
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemo-enzymatic three-step conversion of glucose to kojic acid†
Robert
Lassfolk
a,
Anu
Suonpää
b,
Klara
Birikh
b and
Reko
Leino
 *a
*a
aJohan Gadolin Process Chemistry Centre, Åbo Akademi University, FI-20500 Åbo, Finland. E-mail: reko.leino@abo.fi
bMetGen Oy, Rakentajantie 26, FI-20780 Kaarina, Finland
First published on 13th November 2019
Abstract
Kojic acid is an important biomolecule, currently produced by fermentation and having a wide range of potential applications. A faster and more direct chemical route could open the door for its large-scale production and wider utilization in biorefineries. Here we describe an efficient method for the preparation of kojic acid from D-glucose via glucosone by a three-step chemo-enzymatic route.
Kojic acid (1) (Fig. 1) is a small organic molecule with a range of potential applications, from pharmaceuticals to biopolymers and cosmetics. The compound itself possesses both antiparasitic1 and cytotoxic activity,2 while its derivatives have been investigated for their antimalarial,3 antimicrobial,4 antibacterial,5,6 antifungal7,8 and cytotoxic properties,9,10 acting also as a tyrosinase inhibitor.11,12 Kojic acid also functions as a radioprotective agent,13 antioxidant14,15 and as a radical scavenger.16 In cosmetics, the parent compound and its derivatives are used as whitening agents.6,17 In agriculture, kojic acid has been investigated as a pesticide18 and insecticide.19 In polymer applications, the biological activity of kojic acid has been utilized, for example, in antimicrobial materials,4 while biodegradable polymers have been designed to release the kojic acid monomer to inhibit melanogenesis.20 Besides for biological applications, kojic acid and its derivatives have been tested in dye sensitized solar cells,21 and, among others, in chelation, determination of the concentration and removal of metals, especially iron, from solution.22–24 A kojic acid containing styrene polymer has been evaluated for recovery of heavy metals.25 Industrially, kojic acid is currently obtained as a byproduct from malting rice fermentation in sake manufacturing processes in Japan. It is produced by fungi of the Aspergillus family,26–29 in particular by Aspergillus oryzae. Chemically, kojic acid has been prepared from tetraacetylgalactosone hydrate under acetylating conditions with pyridine and acetic anhydride to yield diacetylkojic acid, followed by deacetylation with ammonia in methanol.30 Preparation of acylated kojic acids has been reported previously also, for example, by oxidation of C2 in tetra-1,3,4,6-O-acetyl-D-galactose and the corresponding glucose analogue;31 dibenzoylkojic acid has been prepared from the glucosone derivative 1-chloro-3,5-dibenzoyl-glucoson-4,5-ene by boiling the starting material in acetic acid and sodium acetate.32 For broader use and supply, the development of efficient chemical synthesis of kojic acid would be desirable.
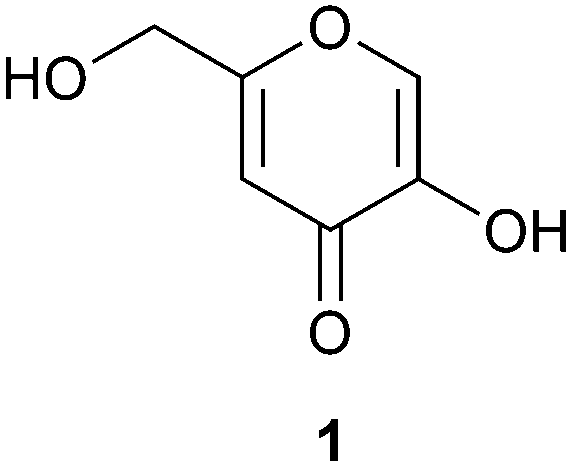 | ||
| Fig. 1 Structure of kojic acid (1). | ||
There is a steadily increasing need to further develop and expand the use of renewable resources for future biorefinery production of commodity and fine chemicals, polymeric materials and building blocks. Here, readily available carbohydrates are anticipated to play a constantly increasing role. Glucosone, a C6 sugar, is an immediate derivative of D-glucose, the most abundant organic molecule on Earth. The additional keto functionality of glucosone, together with its added prochirality, renders it an attractive but yet largely unutilized building block for the preparation of potentially valuable materials or biologically active compounds. Currently unavailable in industrial quantities, glucosone has been largely overlooked as a starting material for biorefinery research.
Here, we describe the high yield bioconversion of D-glucose to glucosone using commercially available industrial enzyme MetZyme® PURECO Pyranose oxidase (MetGen, Finland), followed by chemical conversion of the formed glucosone to kojic acid by a simple acetylation/deacetylation sequence. This new bioconversion process provides commercial opportunities for previously unutilized valorization routes for glucose via glucosone, while the efficient chemical route of conversion of glucosone to kojic acid offers rapid access to this highly interesting biomolecule and building block, potentially enabling its larger-scale future use, independent of fermentation side-streams.
Pyranose oxidase is from the beginning known to oxidize the C2 position in D-glucose to yield glucosone (2-keto-D-glucose).33,34 The enzyme uses oxygen as the electron acceptor, forming hydrogen peroxide as one of the byproducts. In the bioconversion of D-glucose to glucosone, hydrogen peroxide is continuously removed from the reaction by the use of a second enzyme, catalase. Catalase functions by converting the formed hydrogen peroxide to water and oxygen, returning half of the equivalent of the reacted oxygen back to the reaction. MetZyme® PURECO Pyranose Oxidase is an industrial enzyme produced by MetGen Oy, performing equally well at pH = 5.0–7.0 and showing 30% activity at pH = 8, with an optimal temperature range of 40–50 °C. There is no noticeable loss of activity within several hours at 50 °C; however, at 60 °C the enzyme loses 80% of its activity within 1 h. The enzyme is provided as a suspension of active protein aggregates and is not inhibited by the substrate (D-glucose) up to 200 g l−1 concentration. Recovery of the enzyme from the reaction takes place by centrifugation or filtration, enabling simple reuse or incorporation in a continuous process.
In this work, bioconversion of D-glucose (2) to glucosone (3) was performed in a controlled batch reaction (Scheme 1). The conversion reached 80% after 6 h and the reaction was stopped at >95% conversion after 10 h. The glucosone obtained was characterized by HPLC and 1H NMR spectroscopy and the analytical data were compared to those of commercially available products.35 The 1H NMR spectrum of glucosone is rather complex due to configurational equilibrium in solution. The 1H NMR spectrum of the bioconversion product prior to purification is, however, well aligned with the corresponding 1H NMR spectrum of the commercial reference sample. Notably, pure D-glucose was used as the starting material here. The MetZyme® PURECO enzyme also performs well in unpurified hydrolysate of lignocellulosic biomass.
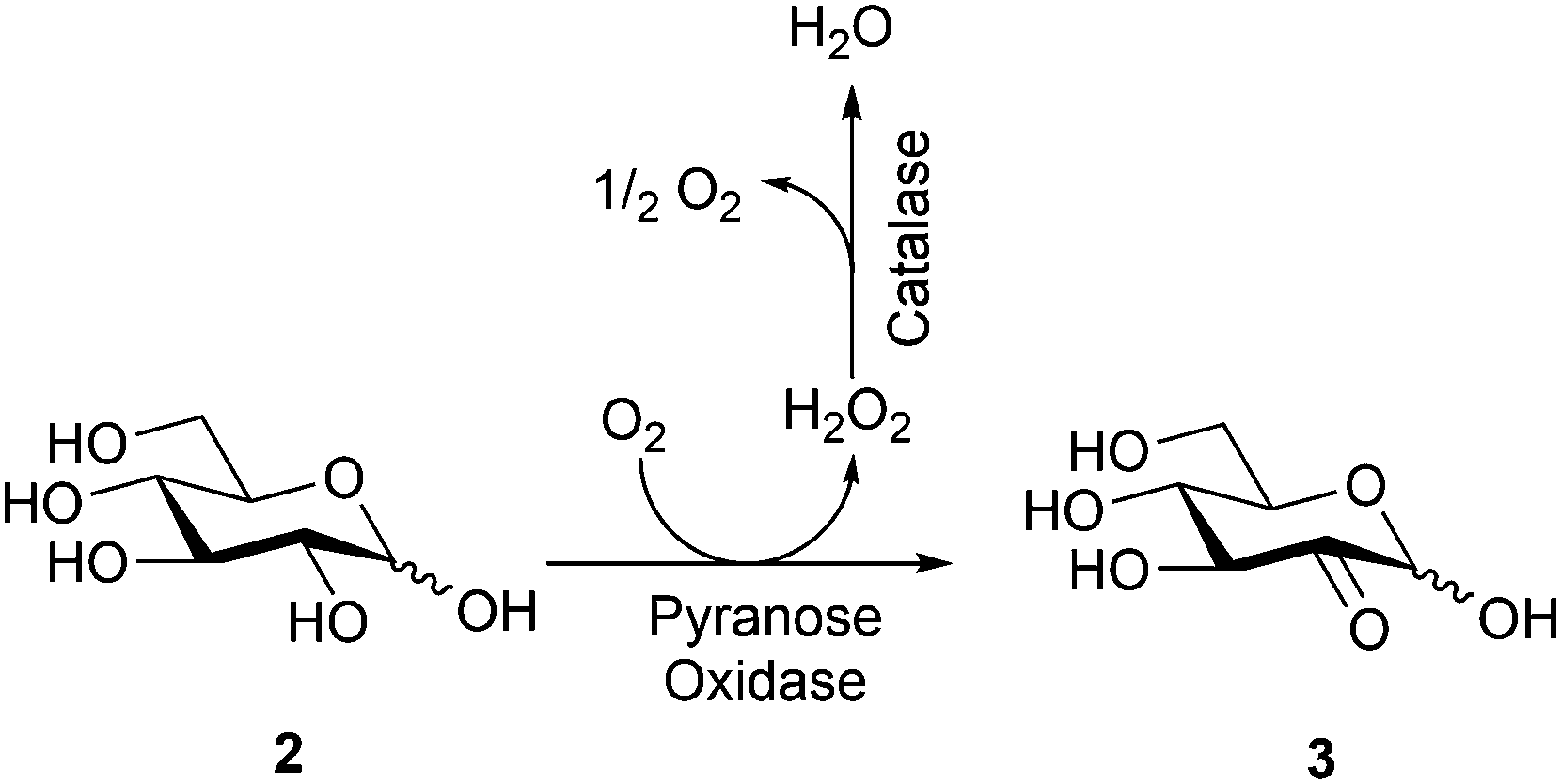 | ||
| Scheme 1 Bioconversion of D-glucose (2) to D-glucosone (3). | ||
MetZyme® PURECO Pyranose oxidase can also accept galactose as a substrate and convert it to 2-keto-galactose. The maximum catalytic rate is approximately 6% of that for glucose conversion. This implies that some galactose present in second generation glucose feedstocks can be directly included in the process described in this work. This also implies that the process could be extended to galactose valorization by future enzyme engineering to optimize the enzymatic oxidation of galactose.
In our initial experiments, acetylation of the enzymatically obtained glucosone with acetic anhydride in pyridine directly provided diacetylkojic acid (4) together with byproduct 5 in 22% and 14% yields (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.6 ratio), respectively, after 3 h of reaction (Scheme 2). Compound 5 is known from earlier literature.36 For optimization of the yield, different solvents and bases were screened with the best results obtained in DMF, while only traces or no product was observed even after 24 h when the reaction was carried out in ethyl acetate, acetone, toluene or dichloromethane, possibly due to the poor solubility of glucosone in these solvents. An increase in the reaction temperature typically resulted in ill-defined polymeric products. Of the NaOAc, lutidine, pyridine and DBU bases screened (Table 1), optimal results in DMF were obtained with NaOAc, which provided acetylated kojic acid and byproduct 5 in 45% and 30% isolated yields, respectively.
:0.6 ratio), respectively, after 3 h of reaction (Scheme 2). Compound 5 is known from earlier literature.36 For optimization of the yield, different solvents and bases were screened with the best results obtained in DMF, while only traces or no product was observed even after 24 h when the reaction was carried out in ethyl acetate, acetone, toluene or dichloromethane, possibly due to the poor solubility of glucosone in these solvents. An increase in the reaction temperature typically resulted in ill-defined polymeric products. Of the NaOAc, lutidine, pyridine and DBU bases screened (Table 1), optimal results in DMF were obtained with NaOAc, which provided acetylated kojic acid and byproduct 5 in 45% and 30% isolated yields, respectively.
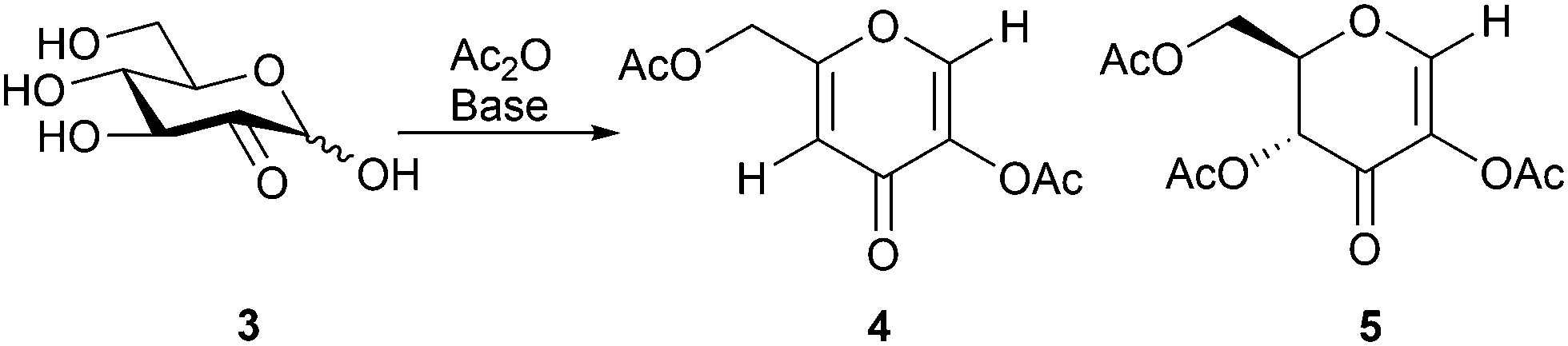 | ||
| Scheme 2 Acetylation of D-glucosone (3) resulting in compounds 4 and 5. | ||
| Base | 4 | 5 |
|---|---|---|
| a Conditions: glucosone, Ac2O (6 eq.), base (5 eq.), DMF. | ||
| Pyridine | 22% | 14% |
| NaOAc | 45% | 30% |
| DBU | 3% | 0% |
| Lutidine | 12% | 5% |
Monitoring of the glucosone acetylation process in DMF by 1H NMR-spectroscopy confirmed that the products of this reaction do not interconvert, but instead competition between the formation of 4 and 5 takes place. The influence of reaction conditions or the underlying kinetic and thermodynamic factors contributing to product distribution are not fully understood at present. A tentative reaction mechanism, similar to that proposed in the literature for the formation of 4,31 is illustrated in Scheme 3. First, a double bond between C2 and C3 is formed, followed by the formation of ketone at C2, forcing the double bond to shift between C3 and C4, subsequently eliminating the AcO group from C4. Next, a new double bond between C4 and C5 is formed, causing the first double bond to shift between C2 and C3. The final step would then involve the formation of ketone at C3, shifting the double bond between C1 and C2 and eliminating AcO from C1, to yield the final product 4. This mechanistic hypothesis is supported by a product formed upon oxidation of methyl 3,4,6-tri-O-acetyl-D-glucopyranoside at C2, which leads to elimination of AcO, forming (2S,6R)-4-acetoxy-2-(acetoxymethyl)-6-methoxy-5,6-dihydro-2H-pyran-5-one, which is a compound similar to the second intermediate in the formation of 4.37 As for the formation of 5, the first step is the same as for 4, followed by the formation of ketone at C3, shifting the double bond to C1 and C2 and eliminating AcO from C1. To investigate the role of acetyl groups in the reaction and the possible direct conversion of glucosone to kojic acid, pure glucosone was dissolved in base in the absence of acetic anhydride. No product was formed in either pyridine or 1 M NaOH, indicating that the presence of acetyl groups in glucosone is necessary for the elimination to products 4 and 5 to take place.
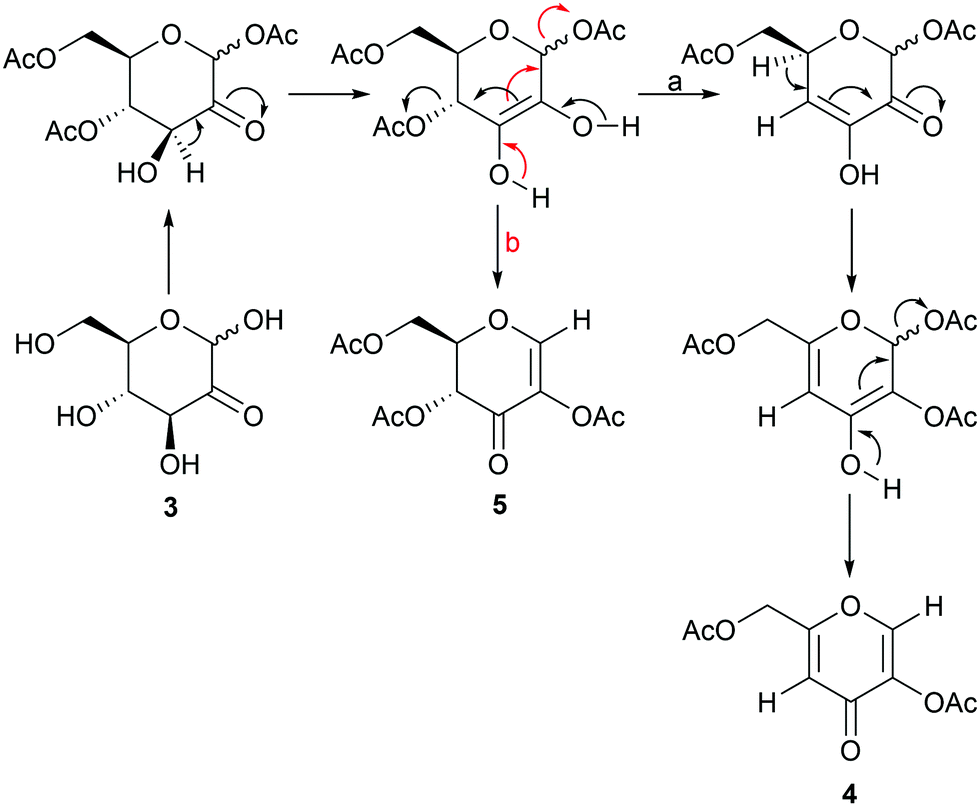 | ||
| Scheme 3 A tentative mechanism for the formation of products 4 (a) and 5 (b). | ||
Next, in order to investigate the influence of purity of the starting material on the yield of the reaction, acetylation of crude glucosone from the enzymatic reaction, subjected only to prior filtration and lyophilization, was compared to acetylation of glucosone, purified by column chromatography, under the previously optimized reaction conditions. Whereas the purified glucosone afforded compounds 4 and 5 in 45% and 30% (1:0.7 ratio), respectively, a higher selectivity towards acetylated kojic acid was obtained by use of the unpurified glucosone, providing 4 and 5 in 47% and 23% yields (1:0.5 ratio), respectively. While challenging to study conclusively, it is possible that salt residues from the enzymatic reaction influence the chemical conversion of glucosone, favoring the formation of acetylated kojic acid instead of the undesired byproduct.
Finally, deacetylation of the purified and isolated diacetylkojic acid with sodium methoxide in methanol provided pure kojic acid in 95% yield. For minimization of the purification steps needed, a direct route from the crude glucosone to kojic acid, depicted in Scheme 4, and based on direct deacetylation of the crude 4/5 compound mixture, was also developed. Conveniently, under the deacetylation conditions employed, byproduct 5 underwent the polymerization reaction, which was seen as polymer type peaks in the 1H NMR spectra of the crude mixture. Deacetylation of pure 5 yielded a mixture of products, although polymer peaks similar to those observed in the direct deacetylation reaction were not observed by NMR spectroscopy. It is possible that unknown impurities in the crude mixture facilitate the polymerization reaction, which does not take place with the pure compound. Kojic acid could then be extracted with ethanol with subsequent precipitation of some impurities by addition of acetone or diethyl ether followed by filtration. After evaporation of the solvents, the remaining impurities could be dissolved in acetone leaving kojic acid as a powder. The isolated kojic acid had a purity of >95% with 20% yield on a 1.3 g scale based on glucosone. While the yield is likely to be improved by future optimization of the purification procedures, as it stands, this method already shows considerable promise for producing kojic acid from glucose with a minimal number of isolation and purification steps.
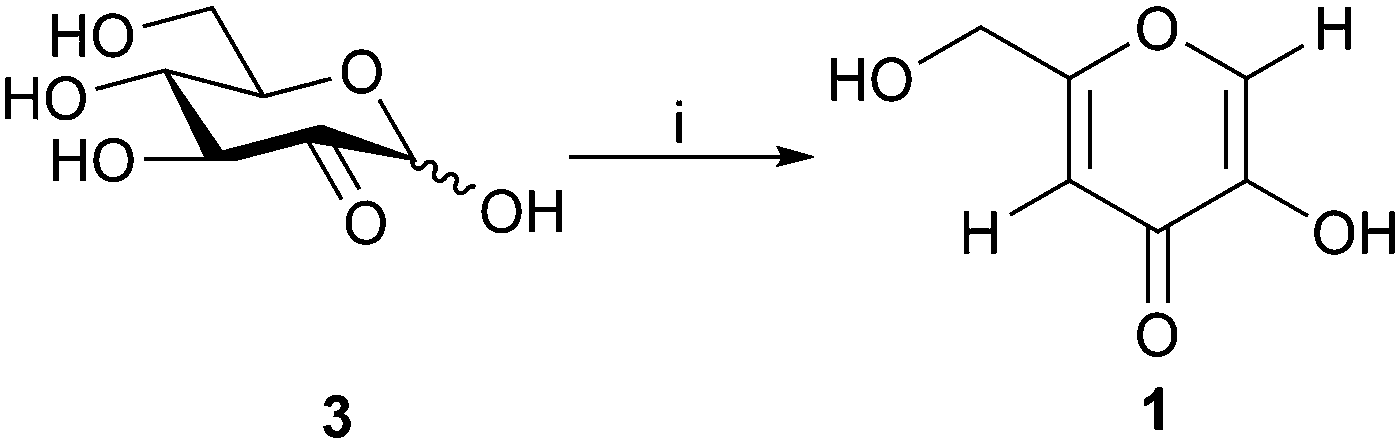 | ||
| Scheme 4 Acetylation/deacetylation of D-glucosone (3) to yield kojic acid (1). i: (a) Ac2O, NaOAc, DMF, 24 h; (b) NaOMe, MeOH, 1 h. | ||
To conclude, we have developed a new chemo-enzymatic preparative route to kojic acid, a valuable biomolecule with a broad range of applications, obtained currently in significant quantities only as a side-stream of a fermentation process. The new method produces crude kojic acid from glucose in a few steps in the multigram scale. Further optimization of the preparative procedure and purification processes will potentially increase the supply of this valuable molecule, independent of fermentation routes, contributing to further research opportunities for biorefineries.
The authors thank the Magnus Ehrnrooth foundation, Waldemar von Frenckell foundation and the EU Horizon 2020 research and innovation program, project SWEETWOODS (792061). PhD Jani Rahkila is gratefully acknowledged for help with NMR analysis. PhD Risto Savela is gratefully acknowledged for HRMS analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Montazeri, S. Emami, H. Asgarian-Omran, S. Azizi, M. Sharif, S. Sarvi, F. Rezaei, M. Sadeghi, S. Gohardehi and A. Daryani, Exp. Parasitol., 2019, 200, 7–12 CrossRef CAS PubMed.
- S. A. Moharib and E. I. Ibrahim, Adv. Food Sci., 2017, 39, 35–43 CAS.
- L. S. Dehkordi, Z. D. Liu and R. C. Hider, Eur. J. Med. Chem., 2008, 43, 1035–1047 CrossRef CAS.
- M. Saraei, G. Zarrini, M. Esmati and L. Ahmadzadeh, Des. Monomers Polym., 2017, 20, 325–331 CrossRef CAS PubMed.
- L. L. Wu, W. Li and Y. J. Mei, Russ. J. Coord. Chem., 2019, 45, 154–162 CrossRef CAS.
- X. R. Wang, H. M. Cheng, X. W. Gao, W. Zhou, S. J. Li, X. L. Cao and D. Yan, Chin. Chem. Lett., 2019, 30, 919–923 CrossRef CAS.
- H. Kayahara, N. Shibata, K. Tadasa, H. Maeda, T. Kotani and I. Ichimoto, Agric. Biol. Chem., 1990, 54, 2441–2442 CAS.
- S. Baláž, M. Uher, J. Brtko, M. Veverka, J. Bransová, J. Dobias, M. Pódová and J. Buchvald, Folia Microbiol., 1993, 38, 387–391 CrossRef PubMed.
- S. H. Hussein-Al-Ali, P. Arulselvan, M. Z. Hussein, S. Fakurazi and B. Saifullah, J. Biomimetics, Biomater. Biomed. Eng., 2018, 36, 45–55 CAS.
- V. P. Peroković, Ž. Car, A. Usenik, T. Opačak-Bernardi, A. Jurić and S. S. Tomić, Mol. Divers., 2019 DOI:10.1007/s11030-019-09948-1.
- R. Saruno, F. Kato and T. Ikeno, Agric. Biol. Chem., 1979, 43, 1337–1338 CAS.
- J. M. Noh, S. Y. Kwak, H. S. Seo, J. H. Seo, B. G. Kim and Y. S. Lee, Bioorg. Med. Chem. Lett., 2009, 19, 5586–5589 CrossRef CAS PubMed.
- S. Emami, S. J. Hosseinimehr, S. M. Taghdisi and S. Akhlaghpoor, Bioorg. Med. Chem. Lett., 2007, 17, 45–48 CrossRef CAS PubMed.
- J. S. Chen, C. I. Wei, R. S. Rolle, W. S. Otwell, M. O. Balaban and M. R. Marshall, J. Agric. Food Chem., 1991, 39, 1396–1401 CrossRef CAS.
- Y. Niwa and Y. Miyachi, Inflammation, 1986, 10, 79–91 CrossRef CAS PubMed.
- Y. Niwa and H. Akamatsu, Inflammation, 1991, 15, 303–315 CrossRef CAS PubMed.
- M. O. Masse, V. Duvallet, M. Borremans and L. Goeyens, Int. J. Cosmet. Sci., 2001, 23, 219–232 CrossRef CAS PubMed.
- P. F. Dowd, Pestic. Biochem. Physiol., 1988, 32, 123–134 CrossRef CAS.
- J. Dobias, P. Nemec and J. Brtko, Biológia, 1977, 32, 417–421 CAS.
- J. J. Faig, A. Moretti, L. B. Joseph, Y. Zhang, M. J. Nova, K. Smith and K. E. Uhrich, Biomacromolecules, 2017, 18, 363–373 CrossRef CAS PubMed.
- C. Z. W. Sie and Z. Ngaini, J. Sol. Energy, 2017, 2017, 1–10 CrossRef.
- R. Bentley, Methods Enzymol., 1957, 3, 238–241 Search PubMed.
- V. M. Nurchi, M. de Guadalupe Jaraquemada-Pelaez, G. Crisponi, J. I. Lachowicz, R. Cappai, L. Gano, M. A. Santos, A. Melchior, M. Tolazzi, M. Peana, S. Medici and M. A. Zoroddu, J. Inorg. Biochem., 2019, 193, 152–165 CrossRef CAS PubMed.
- Z. D. Liu, H. H. Khodr, D. Y. Liu, S. L. Lu and R. C. Hider, J. Med. Chem., 1999, 42, 4814–4823 CrossRef CAS PubMed.
- I. Tomita, K. Mitsuhashi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 271–276 CrossRef CAS.
- R. Mohamad, M. S. Mohamed, N. Suhaili, M. M. Salleh and A. B. Ariff, Biotechnol. Mol. Biol. Rev., 2010, 5, 24–37 CAS.
- W. Feng, J. Liang, B. Wang and J. Chen, Bioprocess Biosyst. Eng., 2019, 42, 753–761 CrossRef CAS PubMed.
- J. Chaudhary, A. N. Pathak and S. Lakhawat, Annu. Res. Rev. Biol., 2014, 4, 3165–3196 CrossRef.
- I. A. El-Kady, A. Naser, A. Zohri and S. R. Hamed, Biotechnol. Res. Int., 2014, 1–10 Search PubMed.
- K. Maurer and R. Bohme, Ber. Dtsch. Chem. Ges. B, 1930, 63, 2069–2073 CrossRef.
- G. J. F. Chittenden, Carbohydr. Polym. Res., 1969, 11, 424–427 CrossRef CAS.
- K. Maurer and R. Bohme, Ber. Dtsch. Chem. Ges. B, 1936, 69, 1399–1410 CrossRef.
- A. Huwig, H. J. Danneel and F. Giffhorn, J. Biotechnol., 1994, 32, 309–315 CrossRef CAS.
- G. Treitz, G. Maria, F. Giffhorn and E. Heinzle, J. Biotechnol., 2001, 85, 271–287 CrossRef CAS PubMed.
- Glucosone from Sigma-Aldrich; cat no 61793.
- F. W. Lichtenthaler, S. Nishiyama and T. Weimer, Liebigs Ann. Chem., 1989, 1163–1170 CrossRef CAS.
- F. W. Lichtenthaler and P. Heidel, Angew. Chem., Int. Ed. Engl., 1969, 8, 978–979 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07405h |
| This journal is © The Royal Society of Chemistry 2019 |