from classical maleimide–thiol conjugates, PD–thiol conjugates comprise a 6-membered ring that may allow for more conformational freedom along the –CH2–CH(SR)– backbone of the ring, giving rise to two possible “pseudoaxial” and “pseudoequatorial” conformers. Using molecular mechanics calculations of Boc-Cys(PD)-OMe 9, applying the MMX force field25 to find the optimised geometries of the potential pseudoaxial/pseudoequatorial conformers, we experimentally determined conformer ratio in a phosphate buffer (PB) pH 7.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3CN (7:3) solvent system at 37 °C by 1H NMR (see ESI†).26,27 We found that the PD ring of Boc-Cys(PD)-OMe 9 adopts both conformers, but with the pseudoaxial conformer, which we assume is more likely to be prone to eventual elimination, dominating the equilibrium (3:1 ratio of pseudoaxial:pseudoequatorial) (Fig. 2b). With small molecule Boc-Cys(PD)-OMe 9 in-hand, we next monitored the potential deconjugation of the PD by 1H NMR at 24 h intervals over the course of 96 h. New peaks in the 1H NMR corresponding to deconjugated PD 11 emerged, with integrations of the newly appearing PD 11 CH protons suggesting gradual PD liberation from the conjugate over time (Fig. 2c). To further confirm that deconjugation was taking place, we next performed an Ellman's test to establish the presence of free thiol generated from deconjugation of conjugate 9. Boc-Cys(PD)-OMe 9 was incubated with DTNB (Ellman's reagent) 12, and anticipated TNB 13 release (from reaction of liberated Boc-Cys-OMe 10 with DTNB 12) was monitored by UV-Vis absorption at 412 nm at 24 h intervals over the course of 96 h (Fig. 2d). Over this time course, we noted a 36% increase in the amount of deconjugated species determined from absorbance at 412 nm when DTNB 12 was incubated with conjugate 9, compared to a control experiment (Fig. 2d, see ESI† for details). These experiments confirmed the presence of a retro-Michael deconjugation pathway being effective on a thiolated PD.
:CD3CN (7:3) solvent system at 37 °C by 1H NMR (see ESI†).26,27 We found that the PD ring of Boc-Cys(PD)-OMe 9 adopts both conformers, but with the pseudoaxial conformer, which we assume is more likely to be prone to eventual elimination, dominating the equilibrium (3:1 ratio of pseudoaxial:pseudoequatorial) (Fig. 2b). With small molecule Boc-Cys(PD)-OMe 9 in-hand, we next monitored the potential deconjugation of the PD by 1H NMR at 24 h intervals over the course of 96 h. New peaks in the 1H NMR corresponding to deconjugated PD 11 emerged, with integrations of the newly appearing PD 11 CH protons suggesting gradual PD liberation from the conjugate over time (Fig. 2c). To further confirm that deconjugation was taking place, we next performed an Ellman's test to establish the presence of free thiol generated from deconjugation of conjugate 9. Boc-Cys(PD)-OMe 9 was incubated with DTNB (Ellman's reagent) 12, and anticipated TNB 13 release (from reaction of liberated Boc-Cys-OMe 10 with DTNB 12) was monitored by UV-Vis absorption at 412 nm at 24 h intervals over the course of 96 h (Fig. 2d). Over this time course, we noted a 36% increase in the amount of deconjugated species determined from absorbance at 412 nm when DTNB 12 was incubated with conjugate 9, compared to a control experiment (Fig. 2d, see ESI† for details). These experiments confirmed the presence of a retro-Michael deconjugation pathway being effective on a thiolated PD.
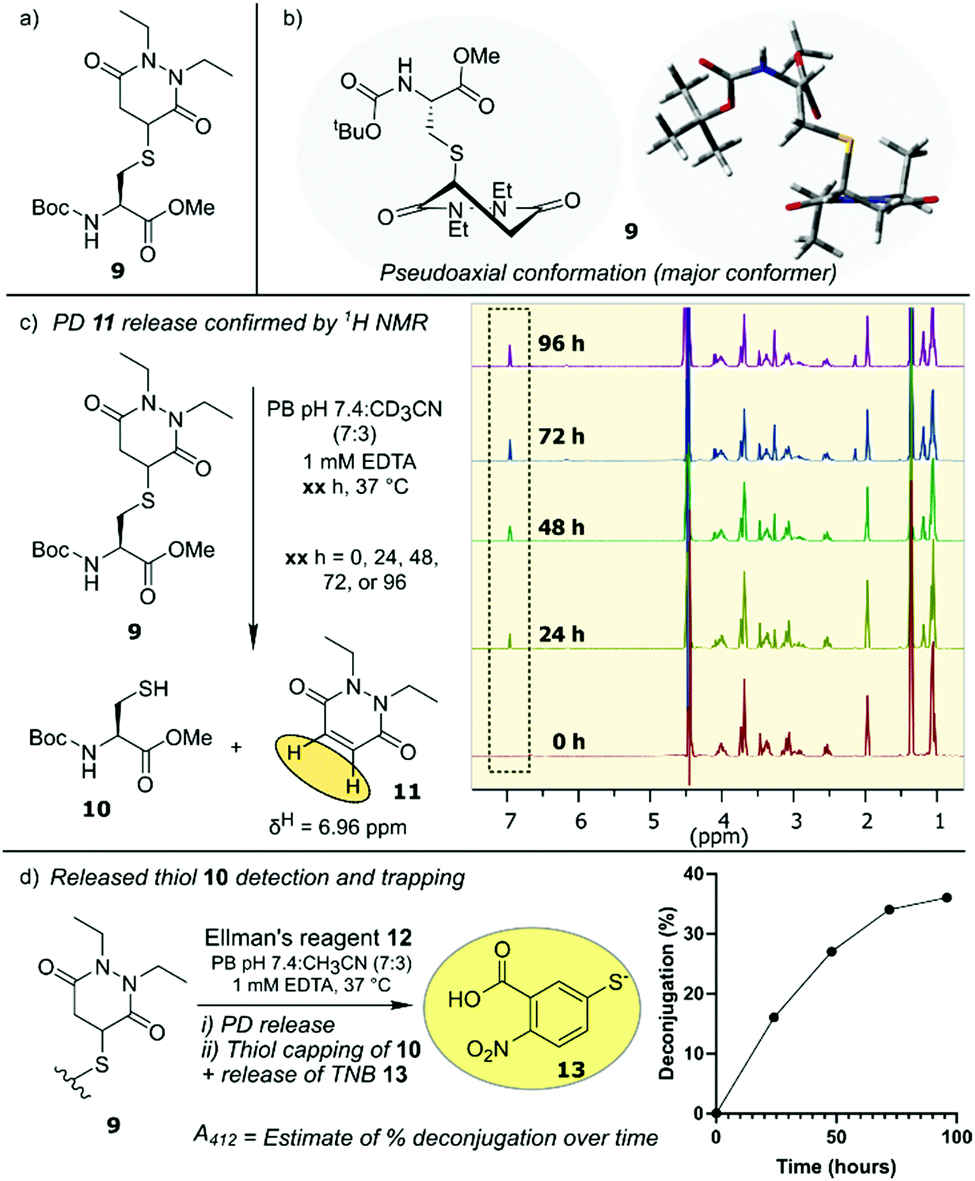 | ||
| Fig. 2 (a) Structure of Boc-Cys(PD)-OMe 9. (b) Structural representation of pseudoaxial conformation of Boc-Cys(PD)-OMe 9. (c) NMR analysis showing slow release of PD from Boc-Cys(PD)-OMe 9 to form diethyl PD 11 and Boc-Cys-OMe 10. (d) Schematic of TNB 13 release from reaction with 9 and DTNB 12 with graph depicting calculated % deconjugation. | ||
Next, we turned our attention towards the reversibility of PDs within the context of protein bioconjugates, utilising a mutant green fluorescent protein containing a solvent-accessible free cysteine as a model system (GFPS147C 14, expressed as described previously).28 Additionally, we also used a therapeutically relevant antibody Fab 15 (Fab, produced through digestion of the corresponding full antibody). Following reduction with tris(2-carboxyethyl)phosphine (TCEP, see ESI,† the GFP single cysteine formed a dimer spontaneously in storage so reduction was required) GFPS147C 14 and an antibody Fab 15 were fully modified with model PD 11 as judged by LC-MS to give GFP–PD 16 and Fab–PD 17 respectively (Fig. 3a-ii, Fab–PD 17 retained binding by ELISA – see ESI†). As a comparative model, GFPS147C 14 and Fab fragment 15 were also modified with a slowly hydrolysing N-methyl maleimide 23 to give GFP–maleimide 18 and Fab–maleimide 19 conjugates (Fig. 3a-iii).29 These bioconjugates were then subjected to a seven-day incubation period in buffer representing physiological conditions (pH 7.4, 37 °C, no EDTA), and deconjugation/ring hydrolysis of the conjugates was monitored by LC-MS analysis (Fig. 3b).
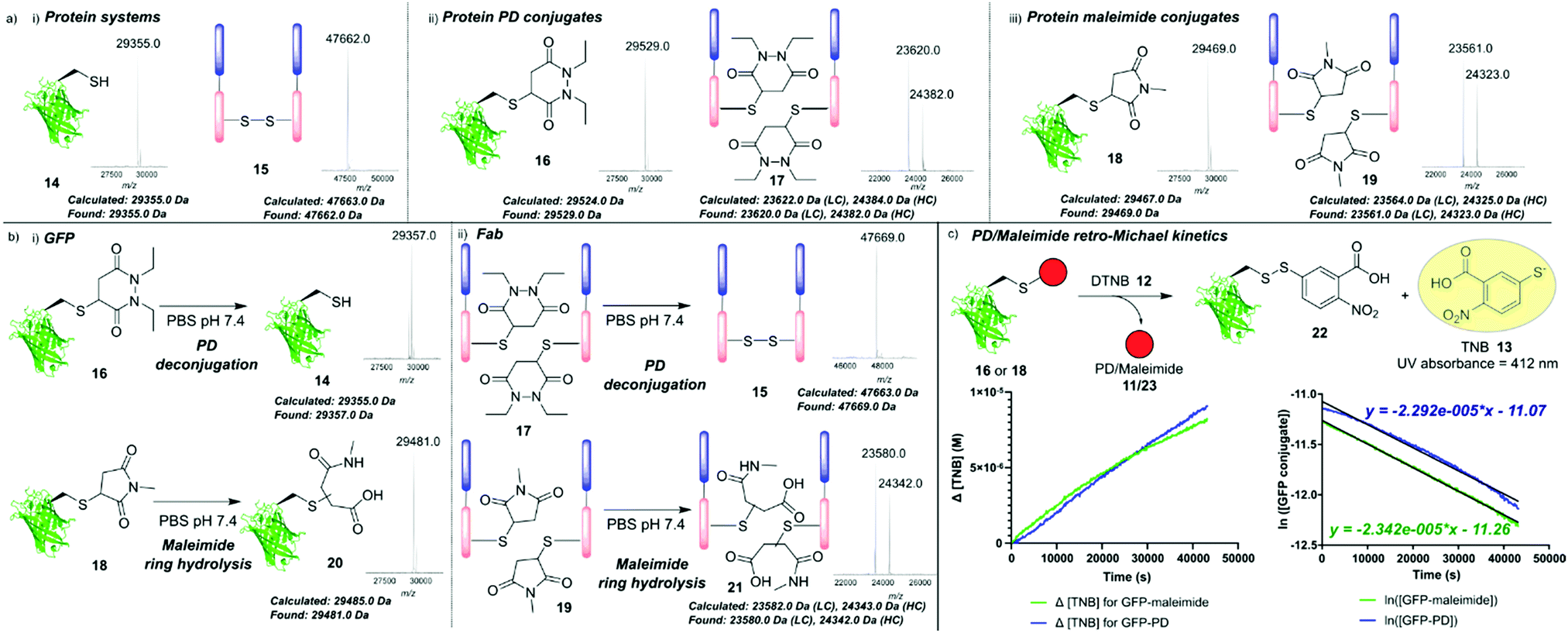 | ||
| Fig. 3 (a) Mass spectrometry data showing (i) GFPS147C 14 and model Fab fragment 15; (ii) GFP–PD species 16 and Fab–PD species 17; (iii) GFP-maleimide species 18 and Fab-maleimide species 19; (b) mass spectrometry analysis of (i) incubation of GFP conjugates 16 and 18 (50 μM) in PBS pH 7.4 for 7 days at 37 °C; (ii) incubation of Fab conjugates 17 and 19 (20 μM) in PBS pH 7.4 for 7 days at 37 °C. (c) Kinetic analysis of reaction between GFP conjugates (16 and 18) and DTNB 12 in PBS pH 7.4 for 12 h at 37 °C to form TNB 13 that was monitored by UV absorbance at A412. Plots show changing [TNB] and ln[GFP conjugate] vs. time. | ||
Significant release of the PD species from both GFP–PD 16 and Fab–PD 17 was noted, yielding GFPS147C 14 and reoxidised Fab 15 respectively with no conjugates bearing ring hydrolysis observed. In sharp contrast, GFP–maleimide 18 and Fab–maleimide 19 afforded only the succinamic acid species 21 under the same conditions; succinamic acids have previously been shown to be stable to retro-Michael deconjugation.30 GFP and Fab conjugates 16–19 were also subjected to incubation with a model monoBr PD species, which would theoretically form an irreversible unsaturated GFP–PD species/Fab–PD species that would act as a “trap” for any available cysteine liberated from a retro-Michael pathway (see ESI†). For the GFP–PD 16 and Fab–PD 17, a newly formed unsaturated GFP–PD conjugate and Fab–PD conjugate was obtained, with LC-MS analysis indicating near-full release of the original PD cargo (see ESI†). In contrast, incubation of monoBr PD with GFP–maleimide 18 and Fab–maleimide 19 showed minor formation of unsaturated GFP–PD species and Fab–PD species with a significant amount of maleimide hydrolysis observed (see ESI†). These results confirm that both Michael acceptors (nonBr PDs and maleimides) exist in an equilibrium between conjugation and retro-Michael deconjugation. It also highlights the competing hydrolysis pathway when using maleimide conjugates in this context. We next investigated the comparative rates of retro-Michael deconjugation PDs and maleimides. GFP conjugates 16 and 18 were incubated with excess DTNB 12 at 37 °C, with any liberated cysteine anticipated to react quantitatively to form TNB 13, giving a characteristic absorbance at 412 nm which could be used to determine PD/maleimide deconjugation (Fig. 3c and ESI†). Approximate rate constants for the pseudo first order retro-Michael deconjugation reactions were found to be 2.34 × 10−5 s−1 for the GFP–maleimide species 18 and 2.29 × 10−5 s−1 for the GFP–PD species 16, suggesting no significant difference between the rate of deconjugation. By comparing retro-Michael deconjugation rates with deconjugation seen by LC-MS, we can infer that the difference in equilibrium shift between maleimides and PDs is likely a consequence of the vastly different rates of Michael addition presented by these two motifs, i.e., maleimide and PD scaffolds are releasing at a similar rate but maleimides immediately react with the released cysteine.
In view of the relatively slow release of PD from the protein conjugates, we thought that it could be exploited as a novel extracellular cleavable linker for use in protein conjugates where the tumour targeting protein carrier is non-internalising (e.g., an alternative to conventional non-internalising ADC cleavable linkers such as disulfide and Val–Cit as discussed in the introduction). Whilst maleimide conjugates have been explored in this context (i.e., as a reversible cleavable linker),31 the PD conjugate does not undergo hydrolysis and the low reactivity of unsaturated PDs with protein thiols would ideally result in any prematurely released cargo (if any) being rapidly cleared from the body as a small molecule.
To this end, Fab–PD conjugate 17 was incubated with common blood thiols glutathione (5 μM, 4 h) and HSA 5 (20 μM, 48 h). An antibody Fab fragment was seen as a relevant model for these studies as various antibodies and antibody fragments have found application as non-internalising ADCs.32,33 To our delight, no transfer of PD to HSA 5 was observed (Fig. 4a) and addition of excess glutathione did not accelerate PD release (see ESI†). In sharp contrast, when Fab–maleimide conjugate 19 was subjected to HSA incubation conditions, significant amounts of HSA–maleimide species 24 was observed; confirming previous reports of blood instability of maleimide conjugates (Fig. 4b).19 Furthermore, incubation of each of HSA 5 (50 μM) and glutathione (500 μM) with diethyl PD 11 (2.5 mM and 500 μM, respectively) for 24 h at pH 7.4 to appraise whether any reaction occurred, even under these forcing conditions, led to no observed reaction in either case (see ESI†). Finally, we wanted to assess the applicability of the PD conjugate as an extracellular cleavable linker, in a partially acidic environment such as that of a tumour (ca. pH 6.5).34 Fab–PD species 17 was incubated under buffered conditions at pH 6.5 for 7 days. Despite acidic conditions slowing down the rate of Michael deconjugation, the majority of the PD was still released from the conjugate (Fig. 4c). We would therefore expect that PD cleavage would occur in the extracellular environment (following antigen binding) resulting in a high local concentration of PD-linked payload that may then diffuse into target cells. By using PDs in conjunction with linkers designed for intracellular triggered release (e.g., Val–Cit, hindered disulfides), efficient delivery of cargo using non-internalising targeting proteins could be achieved. Furthermore, combining a predictable time-dependent release mechanism in a tumour environment with a high tolerance towards HSA may provide a useful cleavage mechanism, such that a PD payload could be slowly released in a target area (e.g., a tumour microenvironment) without being carried away by blood thiols.
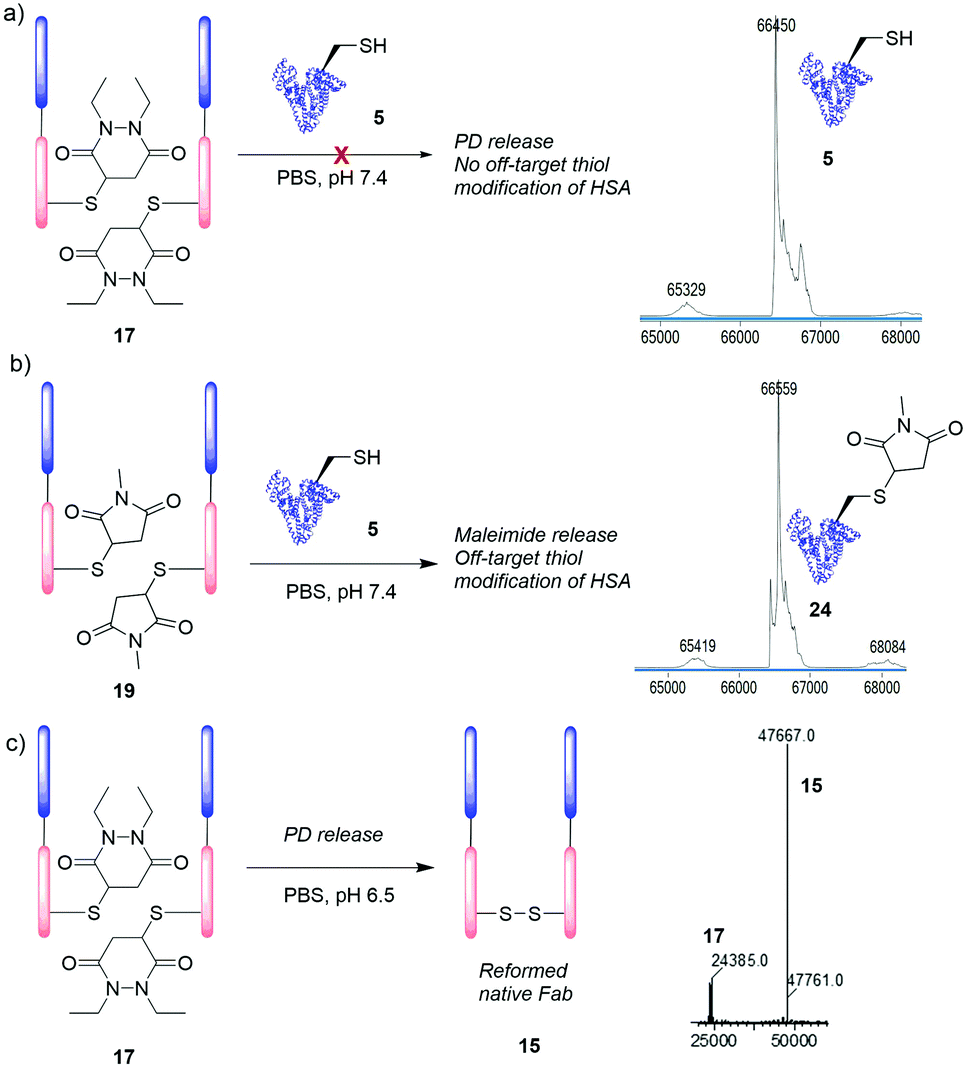 | ||
| Fig. 4 Incubation of Fab–PD conjugate 17 (20 μM) with (a) HSA 5 (20 μM) for 48 h. (b) Incubation of Fab–maleimide species 19 (20 μM) with HSA 5 (20 μM) for 48 h. (c) Incubation of Fab–PD conjugate 17 (20 μM) at pH 6.5 for 7 days. | ||
In this work, we have for the first time explored the use of the pyridazinedione motif in the context of dynamic reversible cysteine modification. When comparing this motif with other Michael acceptors used in the field (e.g., highly thiol-reactive maleimides and cyanoacrylates) we observe slow release of PD over time, the absence of any competing hydrolysis and a lack of reactivity between PDs and blood thiols (particularly with blood thiol serum albumin). The PD scaffold that we report on could have a unique place in the extracellular cleavable linker space by providing an opportunity for consistent slow release of cargo, which can then diffuse into cells and undergo intracellular specific cleavage, as a novel alternative to triggered cleavable linkers applied in the same context. Through rational bioconjugate design, a targeting protein, with a suitable blood half-life, modified with PD linkers could provide a slow release of payload without concern over cleavage in off-target areas. We also believe the time-dependent degradation of pyridazinedione–thiol motif could have a variety of ex vivo applications wherein maleimide–thiol conjugates are used as degradable systems (e.g., synthesis of hydrogels).35
We gratefully acknowledge the EPSRC (CASE Award with LifeArc, 173621) for funding C. B., and the Leverhulme Trust (RPG-2017-288, 176274) for funding R. J. S. A. M. would like to acknowledge the Ramsay Memorial trust for provision of a Ramsay Fellowship. F. J. and P. S. would like to thank the Wellcome Trust for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 1–14 Search PubMed.
- S. Sechi and B. T. Chait, Anal. Chem., 1998, 70, 5150–5158 CrossRef CAS PubMed.
- Y. Kim, S. O. Ho, N. R. Gassman, Y. Korlann, E. V. Landorf, F. R. Collart and S. Weiss, Bioconjugate Chem., 2008, 19, 786–791 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaia, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- A. G. Polson, et al. , Blood, 2007, 110, 616–623 CrossRef CAS PubMed.
- S. M. Ansell, Blood, 2014, 124, 3197–3200 CrossRef CAS PubMed.
- J. D. Bargh, A. Isidro-Llobet, J. S. Parker and D. R. Spring, Chem. Soc. Rev., 2019, 48, 4361–4374 RSC.
- R. Gébleux, M. Stringhini, R. Casanova, A. Soltermann and D. Neri, Int. J. Cancer, 2017, 140, 1670–1679 CrossRef PubMed.
- G. J. L. Bernardes, G. Casi, S. Trüssel, I. Hartmann, K. Schwager, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2012, 51, 941–944 CrossRef CAS PubMed.
- A. Dal Corso, R. Gébleux, P. Murer, A. Soltermann and D. Neri, J. Controlled Release, 2017, 264, 211–218 CrossRef CAS.
- A. Dal Corso, S. Cazzamalli, R. Gébleux, M. Mattarella and D. Neri, Bioconjugate Chem., 2017, 28, 1826–1833 CrossRef CAS PubMed.
- T. H. Pillow and J. A. Flygare, et al. , Chem. Sci., 2017, 8, 366–370 RSC.
- M. Danial and A. Postma, Ther. Delivery, 2017, 8, 359–362 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- H. Wu, P. J. Levalley, T. Luo, A. M. Kloxin and K. L. Kiick, Bioconjugate Chem., 2018, 29, 3595–3605 CrossRef CAS PubMed.
- B. Q. Shen, et al. , Nat. Biotechnol., 2012, 30, 184–189 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645–6655 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS PubMed.
- (a) V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781 RSC; (b) A. Maruani, S. Alom, P. Canavelli, M. T. W. Lee, R. E. Morgan, V. Chudasama and S. Caddick, Chem. Commun., 2015, 51, 5279–5282 RSC.
- I. M. Serafimova, M. A. Pufall, S. Krishnan, K. Duda, M. S. Cohen, R. L. Maglathlin, J. M. McFarland, R. M. Miller, M. Frödin and J. Taunton, Nat. Chem. Biol., 2012, 8, 471–476 CrossRef CAS PubMed.
- M. F. Schlecht, Molecular Modeling on the PC, Wiley-VCH, New York, 1998 Search PubMed.
- K. E. Gilbert, PCMODEL, version 8.5, SerenaSoftware, Bloomington Search PubMed.
- C. A. G. Haasnoot, F. A. A. M. de Leeuw and C. Altona, Tetrahedron, 1980, 36, 2783–2792 CrossRef CAS.
- P. Moody, M. E. B. Smith, C. P. Ryan, V. Chudasama, J. R. Baker, J. Molloy and S. Caddick, ChemBioChem, 2012, 13, 39–41 CrossRef CAS PubMed.
- S. Matsui and H. Aida, J. Chem. Soc., Perkin Trans. 2, 1978, 1277 RSC.
- J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef CAS PubMed.
- R. Gébleux, S. Wulhfard, G. Casi and D. Neri, Mol. Cancer Ther., 2015, 14, 2606–2612 CrossRef PubMed.
- G. J. L. Bernardes, G. Casi, S. Trüssel, I. Hartmann, K. Schwager, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2012, 51, 941–944 CrossRef CAS PubMed.
- G. Hao, Z. P. Xu and L. Li, RSC Adv., 2018, 8, 22182–22192 RSC.
- A. D. Baldwin and K. L. Kiick, Polym. Chem., 2013, 4, 133–143 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: [DETAILS]. See DOI: 10.1039/c9cc08362f |
| ‡ These authors contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2019 |