
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective and reversible interconversion of nanosliders commanded by remote control via metal-ion signaling†
Suchismita
Saha
,
Pronay Kumar
Biswas
,
Indrajit
Paul
and
Michael
Schmittel
 *
*
Center of Micro and Nanochemistry and Engineering, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, D-57068 Siegen, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
First published on 21st November 2019
Abstract
A multi-device network mainly consisting of two two-component nanosliders was formed by self-sorting of six components. Addition/removal of zinc(II) ions reversibly reorganized the network by chemical signaling involving the translocation of copper(I) from a relay station followed by the selective disassembly/assembly of one of both multi-component devices. The thus liberated machine parts served to erect a three-component nanoslider alongside the other unchanged two-component nanoslider.
Whereas life vitally depends on biocybernetic regulation,1,2 the networking of artificial molecular devices is still in its infancy.3–9 It is a great promise of interdependent multi-component ensembles to pave the way toward autonomous and self-regulating systems, i.e., for realizing properties that exceed by far what individual molecules can achieve.
In order to network stand-alone molecular10–12 or supramolecular13–20 devices, the functional entities need to be linked by fast chemical communication without harmful interference. All receptors and emitters involved in communication thus need to be highly dynamic and selective with regard to signal uptake, information processing and signal release (= output). The first examples of networked ensembles allowing ON/OFF switchable catalysis have surfaced recently.21–24 In their key step, a nanoswitch transmitted a defined chemical signal to a functional device (e.g., nanoswitch, nanorotor, or fluorescent receptor) in response to a trigger input. Herein, we demonstrate how a remote control commands the transformation of one nanodevice into another one using the sequential translocation of more than one chemical signal.
In detail, we describe six- and seven-component networks that allow the reversible parallel interconversion of nanosliders. Upon addition of three equiv. of Zn2+ to 3 × [Cu(S)]+ (Fig. 1a and b) the equivalent amount of Cu+, initially deeply buried in nanoswitch [Cu(S)]+, will be translocated from S to the free phenanthroline sites of deck D2. In essence, the trigger signal commands a remote control unit to emit a chemical signal as second messenger. The ensuing complex [Cu3(D2)]3+ now instructs the nanoslider M1 = D1·A1 to disassemble and to transfer its biped A1 for enabling the assembly of the device M2 = [Cu3(D2)(A1)]3+. In other words, a single input (Zn2+) is sufficient for the parallel dismantling and production of nanodevices through successive two-component translocation. To underline the potential of the concept, the network was furthermore extended to a seven-component system, in which the second translocation had to occur selectively with one out of two bipeds (Fig. 1c).
 | ||
| Fig. 1 (a) Chemical structures of switch S, decks D1 and D2 and bipeds A1 and A2 along with their cartoon representations and those of ligands 1, 2 and 3. (b) Cartoon representation of the reversible interconversion of nanosliders in NetState-I and NetState-II by addition and removal of Zn2+. (c) Cartoon representation of the selective and reversible interconversion of nanosliders in NetState-III and NetState-IV by addition and removal of Zn2+. | ||
To probe the required self-sorting, a small set of model studies with various substituted pyridines was undertaken. It turned out that picoline 3 was the optimal terminus in the biped, as it cleanly formed complex 1·3 at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio of zinc porphyrin 1, phenanthroline 2 and picoline 3. In contrast, 3 did bind predominantly to 2 in the form of [Cu(2)(3)]+ through HETPYP-I (HETeroleptic PYridine and Phenanthroline)25,26 complexation when one equiv. of Cu+ was present (Fig. S9, ESI†).27 The bulky aryl groups at the 2,9-position of phenanthroline 2 are needed to prevent homoleptic metal complexation.28,29 The methyl substitution in 2-picoline 3 caused weak binding at the zinc(II) porphyrin (logK = 2.72)30 whereas the Npic → [Cu(2)]+ complexation was strengthened (logK = 3.43).27
:1:1 ratio of zinc porphyrin 1, phenanthroline 2 and picoline 3. In contrast, 3 did bind predominantly to 2 in the form of [Cu(2)(3)]+ through HETPYP-I (HETeroleptic PYridine and Phenanthroline)25,26 complexation when one equiv. of Cu+ was present (Fig. S9, ESI†).27 The bulky aryl groups at the 2,9-position of phenanthroline 2 are needed to prevent homoleptic metal complexation.28,29 The methyl substitution in 2-picoline 3 caused weak binding at the zinc(II) porphyrin (logK = 2.72)30 whereas the Npic → [Cu(2)]+ complexation was strengthened (logK = 3.43).27
Thus, the tris(zinc porphyrin) deck D1, tris-phenanthroline deck D2 and the picoline-terminated biped A1 were selected (Fig. 1a). Due to the anticipated stronger binding of biped A1 to the Cu+-loaded phenanthroline deck D2 than to porphyrin deck D1, it was expected that A1 would prefer binding to [Cu3(D2)]3+. D2 was synthesized by Sonogashira coupling of 2,9-diaryl-3-ethynyl-1,10-phenanthroline and 1,3,5-tris(4-iodophenyl)benzene (ESI†).
The 1:1 mixture of D1 and A1 in CD2Cl2 is known to quantitatively afford the two-component nanoslider M1 = D1·A1 where the picoline feet of the biped are axially bound to the zinc(II) porphyrins of the deck (Fig. 1b).31 As described earlier, due to axial binding, protons t-H and β-H of the tris-porphyrin deck and protons a-H, b-H and c-H of the biped arm A1 diagnostically move upfield.30 The exchange of the biped between the three zinc porphyrin sites occurs at an exchange frequency of k298 = 440 kHz.31
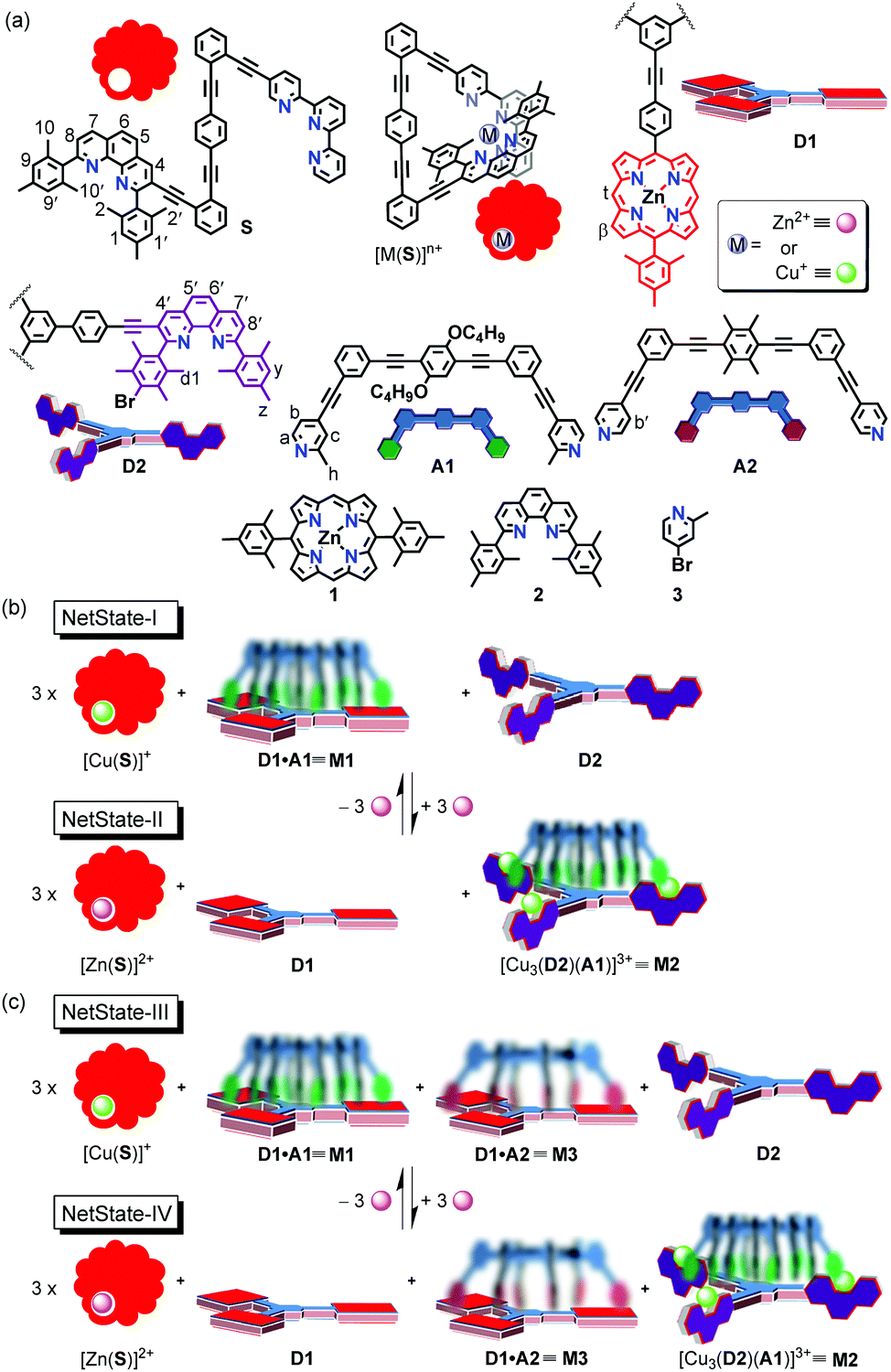
Formation of the slider-on-deck M2 simply required mixing of D2, A1 and [Cu(CH3CN)4]PF6 (1:1:3) in CD2Cl2, as confirmed by the upfield shifts of protons a-H, b-H, and c-H of A1 from 8.48, 7.21, and 7.28 ppm to 7.54, 7.11, and 7.17 ppm, respectively (Fig. 2a). Upfield shifts of protons y-H, d1-H, and z-H from 7.02, 2.53, and 2.36 ppm in the case of [Cu3(D2)]3+ to 6.87, 2.37, and 2.23 ppm in M2, respectively, corroborated the biped binding at the metal phenanthroline stations (Fig. 2a and Fig. S19, ESI†). Due to the HETPYP-I binding, these protons were placed in the shielding zone of the π-ring current.27 The occurrence of a single set of all phenanthroline protons suggested fast exchange of A1 between all three phenanthroline stations of [Cu3(D2)]3+. To determine the exchange frequency, VT-1H NMR of M2 was performed which exhibited splitting and a 2:1 ratio of various phenanthroline protons (4′-H, 5′-H, 6′-H, 7′-H) at lower temperature (−50 °C). The more downfield signals were assigned to the HETPYP-I complexed phenanthroline sites and the upfield ones to the Cu+-loaded phenanthroline station (Fig. S30, ESI†). To calculate the exchange frequency of M2, the splitting of proton 4′-H was analyzed which at 25 °C appeared as a sharp singlet at 8.86 ppm but split at −50 °C into two sets in a 2:1 ratio with a strong coalescence around −40 °C (Fig. 2b). Exchange frequencies (k) at different temperatures were calculated using WinDNMR32 which provided k298 = 20 kHz. Activation parameters were derived from the Eyring plot as ΔH‡ = 61.5 kJ mol−1, ΔS‡ = 44.7 J mol−1 K−1 and ΔG‡298 = 48.2 kJ mol−1 (Table 1 and Fig. S31, ESI†). A single peak at m/z = 915.2 (tri-charged) in ESI-MS confirmed the identity of M2 (Fig. S36, ESI†), which was additionally supported by a single set of diffusion signals in the 1H–1H DOSY (Fig. S32, ESI†).
 | ||
| Fig. 2 (a) Partial 1H NMR (400 MHz, CD2Cl2, 298 K) of D2, A1, [Cu3(D2)]3+ and M2 = [Cu3(D2)(A1)]3+. (b) VT-1H NMR (600 MHz, CD2Cl2) of M2 showing the splitting of 4′-H in a 1:2 ratio. | ||
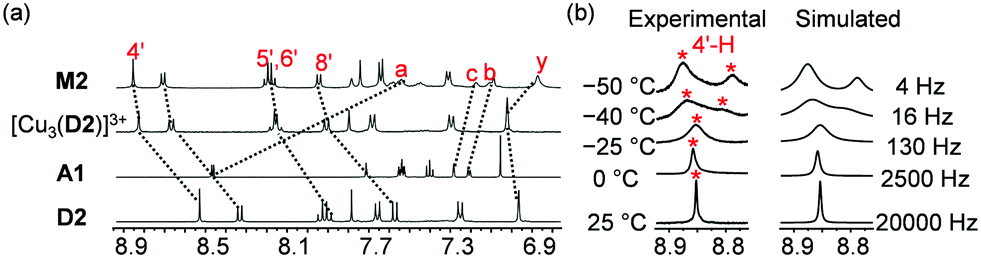
After successful formation of both nanosliders, our next intention was to control the reversible and alternate formation of M1 and M2 by remote control via Cu+ as a signal. For this purpose, the triangular nanoswitch S was selected as a relay because of its ability to capture Cu+ selectively in the presence of the free deck D2 and to rather rapidly release Cu+ onto D2 upon addition of Zn2+. Nanoswitch S, its Cu+ and Zn2+ complexes were unambiguously characterized by 1H NMR, 1H–1H COSY, ESI-MS and elemental analysis (ESI†). Upon complexation with metal ions, protons 1-H, 9-H, 2-H and 10-H split in a 1:1 ratio and moved upfield, with the shift being more manifest in [Zn(S)]2+ than in [Cu(S)]+ (Fig. 3 and Fig. S14, ESI†). In contrast, protons 4-H, 5-H, 6-H, 7-H and 8-H shifted downfield, again more pronouncedly in [Zn(S)]2+ than in [Cu(S)]+ (Fig. 3).
 | ||
| Fig. 3 Partial 1H NMR (400 MHz, CD2Cl2, 298 K) of S, [Cu(S)]+ and [Zn(S)]2+. | ||
Finally the self-sorting of NetState-I was tested. Nanoswitch S, the decks D1, D2, biped A1 and Cu+ were mixed (3:1:1:1:3) in CD2Cl2 to furnish quantitatively 3 × [Cu(S)]+, M1 and free D2 (Fig. 1b). Addition of 3.0 equiv. of Zn2+ to NetState-I afforded thrice complex [Zn(S)]2+ thus liberating 3.0 equiv. of Cu+ which translocated to the three phenanthroline sites of D2 with the effect that biped A1 shifted from M1 to [Cu3(D2)]3+ generating NetState-II. NetState-II was reversed back to NetState-I by addition of 3.0 equiv. of hexacyclen. Two complete cycles between NetState-I and NetState-II were performed to demonstrate the reversible nature of the networked system (Fig. S26, ESI†).
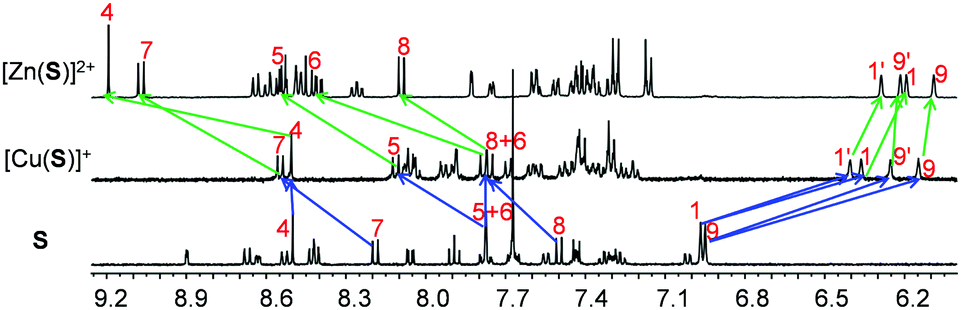
Quantitative formation and interconversion of NetStates-I & II was proven by 1H NMR through comparison with data of the individually prepared devices (Fig. 4a). The fabrication of NetState-II was further confirmed by ESI-MS (Fig. S38, ESI†). Protons 1-H, 1′-H, 9-H and 9′-H of S with their characteristic 1H NMR signals for [Cu(S)]+ and [Zn(S)]2+ allowed tracking of the metal exchange at nanoswitch S whereas protons t-H of D1 and y-H of D2 showed peak shifts being diagnostic for the formation of M1 and M2 (Fig. 4).
 | ||
| Fig. 4 (a) Partial 1H NMR (400 MHz, CD2Cl2) of [Cu(S)]+, [Zn(S)]2+, D1, D2, M1, M2, NetState-I and NetState-II. (b) UV-vis spectra showing conversion of NetState-I to NetState-II in CH2Cl2 (298 K, c = 10−6 M) over time. Inset: Changes in absorbance at 339 nm with time. | ||
The kinetics of the interconversion NetState-I ⇆ II was monitored by UV-vis spectroscopy. As there are negligible shifts in the Soret and Q bands of the zinc porphyrins in D1 during the detachment/attachment of the biped, changes were monitored at the HETPYP-I complexation site in D2, i.e., at the [Cu(phenAr2)(pic)]+ linkage. Upon changing from NetState-I → NetState-II, the absorptions at 339 and 357 nm disappeared with simultaneous increase of a peak at 386 nm due to formation of M2 (Fig. 4b). The peaks at 339 and 357 nm were assigned to transitions of the phenanthroline residues whereas the absorption at 386 nm was attributed to the MLCT band of the HETPYP-I complex. Reciprocally, in the NetState-II → I conversion, new peaks at 339 and 357 nm emerged along with the disappearance of the absorbance at 386 nm in agreement with breaking up the HETPYP-I complexation in M2 (Fig. S44, ESI†).
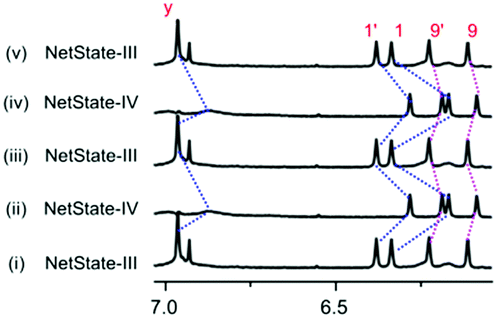 | ||
| Fig. 5 Partial 1H NMR (400 MHz, CD2Cl2, 298 K) shows the reversible interconversion of NetState-III and NetState-IV over two complete cycles. (i) After mixing S, D1, D2, A1, A2 and Cu+ in a 3:2:1:1:1:3 ratio (NetState-III). (ii) Addition of 3.0 equiv. of Zn2+ to (i). (iii) Addition of 3.0 equiv. of hexacyclen to (ii). (iv) Addition of 3.0 equiv. of Zn2+ to (iii). (v) Addition of 3.0 equiv. of hexacyclen to (iv). | ||
Absorbance changes at 339 nm were monitored for both the forward and backward process with time (Fig. 4b and Fig. S45, ESI†). Forward conversion (i.e., NetState-I → II, c = 10−6 M) took 60 min for completion whereas the backward process (i.e., NetState-II → I) was finished within 12 min. The slow forward process was accelerated by adding iodide as a nucleophile.33,34 For instance, addition of 3.0 equiv. of iodide sped up the forward transformation (conversion took 12 min) without affecting the rate of the backward process (Fig. S47 and S49, ESI†).
Since the rate determining step in the forward process is the replacement of Cu+ in [Cu(S)]+ by Zn2+, iodide is supposed to facilitate opening of the switch by coordination to the Cu+ ion. In the backward process, the rate determining step involves removal of zinc(II) from [Zn(S)]2+ by hexacyclen. Since hexacyclen is itself a good nucleophile and in addition the chelate ligand for Zn2+ the effect of added iodide is minor.
To demonstrate biped selectivity in the remote control of the interconverting nanodevices, the complexity was increased by adding the nanoslider M3 = D1·A2. This nanodevice (k298 = 32.2 kHz) has been described and fully characterized in a previous publication.31
In Netstate-III we thus assembled [Cu(S)]+, nanosliders M1, M3 and free deck D2 (Fig. 1c) by self-sorting of nanoswitch S, [Cu(CH3CN)4]PF6, deck D1, bipeds A1 and A2 and phenanthroline deck D2 (3:3:2:1:1:1). Analysis of the 1H NMR confirmed formation of M1 and M3 by the characteristic upfield shifts of protons c-H, h-H and b′-H at 6.16, −0.14 and 5.45 ppm, respectively (Fig. S27, ESI†). In addition, characteristic signals for 1-H, 1′-H, 9-H and 9′-H of [Cu(S)]+ and y-H of free D2 substantiated the quantitative formation of NetState-III. Addition of 3.0 equiv. of Zn(OTf)2 translocated the Cu+ ions from [Cu(S)]+ to the free phenanthroline sites of deck D2. The resultant [Cu3(D2)]3+ has now the option to claim either biped A1 or A2 for generating either M2 or M4 = [Cu3(D2)(A2)]3+. Selective translocation of A1 was proven by the upfield shift of protons y-H, z-H and d1-H at 6.87, 2.23 and 2.37 ppm, respectively, exactly matching with the signals for M2 (Fig. S28, ESI†). If A2 had been translocated fabricating M4 = [Cu3(D2)(A2)]3+, the above mentioned peaks should have shifted to 7.02, 2.39 and 2.57 ppm, respectively (Fig. S21, ESI†). Quantitative formation of M2 over M4 in NetState-IV was supported by the ESI-MS (Fig. S39, ESI†). Two complete cycles between NetState-III and NetState-IV proved the reversibility and selectivity of the seven-component networked system (Fig. 5).
In conclusion, we have demonstrated a six-component cybernetic network of a triangular switch controlling the self-assembly of two alternate nanosliders via chemical signaling.35 In this network, a single chemical input (Zn2+ or hexacyclen) controls two consecutive translocations (of Cu+ ion and sliding biped A1) which lead to the alternate assembly/disassembly of two nanosliders. Furthermore, the observed selectivity in the second translocation, i.e., that of biped A1, demonstrated a high level of control in the seven-component system. Two complete cycles of the assembly/disassembly of nanosliders proved the reversibility of the cybernetic process. The kinetics of translocation was controlled by addition of iodide.
In summary, this manifestation of remote control is a decisive step toward supramolecular systems chemistry integrating multiple stand-alone supramolecular devices into higher-order multicomponent nanomachinery. Using furthermore chemical fuel in such domino translocations should lead to interesting off-equilibrium36–42 applications.
We are indebted to the Deutsche Forschungsgemeinschaft for continued support under Schm 647/19-2 and 647/20-2. We thank Dr Paululat for measuring the VT-1H NMR. Dedicated to Prof. Dr J.-P. Sauvage on the occasion of his 75th birthday and with deep gratitude for his inspiring and ground-breaking work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Korzeniewski, J. Theor. Biol., 2001, 209, 275–286 CrossRef CAS PubMed.
- A. Bielecki, Biol. Cybern., 2015, 109, 401–419 CrossRef PubMed.
- M. Barboiu and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5201–5206 CrossRef CAS PubMed.
- M. Barboiu, G. Vaughan, N. Kyritsakas and J.-M. Lehn, Chem. – Eur. J., 2003, 9, 763–769 CrossRef CAS PubMed.
- D. Ray, J. T. Foy, R. P. Hughes and I. Aprahamian, Nat. Chem., 2012, 4, 757–762 CrossRef CAS PubMed.
- S. Pramanik, S. De and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 4709–4713 CrossRef CAS PubMed.
- S. Pramanik, S. De and M. Schmittel, Chem. Commun., 2014, 50, 13254–13257 RSC.
- A.-M. Stadler, L. Karmazin and C. Bailly, Angew. Chem., Int. Ed., 2015, 54, 14570–14574 CrossRef CAS PubMed.
- S. Pramanik and I. Aprahamian, J. Am. Chem. Soc., 2016, 138, 15142–15145 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS PubMed.
- S. Hiraoka, E. Okuno, T. Tanaka, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 9089–9098 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- M. Schmittel, Chem. Commun., 2015, 51, 14956–14968 RSC.
- Y. Takara, T. Kusamoto, T. Masui, M. Nishikawa, S. Kume and H. Nishihara, Chem. Commun., 2015, 51, 2896–2898 RSC.
- P. K. Biswas, S. Saha, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2018, 140, 9038–9041 CrossRef CAS PubMed.
- M. Schmittel and S. Saha, Adv. Inorg. Chem., 2018, 71, 135–176 CrossRef CAS.
- A. Goswami and M. Schmittel, Coord. Chem. Rev., 2018, 376, 478–505 CrossRef CAS.
- N. Mittal, S. Pramanik, I. Paul, S. De and M. Schmittel, J. Am. Chem. Soc., 2017, 139, 4270–4273 CrossRef CAS PubMed.
- A. Goswami, S. Pramanik and M. Schmittel, Chem. Commun., 2018, 54, 3955–3958 RSC.
- S. Gaikwad, S. Pramanik, S. De and M. Schmittel, Dalton Trans., 2018, 47, 1786–1790 RSC.
- I. Paul, N. Mittal and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 5139–5143 CrossRef CAS PubMed.
- S. Neogi, Y. Lorenz, M. Engeser, D. Samanta and M. Schmittel, Inorg. Chem., 2013, 52, 6975–6984 CrossRef CAS PubMed.
- S. Neogi, G. Schnakenburg, Y. Lorenz, M. Engeser and M. Schmittel, Inorg. Chem., 2012, 51, 10832–10841 CrossRef CAS PubMed.
- N. Mittal, M. S. Özer and M. Schmittel, Inorg. Chem., 2018, 57, 3579–3586 CrossRef CAS PubMed.
- M. Schmittel, C. Michel, S.-X. Liu, D. Schildbach and D. Fenske, Eur. J. Inorg. Chem., 2001, 1155–1166 CrossRef CAS.
- K. Mahata and M. Schmittel, J. Am. Chem. Soc., 2009, 131, 16544–16554 CrossRef CAS PubMed.
- A. Ghosh, I. Paul, S. Saha, T. Paululat and M. Schmittel, Org. Lett., 2018, 20, 7973–7976 CrossRef CAS PubMed.
- I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354–358 CrossRef CAS PubMed.
- H. J. Reich, J. Chem. Educ., Software, 1996, 3D, 2 Search PubMed.
- A. Livoreil, J.-P. Sauvage, N. Armaroli, V. Balzani, L. Flamigni and B. Ventura, J. Am. Chem. Soc., 1997, 119, 12114–12124 CrossRef CAS PubMed.
- S. Saha, P. K. Biswas and M. Schmittel, Inorg. Chem., 2019, 58, 3466–3472 CrossRef CAS PubMed.
- P. Remón and U. Pischel, ChemPhysChem, 2017, 18, 1667–1677 CrossRef PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- C. S. Wood, C. Browne, D. M. Wood and J. R. Nitschke, ACS Cent. Sci., 2015, 1, 504–509 CrossRef CAS PubMed.
- M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534, 235–240 CrossRef CAS PubMed.
- B. S. L. Collins, J. C. M. Kistemaker, E. Otten and B. L. Feringa, Nat. Chem., 2016, 8, 860–866 CrossRef CAS.
- J. A. Berrocal, C. Biagini, L. Mandolini and S. Di Stefano, Angew. Chem., Int. Ed., 2016, 55, 6997–7001 CrossRef CAS PubMed.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358, 340–343 CrossRef CAS PubMed.
- A. Ghosh, I. Paul, M. Adlung, C. Wickleder and M. Schmittel, Org. Lett., 2018, 20, 1046–1049 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterizations, spectral data, UV-vis titrations and computational data. See DOI: 10.1039/c9cc07415e |
| This journal is © The Royal Society of Chemistry 2019 |