
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium stannyl formation by organostannane dehydrogenation†
Louis J.
Morris
a,
Michael S.
Hill
 *a,
Ian
Manners
b,
Claire L.
McMullin
*a,
Mary F.
Mahon
a and
Nasir A.
Rajabi
a
*a,
Ian
Manners
b,
Claire L.
McMullin
*a,
Mary F.
Mahon
a and
Nasir A.
Rajabi
a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
bDepartment of Chemistry, University of Victoria, Victoria, BC V8P 5C2, Canada
First published on 3rd October 2019
Abstract
Reaction of the dimeric calcium hydride, [(BDI)CaH]2 (1), with Ph3SnH ensues with elimination of H2 to provide [(BDI)Ca-μ2-H-(SnPh3)Ca(BDI)] (3) and [(BDI)Ca(SnPh3)]2 (4) alongside dismutation to Ph4Sn, H2 and Sn(0). DFT analysis indicates that stannyl anion formation occurs through deprotonation of Ph3SnH and with retention of dinuclear species throughout the reactions.
The stoichiometric and catalytic reaction chemistry of heavier alkaline earth species, particularly those of calcium, continues to attract increasing attention.1 Central to these advances have been a suite of highly nucleophilic molecular hydride derivatives,2,3 which may be applied to an array of reductive multiple bond heterofunctionalisation and cross-metathetical processes.4 Our own recent research has concentrated on the coordinatively unsaturated β-diketiminato derivative [(BDI)CaH]2 (1; BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2: Scheme 1), which reacts directly with a wide range of otherwise unactivated alkenyl C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.5,6 The resultant calcium σ-organometallics have proven to be incredibly potent reagents in their own right and are capable of effecting the direct alkylation of benzene via an unprecedented displacement of hydride (to reform 1), and in which the polarising qualities of the calcium centre itself perform a key role (Scheme 1).5,7
C bonds.5,6 The resultant calcium σ-organometallics have proven to be incredibly potent reagents in their own right and are capable of effecting the direct alkylation of benzene via an unprecedented displacement of hydride (to reform 1), and in which the polarising qualities of the calcium centre itself perform a key role (Scheme 1).5,7
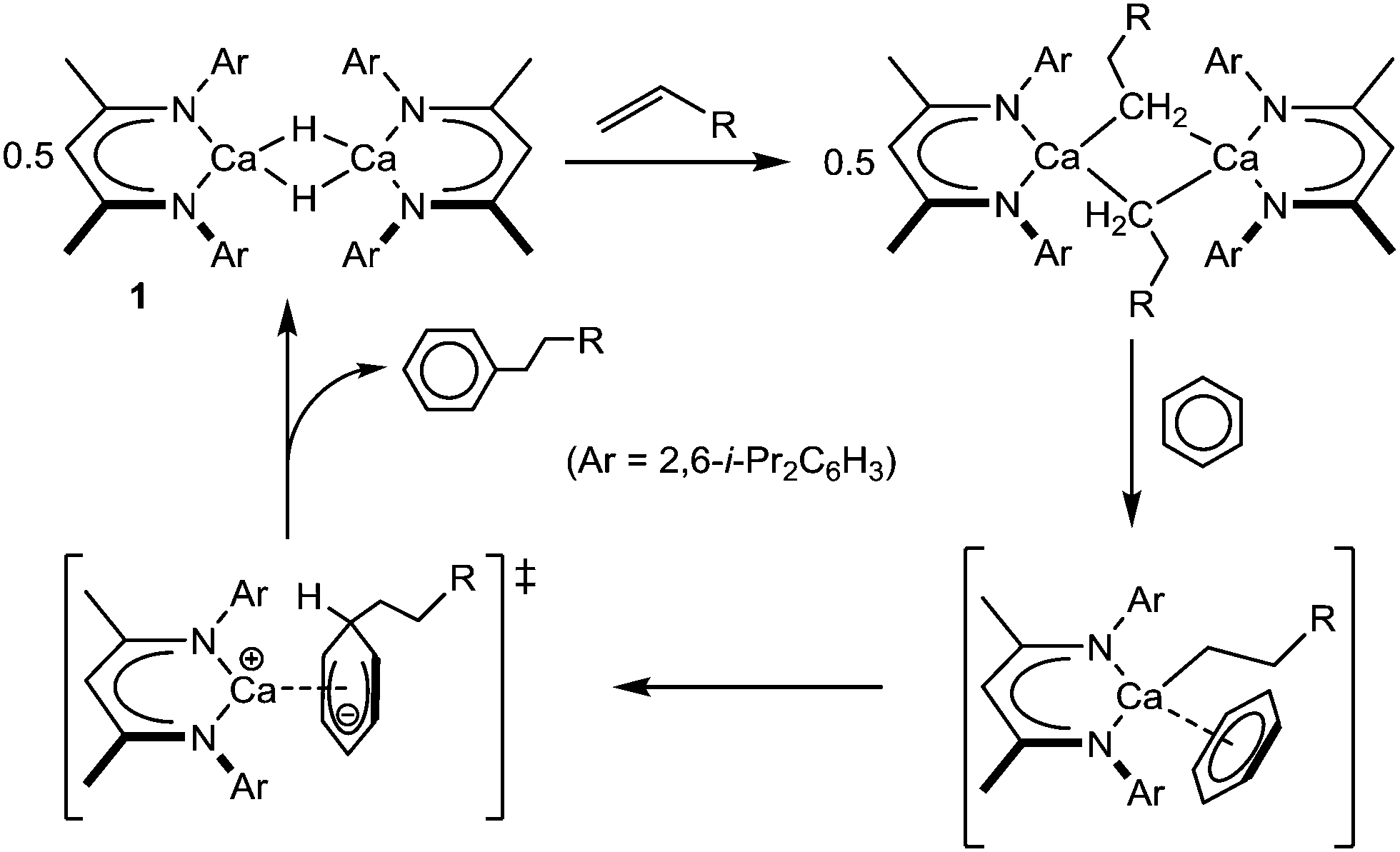 | ||
| Scheme 1 Reaction of compound 1 with alkenes and the calcium-mediated nucleophilic alkylation of benzene. | ||
As an extension of this work, we have turned our attention to the potential of calcium derivatives of the heavier tetrels to effect similarly challenging arene activation. The realisation of such processes would enable the direct nucleophilic access to triorganosilylated and triorganostannylated aryl derivatives which would themselves be valuable substrates for onward elaboration in, for example, catalytic Hiyama-8 or Stille-type cross coupling protocols.9 Although organostannyl anions of the group 1 metals have received significant attention,10 calcium compounds containing bonds to heavier p-block elements are, in general, very rare,3,11 while the sole example of a direct calcium-to-tin bond is provided by Westerhausen's bis(trimethylstannyl), [(Me3Sn)2Ca(THF)4] (2).12 Although the route to this compound, by treatment of calcium metal with hexamethyldistannane, provided a reasonable yield of the stannyl product its heterogeneous nature necessitated a long reaction time. For phenyl–tin derivatives, the intermolecular migration of phenyl substituents can also provide a significant complication. Of most relevance to the current work, Uhlig and co-workers have reported that reaction of metallic calcium with hexaphenyldistannane provided the charge separated species, [Ca(18-crown-6)(HMPA)2][Sn(SnPh3)3]2.13 Herein, we report facile access to triphenylstannyl-calcium derivatives through the reaction of compound 1 and commercially available Ph3SnH.
Addition of 2–3 equivalents of Ph3SnH to a C6D6 solution of compound 1 induced a rapid bubbling and the gradual development of a pale orange colouration. Analysis by 119Sn{1H} NMR spectroscopy after 60 minutes revealed quantitative consumption of the tin hydride and the appearance of a major new resonance at δ −139.8 ppm, which appeared alongside a lower intensity signal at δ −126 ppm and was readily identified as Ph4Sn by comparison to literature values.14 The corresponding 1H NMR spectrum evidenced the formation of a predominant BDI-containing compound (3, Scheme 2) that was most readily characterised by the emergence of a new BDI γ-methine signal at δ 4.75 ppm and a singlet, resonating with half the relative intensity, at δ 3.83 ppm. This latter signal also displayed characteristic satellites separated by 94 Hz, within the typical range for a 2J(119/117Sn–1H) coupling.14
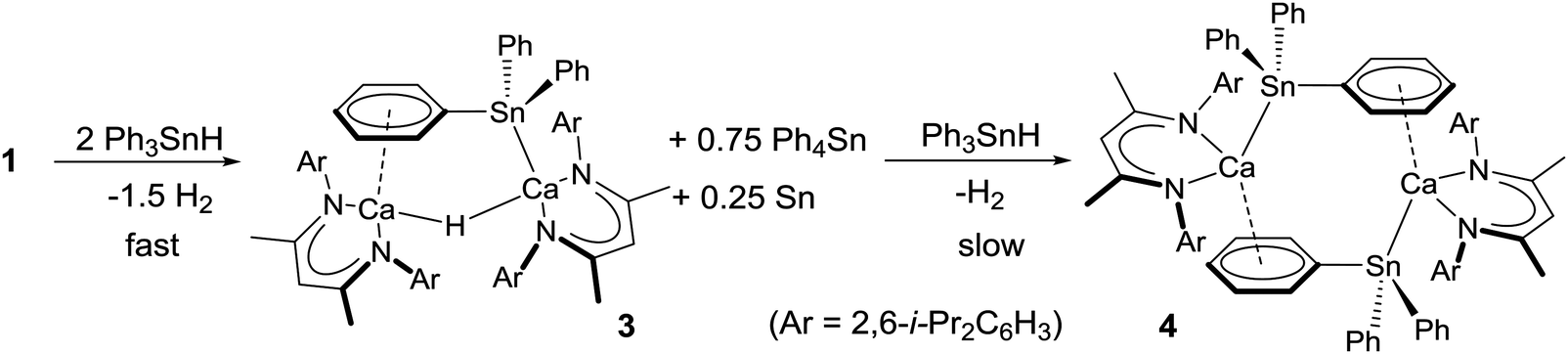 | ||
| Scheme 2 Synthesis of compounds 3 and 4 and the calcium-centred dismutation of Ph3SnH. | ||
While no significant changes to the NMR spectra were observed on standing overnight, the origin of these observations was resolved by an X-ray diffraction analysis, which was performed on a colourless single crystal grown from a hexane/toluene solution at −30 °C. The results of this analysis (Fig. 1) confirmed that compound 3 is an unsymmetrical dinuclear calcium species in which two {(BDI)Ca} units are connected by μ2-bridging hydride and triphenylstannyl ligands.
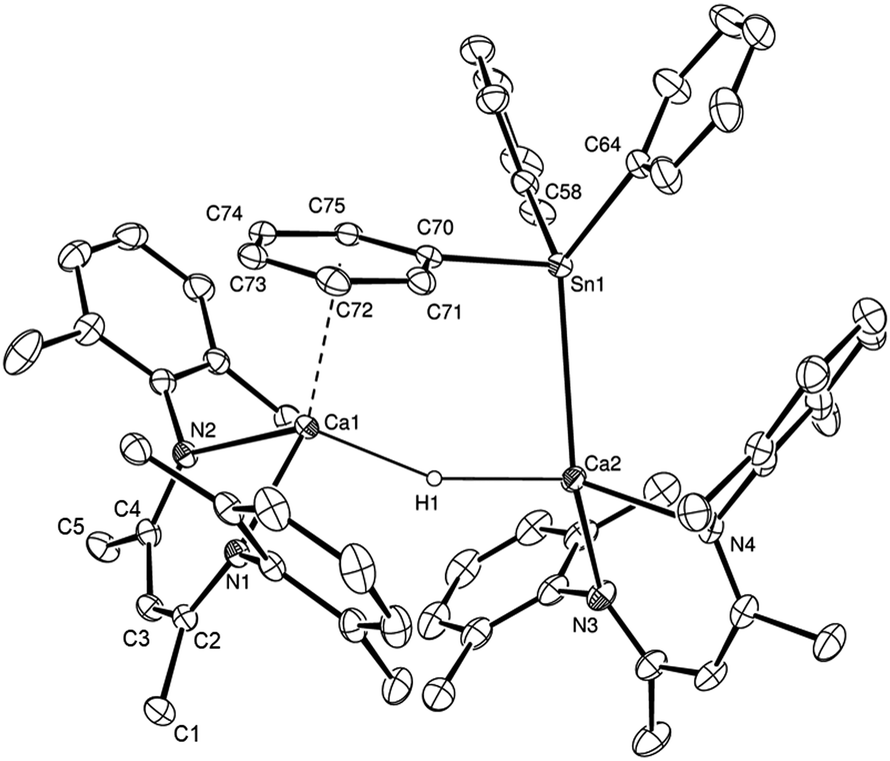 | ||
| Fig. 1 ORTEP representation of compound 3 (25% probability ellipsoids). Hydrogen atoms, except H1, iso-propyl methyl groups and occluded solvent molecules are removed for clarity. Selected bond lengths (Å) and angles (°): Sn1–Ca2 3.2137(4), Sn1–C58 2.174(2), Sn1–C64 2.190(2), Sn1–C70 2.2258(19), Ca1–N1 2.3443(16), Ca1–N2 2.3318(15), Ca1–C70 3.0649(18), Ca1–C71 2.9837(19), Ca1–C72 2.904(2), Ca1–C73 2.864(2), Ca1–C74 2.898(2), Ca1–C75 2.9815(19), Ca2–N3 2.3275(18), Ca2–N4 2.3306(17), C58–Sn1–Ca2 127.45(6), C58–Sn1–C64 98.00(8), C58–Sn1–C70 98.48(8), C64–Sn1–Ca2 129.08(6), C64–Sn1–C70 98.61(7), C70–Sn1–Ca2 96.16(5), N1–Ca1–N2 79.59(5), N3–Ca2–N4 81.18(6). | ||
The tin-centred ligand is bonded to Ca2 through Sn1 and interacts with Ca1 via an η6-interaction of its (C70–C75) phenyl substituent. The significance of secondary interactions in the stabilisation of alkaline-earth complexes is well established.15 Although unsupported η6-arene interactions between benzene and toluene and calcium-centred cations have recently been observed by our own group16 and by Harder and co-workers,17 these structures display significantly longer Ca–arene centroid distances (ca. 2.9 Å) than that observed in 3 [2.5999(9) Å]. This latter distance is, thus, more typical of those observed in a variety of N-mesityl- and N-di-isopropylphenyl calcium amidinate and triazenide derivatives in which the arene substituent is a tethered component of the coordinated anion.18 Although the Ca2–Sn1 bond length [3.2137(4) Å] is similar to that observed in Westerhausen's calcium trimethylstannyl [2, 3.2721(3) Å],12 the strength of this Ca–arene interaction ensures that the coordination environment of the tin centre is significantly perturbed toward a trigonal pyramidal geometry. The C70–Sn1–Ca2 bond angle subtended by the apical C70–C75- containing phenyl substituent is just 96.16(5)°, whereas the sum of the C58–Sn1–Ca2, C58–Sn1–C64 and C64–Sn1–Ca2 angles amounts to 354.5°, with Sn1 lying only 0.331(1) Å out of the basal plane of the pyramid defined by Ca2, C58 and C64.
Repetition of the reaction in d8-toluene provided similar observations at 298 K. Cooling of this sample to 246 K, however, resulted in significant broadening of the BDI γ-methine resonance at δ 4.71 ppm, which was observed to split into two well resolved signals at 4.60 and 4.83 ppm below 211 K. Although a single set of phenyl–tin resonances was also observed at 298 K, below 220 K a series of resonances at δ 6.05, 6.72, and 8.10 ppm, and integrating in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 ratio relative to the two 1H BDI γ-methine signals, could be discriminated. Although the Ca–Sn bond apparently rotates freely at room temperature, these latter signals may be attributed to the persistence of the Ca-η6-phenyl interaction observed in the solid state structure (ΔG‡ = 48 kJ mol−1).
:1:2 ratio relative to the two 1H BDI γ-methine signals, could be discriminated. Although the Ca–Sn bond apparently rotates freely at room temperature, these latter signals may be attributed to the persistence of the Ca-η6-phenyl interaction observed in the solid state structure (ΔG‡ = 48 kJ mol−1).
When the reaction was left to stand for a further three days at room temperature the reaction mixture became opaque with the deposition of a brown-grey solid, which was tentatively assigned as a precipitate of Sn metal. Crystals of Ph4Sn (confirmed by a unit cell check), were also observed to form, coinciding with the emergence of a new signal at δ −160.6 ppm in the 119Sn{1H} NMR spectrum. Removal of solvent from the reaction mixture, extraction of the solid residue into hexane/toluene and crystallisation at −30 °C yielded single crystals of the hydride-free dimeric calcium triphenylstannyl, compound 4. Compound 4 (Fig. 2) is a centrosymmetric dimer, in which each pseudo-four-coordinate calcium centre is bound by a BDI ligand and a direct Ca–Sn σ bond to a triphenylstannyl anion. This Ca–Sn distance is closely comparable to that of compound 3 [3.3221(6) Å]. Dimer propagation occurs in a similar manner, through twofold η6-phenyl-to-calcium interactions, albeit the Ca-centroid distance in 4 is slightly shorter at 2.5376(19) Å. The presence of the more sterically demanding triphenylstannyl ligand in the place of the bridging hydride of 3 also induces some significant geometric adjustments at the calcium centre, which projects some 1.339(3) Å out of the mean plane defined by N1/N2/C1–C5 of the BDI ligand. In addition, although the geometry at tin is somewhat distorted from a perfect tetrahedron, the three C–Sn–C and Ca–Sn–C angles fall within a much closer range (92.9–97.3° and 111.4–130.8°, respectively) than those imposed by the trigonal pyramidal geometry of compound 3.
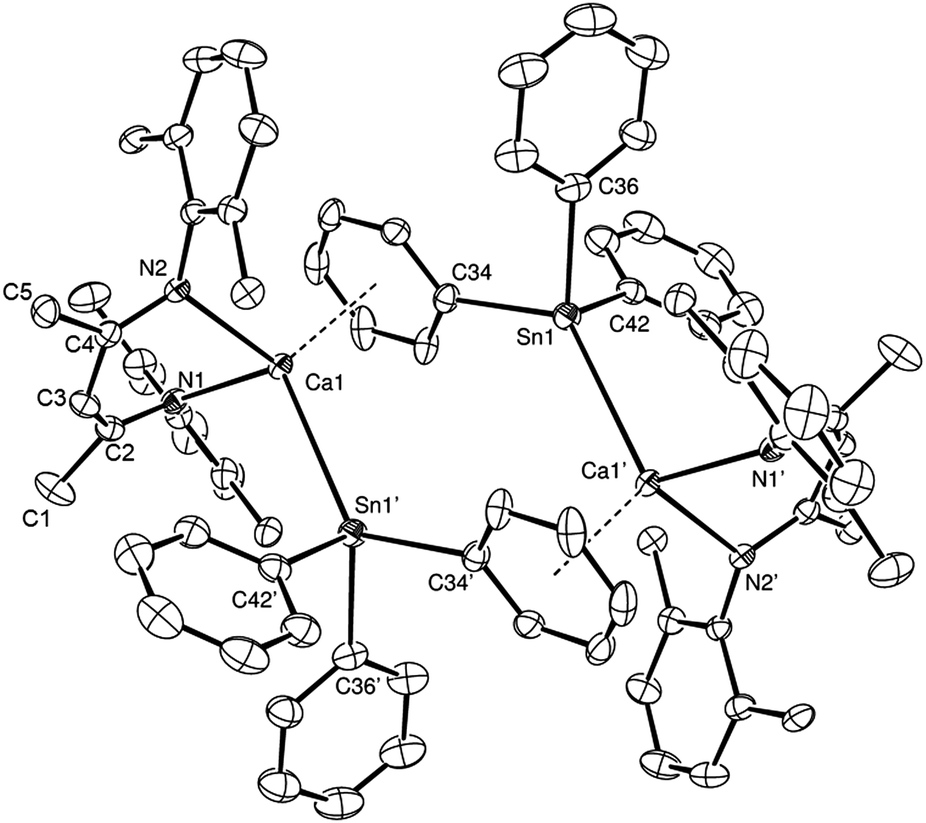 | ||
| Fig. 2 ORTEP representation of compound 4 (25% probability ellipsoids). Hydrogen atoms and iso-propyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): Sn1–Ca1′ 3.3221(5), Sn1–C34 2.222(3), Sn1–C36 2.218(3), Sn1–C42 2.196(3), Ca1–N1 2.319(2), Ca1–N2 2.330(2), Ca1–C34 2.987(3), Ca1–C35 2.917(3), C34–Sn1–Ca1′ 130.80(8), C36–Sn1–Ca1′ 122.05(9), C36–Sn1–C34 94.66(12), C42–Sn1–Ca1′ 111.37(9), C42–Sn1–C34 92.91(11), C42–Sn1–C36 97.30(12), N1–Ca1–N2 82.69(8). Symmetry operations to generate primed atoms; 1 − x, 1 − y, 1 − z. | ||
The electronic structures of compounds 3 and 4 were assessed by Density Functional Theory (DFT, see ESI† for full details of methodology). The HOMOs of both compounds are of a similar energy (ca. −4.45 eV, see Fig. S20, ESI†) and, emphasising the highly ionic nature of the Ca–Sn bonding, effectively comprise donation of the filled tin 5p orbitals to the cationic calcium centre(s) (Fig. 3). The Ca–arene interactions (3: HOMO−27, HOMO−28; 4: HOMO−21, HOMO−22) are also largely electrostatic in nature and originate from interactions between the Lewis acidic tin centres and the almost degenerate pair of highest energy filled π-orbitals associated with the coordinated C6H5 rings (Fig. S20, ESI†).
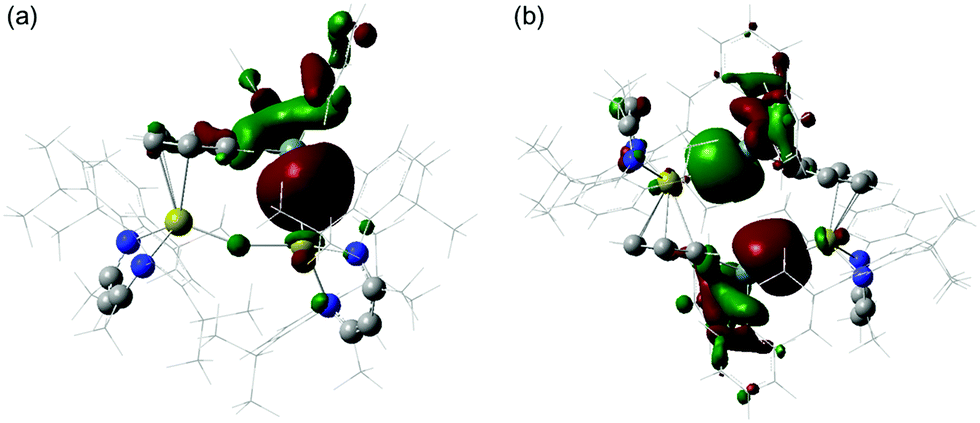 | ||
| Fig. 3 Computed HOMOs (BP86/BS1)19 for (a) 3 and (b) 4. | ||
The reaction between compound 1 and Ph3SnH was also investigated by DFT. Although a pathway (Fig. S22, ESI†) invoking fragmentation of the dimeric hydride (1) was considered, the initial monomerisation process was found to be significantly endergonic (ΔG = +33.7 kcal mol−1). A more viable pathway was computed to occur without any necessary dimer fragmentation (Fig. 4a). In this case, although formation of encounter complex A is endergonic by +7.6 kcal mol−1, consistent with the observed facility of the reaction at room temperature, the Sn–Ha bond and Ca–Hb bonds are broken via a dinuclear transition state TSAB at only +8.3 kcal mol−1. Natural Bond Orbital (NBO) analysis of TSAB indicated partial charges of −0.03 and −0.25 on Ha and Hb, respectively (Fig. 4b), emphasising that H2 elimination results from what is essentially an acid–base reaction where Ph3SnH is deprotonated by the calcium hydride. The newly formed H–H bond in the subsequent intermediate B remains substantially polarised, with partial charges of +0.01 (Ha) and −0.27 (Hb). Isomerisation viaC and dihydrogen loss provides species D at −11.9 kcal mol−1, from which 3 is formed (ΔG = −26.7 kcal mol−1) through realisation of the Ca–π arene interaction. Assessment of the reaction of 3 with a second molecule of Ph3SnH (Fig. S23, ESI†) indicated that the formation of 4 occurs via a similarly polarised and kinetically accessible transition state leading to H2 elimination (ΔG‡ = −12.1 kcal mol−1).
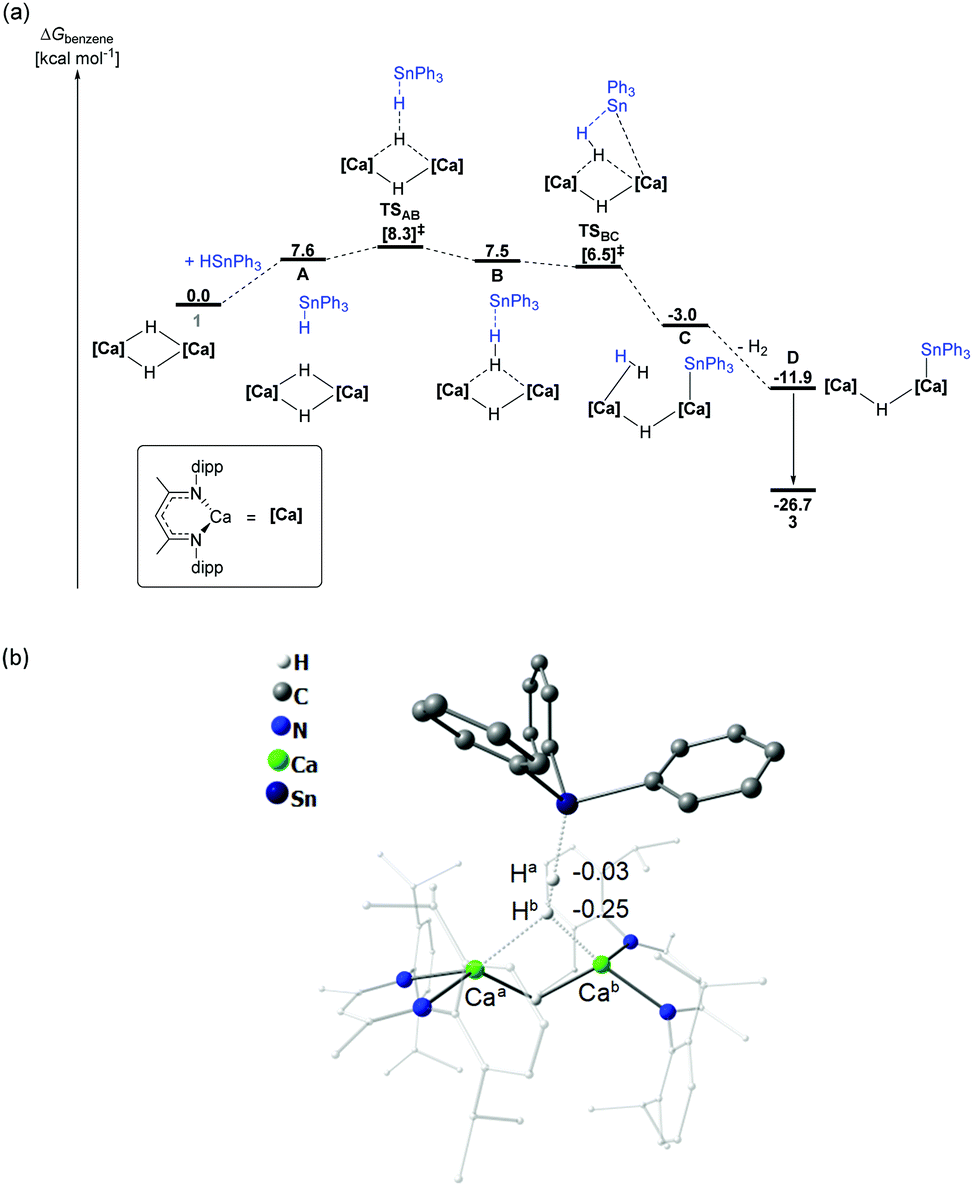 | ||
| Fig. 4 (a) DFT calculated (BP86-D3(BJ)-benzene/BS2//BP86/BS1)19 free energy (kcal mol−1) profile for the reaction of 1 with HSnPh3 to form 3, relative to 1 + HSnPh3; (b) transition state TSAB showing calculated NBO charges of Ha and Hb. BDI ligand frameworks shown as wireframe for clarity. | ||
Further experiments to scale up and optimise the synthesis of 3 and 4 were hampered by the co-crystallisation of both compounds and the facility of the system toward redistribution of the triphenylstannane reagent to Ph4Sn and metallic tin. We tentatively suggest that the production of elemental tin may be ascribed to the dismutation of the Ph3SnH reagent to Ph4Sn and unstable SnH4, which further eliminates H2. Under this regime, compound 3 undergoes Sn–Ph/Sn–H metathesis, possibly facilitated by the polarisation of a Sn–C bond through its interaction with the highly electropositive calcium centre, more rapidly than further stannane deprotonation to form 4. This process provides Ph4Sn and a calcium diphenylstannyl derivative, [(BDI)Ca-μ2-H-(SnPh2H)Ca(BDI)], which is unobservable due to its rapid consumption by analogous reaction with Ph3SnH. Consecutive and increasingly facile reactions of this nature ultimately provide a parent calcium stannane derivative, [(BDI)Ca-μ2-H-(SnH3)Ca(BDI)]. This species will be prone to further Sn–H/Ca–Sn metathesis to reform compound 1 and produce unstable SnH4 with the consequent generation of H2 and elemental tin. Although no further calcium- or tin-centred intermediates could be observed, support for this hypothesis was provided by a further reaction between compound 3 and n-Bu3SnH. Monitoring by 1H and 119Sn{1H} NMR spectroscopy over 40 hours, during which time the reaction again became black and opaque, confirmed that quantitative conversion of Bu3SnH to Bu3SnPh had occurred.
We are continuing to assess these possibilities and to study the reactivity of the resultant calcium stannyl and related p-block centred anions.
We acknowledge financial support from the EPSRC Centre for Doctoral Training in Catalysis (EP/L016443/1) and research grant EP/R020752/1. This research made use of the Balena High Performance Computing (HPC) Service at the University of Bath. I. M. thanks the University of Bristol for support and the Canadian Government for a Canada 150 Research Chair.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. A, 2010, 466, 927–963 CrossRef CAS; (b) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed; (c) M. R. Crimmin and M. S. Hill, in Alkaline-Earth Metal Compounds: Oddities and Applications, ed. S. Harder, 2013, vol. 45, pp. 191–241 Search PubMed; (d) M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC; (e) Y. Sarazin and J. F. Carpentier, Chem. Rec., 2016, 16, 2482–2505 CrossRef CAS PubMed.
- (a) D. Mukherjee, D. Schuhknecht and J. Okuda, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS PubMed; (b) S. Harder and J. Brettar, Angew. Chem., Int. Ed., 2006, 45, 3474–3478 CrossRef CAS PubMed; (c) P. Jochmann, J. P. Davin, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2012, 51, 4452–4455 CrossRef CAS PubMed; (d) V. Leich, T. P. Spaniol and J. Okuda, Inorg. Chem., 2015, 54, 4927–4933 CrossRef CAS PubMed; (e) D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS PubMed; (f) B. Maitland, M. Wiesinger, J. Langer, G. Ballmann, J. Pahl, H. Elsen, C. Farber and S. Harder, Angew. Chem., Int. Ed., 2017, 56, 11880–11884 CrossRef CAS PubMed; (g) A. Causero, G. Ballmann, J. Pahl, H. Zijlstra, C. Farber and S. Harder, Organometallics, 2016, 35, 3350–3360 CrossRef CAS.
- V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2016, 55, 4794–4797 CrossRef CAS PubMed.
- For example, (a) J. Spielmann, F. Buch and S. Harder, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS PubMed; (b) J. Spielmann and S. Harder, J. Am. Chem. Soc., 2009, 131, 5064–5065 CrossRef CAS PubMed; (c) M. D. Anker, M. S. Hill, J. P. Lowe and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 10009–10011 CrossRef CAS PubMed; (d) M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Science, 2017, 358, 1168–1171 CrossRef CAS PubMed.
- (a) A. S. S. Wilson, M. S. Hill and M. F. Mahon, Organometallics, 2019, 38, 351–360 CrossRef CAS; (b) A. S. S. Wilson, C. Dinoi, M. S. Hill, M. F. Mahon and L. Maron, Angew. Chem., Int. Ed., 2018, 57, 15500–15504 CrossRef CAS PubMed; (c) M. S. Hill, M. F. Mahon, A. S. S. Wilson, C. Dinoi, L. Maron and E. Richards, Chem. Commun., 2019, 55, 5732–5735 RSC.
- B. Rosch, T. X. Gentner, H. Elsen, C. A. Fischer, J. Langer, M. Wiesinger and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 5396–5401 CrossRef PubMed.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- (a) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS; (b) M. M. Heravi and L. Mohammadkhani, J. Organomet. Chem., 2018, 869, 106–200 CrossRef CAS.
- (a) D. Reed, D. Stalke and D. S. Wright, Angew. Chem., Int. Ed. Engl., 1991, 30, 1459–1460 CrossRef; (b) P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1993, 554–555 RSC; (c) B. E. Eichler, A. D. Phillips and P. P. Power, Organometallics, 2003, 22, 5423–5426 CrossRef CAS; (d) T. Schollmeier, U. Englich, R. Fischer, I. Prass, K. Ruhlandt-Senge, M. Schurmann and F. Uhlig, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1462–1470 CAS; (e) C. Kleeberg, J. Grunenberg and X. L. Xie, Inorg. Chem., 2014, 53, 4400–4410 CrossRef CAS; (f) J. Jastrzebski, M. van Klaveren and G. van Koten, Organometallics, 2015, 34, 2600–2607 CrossRef CAS.
- (a) W. J. Teng and K. Ruhlandt-Senge, Organometallics, 2004, 23, 2694–2700 CrossRef CAS; (b) W. J. Teng and K. Ruhlandt-Senge, Organometallics, 2004, 23, 952–956 CrossRef CAS; (c) V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2014, 50, 2311–2314 RSC; (d) N. L. Lampland, A. Pindwal, K. K. Yan, A. Ellern and A. D. Sadow, Organometallics, 2017, 36, 4546–4557 CrossRef CAS.
- M. Westerhausen, Angew. Chem., Int. Ed. Engl., 1994, 33, 1493–1495 CrossRef.
- U. Englich, K. Ruhlandt-Senge and F. Uhlig, J. Organomet. Chem., 2000, 613, 139–147 CrossRef CAS.
- B. Wrackmeyer, Annu. Rep. NMR Spectrosc., 1999, 38, 203–264 CrossRef CAS.
- (a) S. C. Rosca, C. Dinoi, E. Caytan, V. Dorcet, M. Etienne, J. F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2016, 22, 6505–6509 CrossRef CAS PubMed; (b) S. C. Rosca, E. Caytan, V. Dorcet, T. Roisnel, J. F. Carpentier and Y. Sarazin, Organometallics, 2017, 36, 1269–1277 CrossRef CAS; (c) S. C. Rosca, V. Dorcet, T. Roisnel, J. F. Carpentier and Y. Sarazin, Dalton Trans., 2017, 46, 14785–14794 RSC; (d) S. C. Rosca, V. Dorcet, J. F. Carpentier and Y. Sarazin, Inorg. Chim. Acta, 2018, 475, 59–64 CrossRef CAS; (e) M. F. Zuniga, G. B. Deacon and K. Ruhlandt-Senge, Inorg. Chem., 2008, 47, 4669–4681 CrossRef CAS PubMed; (f) W. D. Buchanan, D. G. Allis and K. Ruhlandt-Senge, Chem. Commun., 2010, 46, 4449–4465 RSC.
- (a) L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684–12693 RSC; (b) L. Garcia, M. S. Hil and M. F. Mahon, Organometallics, 2019 DOI:10.1021/acs.organomet.9b00493.
- (a) J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC; (b) J. Pahl, A. Friedrich, H. Eisen and S. Harder, Organometallics, 2018, 37, 2901–2909 CrossRef CAS.
- (a) C. Loh, S. Seupel, H. Görls, S. Krieck and M. Westerhausen, Organometallics, 2014, 33, 1480–1491 CrossRef CAS; (b) C. Loh, S. Seupel, A. Koch, H. Görls, S. Krieck and M. Westerhausen, Dalton Trans., 2014, 43, 14440–14449 RSC; (c) S. O. Hauber, F. Lissner, G. B. Deacon and M. Niemeyer, Angew. Chem., Int. Ed., 2005, 44, 5871–5875 CrossRef CAS PubMed.
- BS1 = SDDALL for Ca and Sn, 6-31G** for all other atoms, BS2 = cc-pVTZ for Ca, cc-pVTZ-PP for Sn and 6-311++G** for all other atoms.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic experimental details, NMR spectra, X-ray analysis of compounds 3 and 4, details of the computational analysis and atomic coordinates of the DFT computed structures. CCDC 1951246 and 1951247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07289f |
| This journal is © The Royal Society of Chemistry 2019 |