
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantifying the binding between proteins and open chromatin-like DNA sequences with gold nanorods†
Roger M.
Pallares
 abc,
Nguyen Thi Kim
Thanh
*ab and
Xiaodi
Su
*cde
abc,
Nguyen Thi Kim
Thanh
*ab and
Xiaodi
Su
*cde
aBiophysics Group, Department of Physics and Astronomy, University College London, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk
bUCL Healthcare Biomagnetic and Nanomaterials Laboratories, 21 Albemarle Street, London W1S 4BS, UK
cInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), Innovis, #8-03, 2 Fusionopolis Way, 138634, Singapore. E-mail: xd-su@imre.a-star.edu.sg
dDepartment of Chemistry, National University of Singapore, Block S8, Level 3, 3 Science Drive 3, 117543, Singapore
eSchool of Science and Engineering, University of the Sunshine Coast, Locked Bag 4, Maroochydore DC, Queensland 4558, Australia
First published on 21st October 2019
Abstract
The binding of transcription factors to DNA is one of the main mechanisms in gene regulation. While transcription factors frequently bind to unwrapped long DNA sequences known as open chromatin structures, most bioassays that study protein–DNA binding rely on short oligonucleotide probes. In this work, we develop a gold nanorod-based colorimetric assay for the binding of transcription factors to DNA in long open chromatin-like structures. After the determination of the binding affinity and stoichiometry, we explored the effect of the probe length on the assay performance and compared it to other established techniques.
Transcription factors are proteins that regulate gene expression through specific binding to DNA.1 Because these proteins participate in a wide variety of cellular processes,2 their malfunction has been associated with different diseases.1 Hence, the interaction between transcription factors and DNA is being used to understand the mechanism of cancer and other pathologies, as well as in the screening of new therapeutic agents.
One of the mechanisms by which the body controls the transcription factor activity is the recombination of the chromatin architecture.3 DNA chains inside chromosomes are wrapped around histone proteins, forming nucleosomes.4 This packaging allows the DNA materials to be stored in a more compact way and prevents oligonucleotide entangling. The DNA sequences coiled around the histones are less accessible to external molecules, hindering the binding of transcription factors.5 Under different chemical stimuli, the architecture of the chromatin changes to more open structures that expose the DNA recognition sequences.5–7 These open structures can be as long as 100 base pairs (bp) and favor the binding of the transcription factors and their control over gene expression.6 Although the interactions between transcription factors and these long DNA strands are very important in many regulatory processes, most research studies focus solely on short DNA sequences. Surface plasmon resonance (SPR) and electrophoretic mobility shift assay (EMSA) are the main techniques used to study protein–DNA binding.8–10 SPR provides a low intensity response when using long DNA probes, and the non-specific interaction between the analyte and substrate also hampers the measurements.11 EMSA can be used to study protein–DNA interactions in longer oligonucleotide sequences, but the electrophoresis can disrupt the complex integrity.12 Several alternatives based on fluorophores and nanomaterials have been developed,13–15 but they were also limited to short DNA strands. A sensing principle capable of characterizing DNA–protein binding in long oligonucleotide sequences would be highly beneficial to understand the activity of transcription factors on open chromatin structures.
Colorimetric assays based on gold nanoparticles have been developed as straightforward and rapid sensing alternatives for protein–DNA binding studies.16–18 These assays exploit the unique physical and chemical properties of gold nanoparticles, including the size and shape-dependent optical responses,19–23 and the ease to biofunctionalize.24–26 We recently developed an assay that relies on the binding of transcription factors to short double-stranded DNA (dsDNA) sequences (35 bp) adsorbed on gold nanorods (AuNRs).16 The binding of the proteins induced AuNR aggregation, which could be tracked by UV-Vis spectroscopy and provided information of the binding events, such as stoichiometry and binding affinity. It was unclear, however, whether increasing the length of the DNA probe (≥100 bp) to mimic the open chromatin architecture would hamper the assay sensitivity as it occurs in other common analytical tools, such as SPR.11
Here we address the above concerns by studying protein–DNA binding in long chromatin-like DNA sequences with the AuNR colorimetric assay. We found that the DNA binding affinity for the transcription factors was preserved even when long oligonucleotides were electrostatically adsorbed on the AuNRs. We show that using longer DNA probes increased the standard deviation of the measurements but the assay accuracy was preserved. The binding characteristics, such as the binding affinity and stoichiometry, determined by our assay were similar to the ones estimated by more complex and time-consuming techniques under similar experimental conditions. All these results indicate that the AuNR-based colorimetric assay is a good alternative to the slower and costlier standard techniques for the characterization of protein–DNA binding events.
Fig. 1 illustrates the design of the colorimetric assay with long open chromatin-like dsDNA chains. In a previous work, we identified that the binding sequences of dsDNA electrostatically adsorbed on AuNRs remain active,16 most likely because the interactions between the oligonucleotides and particles are not rigid, allowing the DNA to rearrange on the rod surface. In this current study, the dsDNA used was 180 bp long (similar to the length of the dsDNA in the nucleosome4) and contained a binding sequence for estrogen receptors (Table S1, ESI†). The estrogen receptor α (ERα) is a clinically relevant oncogenic transcription factor involved in the regulation of multiple biological functions, including the reproductive system and bone formation.27 The AuNRs (synthesized following our published protocol22) had an aspect ratio of 4.9 ± 0.8 (length of 51 ± 10 nm, and width of 10 ± 1 nm) and a longitudinal localized surface plasmon (LSP) resonance band centered at 890 nm (Fig. 2). The length of the as-prepared AuNRs was comparable to the 180 bp dsDNA probes (59 nm) in order to maximize their interaction. The optical properties of the AuNRs showed two different trends when interacting with dsDNA. At concentrations below 1 nM dsDNA, increasing the amount of oligonucleotides induced a red shift of the LSP band and decreased its intensity (Fig. 3a). Increasing the concentration of dsDNA above 1 nM, however, induced a blue shift of the LSP band (Fig. 3b). Because AuNRs are positively charged, the addition of small amounts of negative dsDNA caused the rod aggregation by charge neutralization.28,29 As the concentration of DNA increased (above 1 nM), the dsDNA started to adsorb on the AuNR surface, making the nanorods negatively charged, and thus providing electrostatic repulsion and re-dispersing them.28,29 The rods fully re-dispersed at 5 nM dsDNA. The two different trends were clearly distinguishable when plotting the extinction ratios at 510 and 890 nm (Fig. 3c).
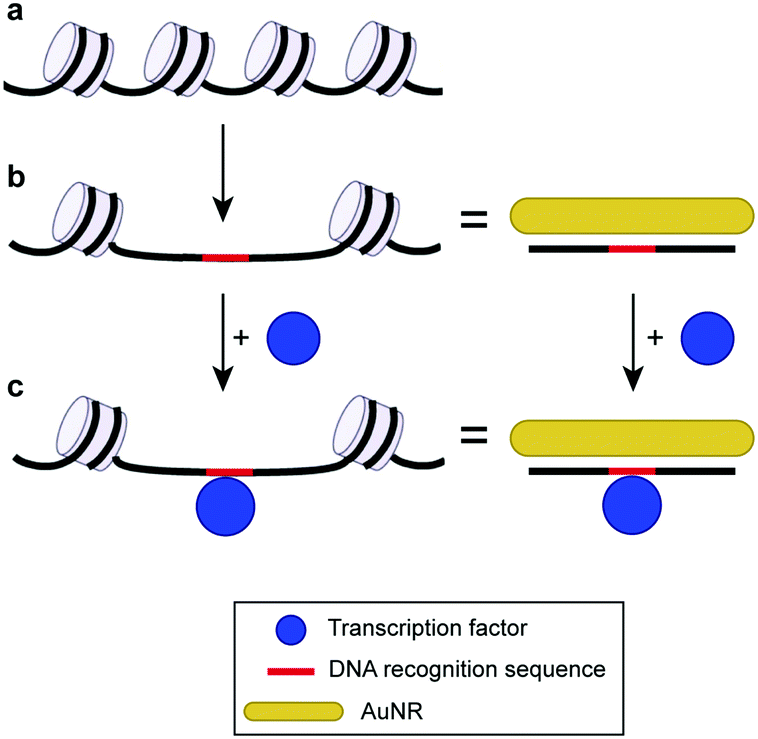 | ||
| Fig. 1 Scheme of the AuNR-based colorimetric assay with a long chromatin-like DNA probe. (a) Chromatin closed structure. (b) Open chromatin architecture and DNA with similar lengths adsorbed on AuNRs. (c) Similarity between DNA in an open chromatin architecture interacting with the transcription factor and AuNR–DNA probe interacting with the same protein. The chromatin scheme has been adapted with permission from ref. 7. Copyright 2013 The Royal Society. | ||
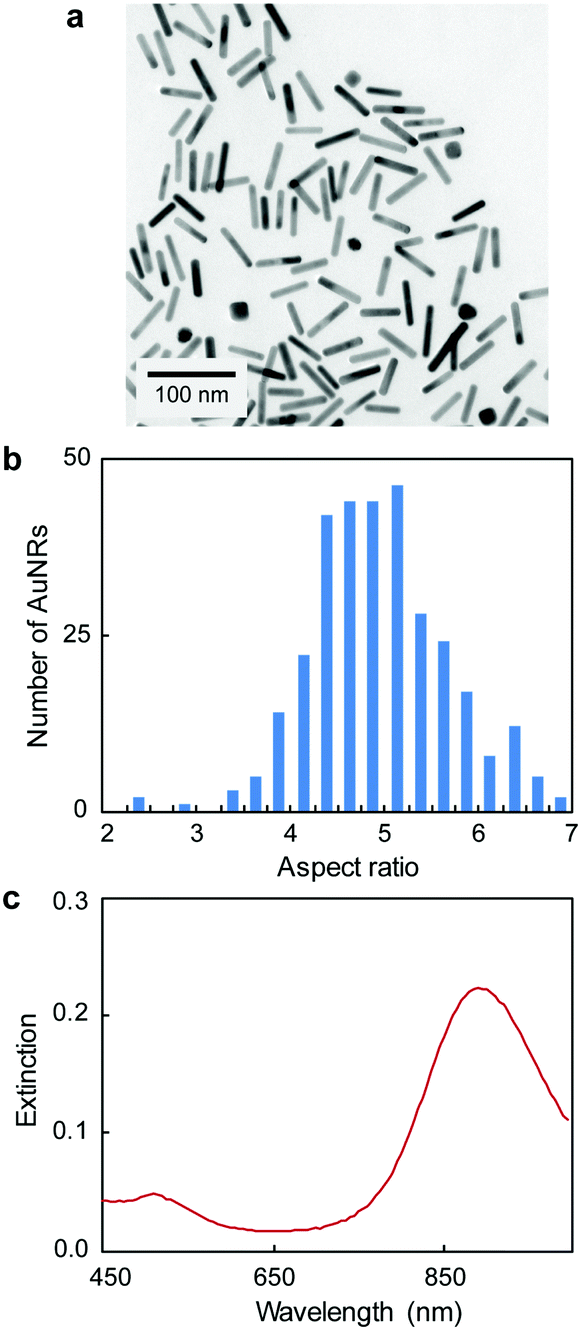 | ||
| Fig. 2 (a) TEM, (b) aspect ratio distribution and (c) UV-Vis spectrum of AuNRs (length of 51 ± 10 nm, and width of 10 ± 1 nm). | ||
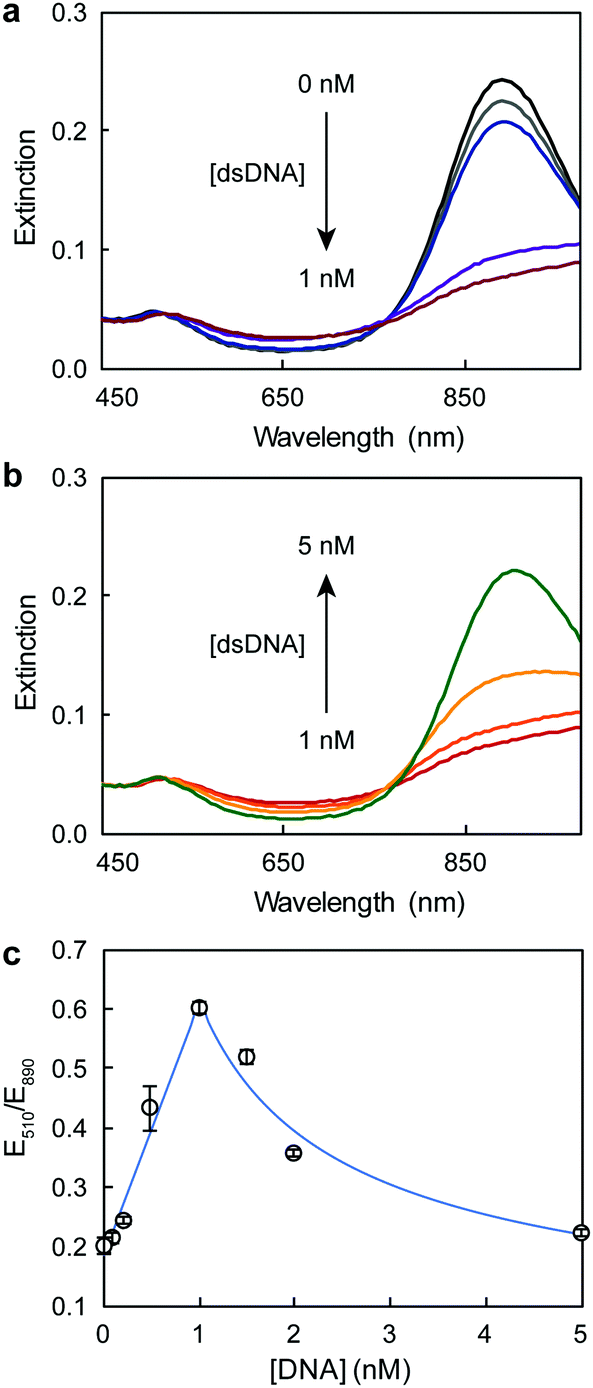 | ||
| Fig. 3 UV-Vis spectra of AuNRs after the addition of (a) low and (b) high dsDNA concentrations. (c) Response curve of AuNRs at different dsDNA concentrations. | ||
To study the protein–DNA binding, we chose 5 nM dsDNA because the AuNRs were dispersed at this oligonucleotide concentration. The colorimetric assay relied on the rapid (within 10 min) aggregation of the dsDNA–AuNR composite upon protein binding (Fig. 4a). Because the dsDNA–AuNRs were negatively charged,28 the binding of positive transcription factors decreased the repulsion between particles, causing their aggregation. This behavior is consistent with previous work using DNA-coated spherical gold nanoparticles and conjugated polymers.15 Tris buffer (8 mM) was used to fix the assay solution pH at 7.3, ensuring the positive charge of ERα (isoelectric point of 8.330). As the concentration of the protein increased in the solution, the LSP band decreased in intensity and slightly red-shifted (Fig. 4b). These changes of the LSP band were consistent with those previously reported for DNA- and protein-induced AuNR aggregation.16,28 The binding curve between the ERα and the 180 bp dsDNA was plotted as the variation of extinction ratios at 510 and 890 nm (Fig. 4c). The curve indicated that ERα binds to the dsDNA as a dimer, because E510/E890 reached saturation at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ERα:DNA concentration ratio, which was in agreement with previous published studies.13,15 The binding stoichiometry was confirmed by fitting the data to the Hill equation (Fig. S1, ESI†), which yielded a Hill coefficient (nH) of 2.3. Thus, the 2:1 binding stoichiometry and the negligible affinity of ERα for DNA controls,31,32 which have the binding site scrambled, indicate that the protein interacts specifically with the oligonucleotide, inducing AuNR aggregation. An apparent dissociation constant (Kd) of 3.0 nM was calculated with the Hill equation. This Kd value was consistent with previous studies that characterized the ERα–DNA binding under similar conditions (pH, buffer type and DNA concentration), however with more complex and/or instrument intensive techniques (Table S3, ESI†).33,34
:1 ERα:DNA concentration ratio, which was in agreement with previous published studies.13,15 The binding stoichiometry was confirmed by fitting the data to the Hill equation (Fig. S1, ESI†), which yielded a Hill coefficient (nH) of 2.3. Thus, the 2:1 binding stoichiometry and the negligible affinity of ERα for DNA controls,31,32 which have the binding site scrambled, indicate that the protein interacts specifically with the oligonucleotide, inducing AuNR aggregation. An apparent dissociation constant (Kd) of 3.0 nM was calculated with the Hill equation. This Kd value was consistent with previous studies that characterized the ERα–DNA binding under similar conditions (pH, buffer type and DNA concentration), however with more complex and/or instrument intensive techniques (Table S3, ESI†).33,34
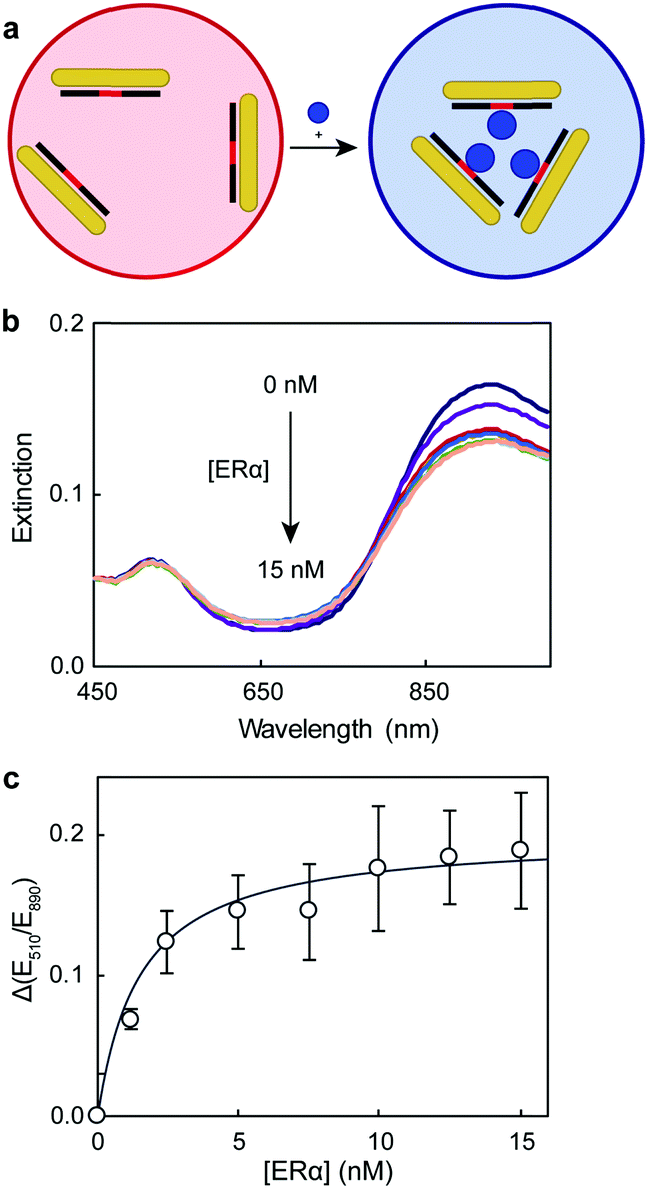 | ||
| Fig. 4 (a) Schematic representation of the AuNR aggregation principle, (b) UV-Vis spectra of dsDNA–AuNR at different ERα concentrations, and (c) the dependence of the extinction ratios at 510 and 890 nm on ERα concentration. | ||
Finally, we studied the effect of dsDNA length on the performance of the assay. Fig. 5 compares the binding curves of 59 bp and 180 bp dsDNA (plotted as the variation of extinction ratios at 510 nm and LSP maximum as a function of protein concentration). We chose 59 bp dsDNA because it is a common oligonucleotide probe length used in nano- and biosensing.29,35,36 Since the 59 bp dsDNA probe was shorter, we also used shorter aspect ratio rods (3.3 ± 0.6) with the LSP band centered at 805 nm (TEM image in Fig. S2, ESI†); and a higher concentration of dsDNA was also necessary to re-disperse the AuNRs (Fig. S3, ESI†). The shape of both ERα binding curves as well as the binding stoichiometry determined with the hill equation (nH of 2.2 and 2.3 for 59 bp and 180 bp dsDNA, respectively) were consistent for the shorter and longer DNA probes. Increasing the probe length, however, caused larger standard deviations of the data points (average standard deviations of 7.8% and 19.2% for 59 bp and 180 bp dsDNA probes, respectively). Hence, although using 180 bp dsDNA decreased the assay precision (higher standard deviations), the assay accuracy was retained, with the estimated Kd and stoichiometry values consistent with those obtained by more complicated techniques, such as radiometric assays, performed under comparable conditions.33,34
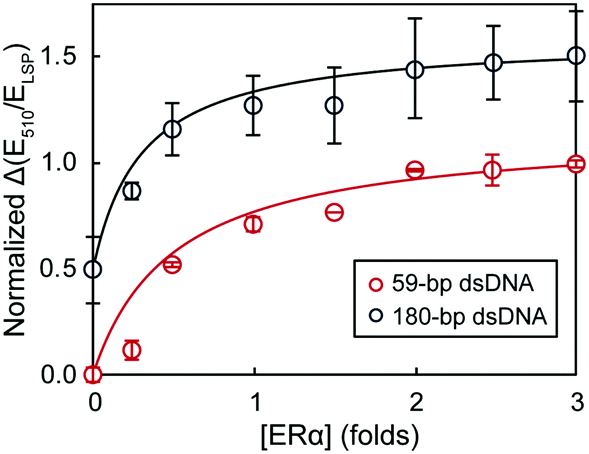 | ||
| Fig. 5 The dependence of extinction ratios at 510 nm and LSP maximum on ERα concentration using different lengths of dsDNA. The binding curves have been offset for clarity. | ||
In summary, we have characterized the protein–DNA binding in open chromatin-like sequences with an AuNR-based colorimetric assay. 180 bp long dsDNA (length of dsDNA in nucleosomes) that displayed a binding sequence for ERα was used as a probe. Although the oligonucleotides were electrostatically adsorbed on the AuNR surface, they preserved their affinity for the proteins. Upon protein binding, the AuNRs aggregated causing a decrease in their LSP band intensity that was proportional to the amount of bound ERα. The binding characteristics, including stoichiometry and Kd, were estimated from the binding curve data. Although using long dsDNA probes caused larger standard deviations, the assay accuracy was not significantly affected. To the best of our knowledge, this is the first colorimetric assay capable of studying protein–DNA interactions with long open chromatin-like DNA sequences. Because this assay is rapid and straight forward, we expect that new opportunities will follow this work in the high throughput screening of new therapeutic agents.
RMP thanks UCL and A*STAR for his PhD fellowship. NTKT thanks EPSRC funding EP/M018016/1 and EP/M015157/1. XS thanks A*STAR JCO funding 14302FG096.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. S. Latchman, Int. J. Biochem. Cell Biol., 1997, 29, 1305–1312 CrossRef CAS PubMed.
- A. Nepveu, Gene, 2001, 270, 1–15 CrossRef CAS PubMed.
- N. Hah, S. Murakami, A. Nagari, C. G. Danko and W. L. Kraus, Genome Res., 2013, 23, 1210–1223 CrossRef CAS PubMed.
- P. N. Cockerill, FEBS J., 2011, 278, 2182–2210 CrossRef CAS PubMed.
- B. R. Cairns, Nature, 2009, 461, 193–198 CrossRef CAS PubMed.
- L. Manelyte, R. Strohner, T. Gross and G. Längst, PLoS Genet., 2014, 10, e1004157 CrossRef PubMed.
- S. Ghisletti and G. Natoli, Philos. Trans. R. Soc., B, 2013, 368, 20120370 CrossRef PubMed.
- C. Song, S. Zhang and H. Huang, Front. Microbiol., 2015, 6, 1049 Search PubMed.
- X. Su, C.-Y. Lin, S. J. O'Shea, H. F. Teh, W. Y. X. Peh and J. S. Thomsen, Anal. Chem., 2006, 78, 5552–5558 CrossRef CAS PubMed.
- H. F. Teh, W. Y. X. Peh, X. Su and J. S. Thomsen, Biochemistry, 2007, 46, 2127–2135 CrossRef CAS PubMed.
- J. Homola, Anal. Bioanal. Chem., 2003, 377, 528–539 CrossRef CAS PubMed.
- P. L. Molloy, Nat. Methods, 2000, 130, 235–246 CAS.
- R. M. Pallares, L. Sutarlie, N. T. K. Thanh and X. Su, Sens. Actuators, B, 2018, 271, 97–103 CrossRef CAS.
- K. M. M. Aung, S. Y. New, S. Hong, L. Sutarlie, M. G. L. Lim, S. K. Tan, E. Cheung and X. Su, Anal. Biochem., 2014, 448, 95–104 CrossRef CAS PubMed.
- S. Lukman, K. M. M. Aung, J. Liu, B. Liu and X. Su, ACS Appl. Mater. Interfaces, 2013, 5, 12725–12734 CrossRef CAS PubMed.
- R. M. Pallares, M. Bosman, N. T. K. Thanh and X. Su, Nanoscale, 2016, 8, 19973–19977 RSC.
- Y. N. Tan, X. Su, Y. Zhu and J. Y. Lee, ACS Nano, 2010, 4, 5101–5110 CrossRef CAS PubMed.
- Y. N. Tan, X. Su, E. T. Liu and J. S. Thomsen, Anal. Chem., 2010, 82, 2759–2765 CrossRef CAS PubMed.
- R. M. Pallares, T. Stilson, P. Choo, J. Hu and T. W. Odom, ACS Appl. Nano Mater., 2019, 2, 5266–5271 CrossRef.
- R. M. Pallares, Y. Wang, S. H. Lim, N. T. K. Thanh and X. Su, Nanomedicine, 2016, 11, 2845–2860 CAS.
- S. Link and M. a. El-Sayed, Int. Rev. Phys. Chem., 2000, 19, 409–453 Search PubMed.
- R. M. Pallares, X. Su, S. H. Lim and N. T. K. Thanh, J. Mater. Chem. C, 2016, 4, 53–61 RSC.
- R. M. Pallares, N. T. K. Thanh and X. Su, Nanoscale, 2019 10.1039/C9NR03040A.
- S. Zeng, K.-T. Yong, I. Roy, X.-Q. Dinh, X. Yu and F. Luan, Plasmonics, 2011, 6, 491 CrossRef CAS.
- J. Yue, R. M. Pallares, L. E. Cole, E. E. Coughlin, C. A. Mirkin, A. Lee and T. W. Odom, ACS Appl. Mater. Interfaces, 2018, 10, 21920–21926 CrossRef CAS PubMed.
- R. M. Pallares, P. Choo, L. E. Cole, C. A. Mirkin, A. Lee and T. W. Odom, Bioconjugate Chem., 2019, 30, 2032–2037 CrossRef CAS PubMed.
- J. R. Schultz, L. N. Petz and A. M. Nardulli, Mol. Cell. Endocrinol., 2003, 201, 165–175 CrossRef CAS PubMed.
- R. M. Pallares, S. L. Kong, T. H. Ru, N. T. K. Thanh, Y. Lu and X. Su, Chem. Commun., 2015, 51, 14524–14527 RSC.
- R. M. Pallares, N. T. K. Thanh and X. Su, Chem. Commun., 2018, 54, 11260–11263 RSC.
- S. Yamashita and Y. Okada, J. Histochem. Cytochem., 2005, 53, 13–21 CrossRef CAS PubMed.
- S. J. Neo, X. Su and J. S. Thomsen, Anal. Chem., 2009, 81, 3344–3349 CrossRef CAS PubMed.
- W. Y. X. Peh, E. Reimhult, H. F. Teh, J. S. Thomsen and X. Su, Biophys. J., 2007, 92, 4415–4423 CrossRef CAS PubMed.
- S. Aliau, T. Groblewski and J.-L. Borgna, Eur. J. Biochem., 1995, 231, 204–213 CrossRef CAS PubMed.
- M. Boyer, N. Poujol, E. Margeat and C. A. Royer, Nucleic Acids Res., 2000, 28, 2494–2502 CrossRef CAS PubMed.
- L. Franceschini, M. Soskine, A. Biesemans and G. Maglia, Nat. Commun., 2013, 4, 2415 CrossRef PubMed.
- L. Sun, K. Frykholm, L. H. Fornander, S. Svedhem, F. Westerlund and B. Åkerman, J. Phys. Chem. B, 2014, 118, 11895–11904 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07511a |
| This journal is © The Royal Society of Chemistry 2019 |