
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-O-Benzyloxycarbonyl protected glycosyl donors: a revival of carbonate-mediated anchimeric assistance for diastereoselective glycosylation†
Julia
Weber
a,
Dennis
Svatunek
 a,
Simon
Krauter
ab,
Gregor
Tegl
ab,
Christian
Hametner
a,
Paul
Kosma
b and
Hannes
Mikula
*a
a,
Simon
Krauter
ab,
Gregor
Tegl
ab,
Christian
Hametner
a,
Paul
Kosma
b and
Hannes
Mikula
*a
aInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9/163, 1060 Vienna, Austria. E-mail: hannes.mikula@tuwien.ac.at
bDivision of Organic Chemistry, University of Natural Resources and Life Sciences, Muthgasse 18, Vienna (BOKU), 1190 Vienna, Austria
First published on 25th September 2019
Abstract
By reviving an old idea, we demonstrate that alkoxycarbonyl groups can be used in glycosylation reactions to achieve full stereocontrol through participation of a carbonate moiety at O-2. Various benzyloxycarbonyl-protected glycosyl donors were prepared and used for efficient 1,2-trans glycosylation of base-labile compounds and the synthesis of glycosyl esters.
A crucial step in the synthesis of glycoconjugates (e.g. glycosylated natural products) is the stereoselective formation of glycosidic linkages.1–4 The synthesis of 1,2-trans glycosides (e.g. β-glucosides) applying neighboring participating protective groups is well established,5–7 but still problematic considering the incompatibility of many natural products with reaction conditions usually applied for the removal of the most common participating groups, i.e. ester functionalities at O-2 of the glycosyl donor. Furthermore, often observed side reactions when using acyl-protected glycosyl donors such as orthoester formation8,9 and acyl transfer7,10 can result in low yields and necessitate lengthy purification of product mixtures.
Several methods have been reported that can be applied to avoid stability problems and side reactions, including chloroacetyl,11 levulinoyl,12 substituted phenylacetyl,13,14 modified benzoyl15–17 or substituted benzyl18 groups. The 2-O-picolinyl moiety, an arming participating group,19 and the 3-(2′-benzyloxyphenyl)-3,3-dimethylpropanoyl group20 can be cleaved by palladium-catalyzed hydrogenation, while the cyanopivaloyl group can be removed by reduction followed by mild base treatment.21 Further studies include the investigation of new strategies to influence the diastereoselectivity of a glycosylation reaction by applying a linker effect,22 remote protecting groups,23 specific promotor-systems,24 catalysts,4,25 or steric effects.26 However, all these methods have a limited scope in terms of diastereoselectivity, reactivity, and versatility of glycosylation and/or deprotection. Moreover, straight-forward preparation of the donor requires commercially available reagents for the introduction of the participating group.
We aimed to use protecting groups that can be selectively removed under mild reaction conditions compatible with base-labile natural products (e.g. compounds containing ester functionalities), and thus furthermore enable the synthesis of glycosyl esters. To this end, we focused on the application of 2-O-alkyloxycarbonyl-protected donors for diastereoselective glycosylation through anchimeric assistance of the carbonate moiety, while deprotection can be achieved by cleavage of the alkyl substituent leading to the formation of an unstable intermediate that spontaneously releases CO2 affording the final product (Fig. 1a).
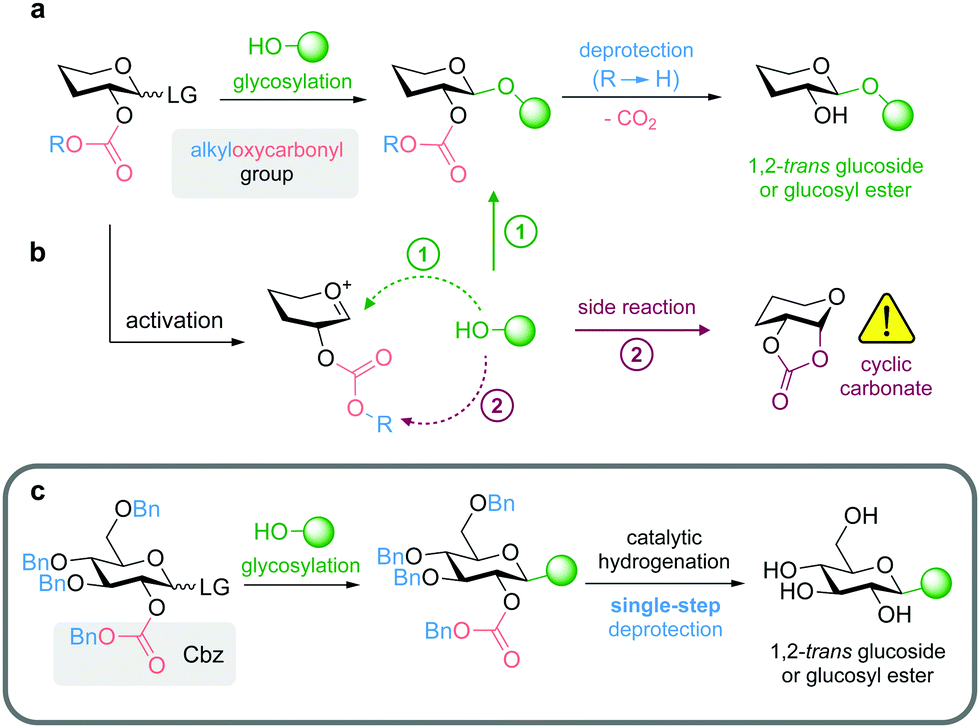 | ||
| Fig. 1 (a) Diastereoselective 1,2-trans glycosylation mediated via neighboring group participation of a 2-O-alkyloxycarbonyl group; (b) in competition to glycosylation ① formation of cyclic carbonates has been reported as undesired side reaction ②; (c) 2-O-Cbz-3,4,6-tri-O-Bn protected donors for the synthesis of glucosides of base-labile products and glucosyl esters; LG = leaving group. | ||
So far, alkyloxycarbonyl groups have found only limited application as participating moieties in glycosylation reactions, although the respective chloroformate reagents (e.g. Cbz-Cl, Alloc-Cl, Fmoc-Cl) are readily available. In the studies of Gentil et al.27 already published in 1990 and Morère et al.28 more than a decade ago, several drawbacks such as formation of cyclic carbonates (Fig. 1b) or the need for mercury salts as promotors of unstable glycosyl bromides have been reported. While Calosso et al.29 achieved diastereoselective synthesis of 1,2-trans glycosides (two examples with yields of approximately 60%) using 2-O-Cbz protection, Ingram and Desoky reported only mixed results in their PhD theses, showing examples of successful glycosylation using 2-O-Cbz-protected acetimidoyl donors, but mainly observed the formation of cyclic carbonates (Fig. 1b), especially in case of 2-O-Cbz-protected thioglycosyl donors.30,31 Further examples reported in the literature include the 2-O-methylsulfonylethoxycarbonyl group (which is only semiorthogonal to ester functionalities and requires a two-step procedure for deprotection under basic conditions)32 and 2-O-Fmoc-protected mannosyl donors.33,34
We focused on using 2-O-benzyloxycarbonyl (Cbz) protected glycosyl donors in combination with benzyl (Bn)-protection of the remaining OH-functionalities to obtain a protecting group pattern that enables mild deprotection in a single step (Fig. 1c).
Due to the excellent properties of thioglycosides, including versatile methods for selective activation and high stability,6 we considered different thiols as leaving groups (SEt, STol, STaz, SPym) for the development of 2-O-Cbz glycosyl donors. Starting from the thioorthoester 135 the 2-O-acetyl protected intermediates 2–5 were prepared followed by deprotection of the acetyl group to obtain the corresponding 2-OH thioglucosides 6–9. Alternatively, DMDO (dimethyldioxirane) epoxidation of 3,4,6-tri-O-benzyl-D-glucal (10) and subsequent nucleophilic addition of the corresponding thiol directly afforded 6 and 7.36 Introduction of the Cbz group was accomplished by reaction with Cbz-Cl in the presence of TMEDA (Fig. 2).37 Noteworthy, we did not observe full conversion of the starting material even after addition of further reagents and/or longer reaction times. However, based on recovered starting material, yields of 77–90% were achieved (see ESI†). Other previously reported methods did not lead to any product formation, including the use of pyridine, triethylamine or DMAP as bases in various solvents.38
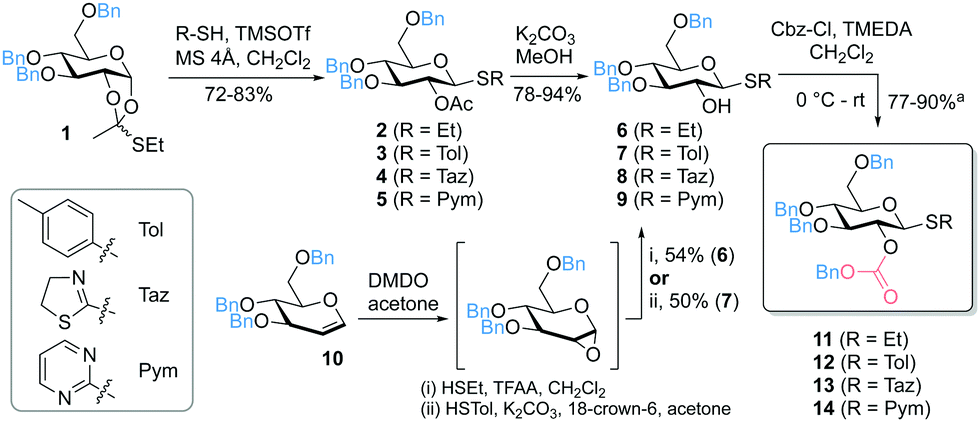 | ||
Fig. 2 Synthesis of 2-O-benzyloxycarbonyl protected thioglucosides (TMSOTf = trimethylsilyl trifluoromethanesulfonate, MS = molecular sieves, TMEDA = N,N,N′,N′-tetramethylethylene diamine, DMDO = dimethyldioxirane); a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) based on recovered starting material. based on recovered starting material. | ||
In addition, 2-O-benzyloxycarbonyl protected glucosyl imidates were prepared that can be activated using a catalytic amount of Lewis acid as promotor. Starting from thioglucoside 11, the anomeric position was modified to obtain 1-hydroxysugar 15. The attempt to prepare the trichloroacetimidate 16 by DBU-catalyzed addition of the anomeric hydroxyl group to Cl3CCN mainly resulted in the formation of cyclic carbonate 17 (Fig. 3). In an alternative approach, glucosyl imidate 18 could successfully be prepared with a yield of 81% by using N-phenyltrifluoroacetimidoyl chloride (ClC(NPh)CF3).
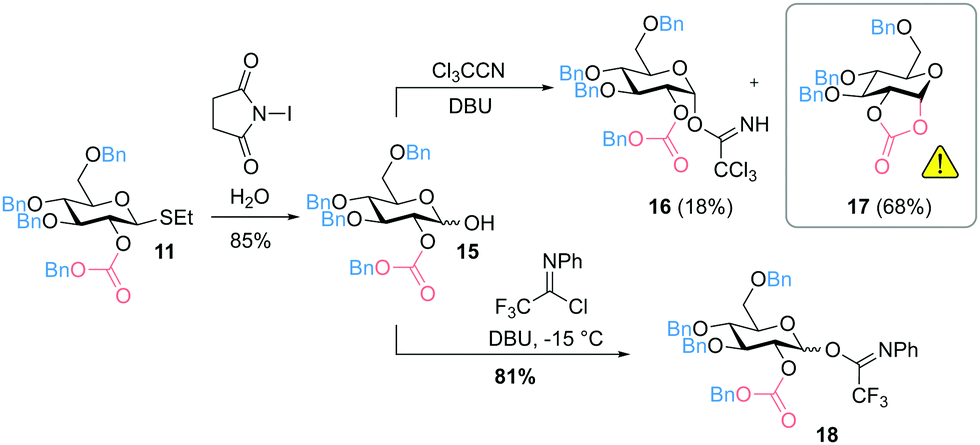 | ||
| Fig. 3 Synthesis of 2-O-benzyloxycarbonyl protected glucosyl imidates (DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene). | ||
The results of selected glycosylation reactions are shown in Table 1. First experiments were carried out using 2-phenylethanol (19) as a simple model compound for a primary alcohol (Table 1, entries 1–7). Activation of the SEt donor 11 and STol donor 12 with NIS (N-iodosuccinimide) and TfOH (trifluoromethanesulfonic acid) yielded the respective 1,2-trans glucoside 24 in almost quantitative yield with complete β-diastereoselectivity, while the cyclic carbonate 17 was not detected (Table 1, entries 1 and 3). When activating 11 under milder conditions using iodine, we observed favored formation of 17 as undesired side reaction and only a low yield of the product 24 (Table 1, entry 2). Thioglucoside 13 (STaz) turned out to be the most labile reagent during storage and showed the lowest yields in glycosylation reactions (e.g.Table 1, entry 4). In contrast, the SPym donor 14 was stable at −20 °C for at least 12 months and could be efficiently activated with AgOTf for the glycosylation of 19 affording 24 in 97% yield (Table 1, entry 5). Noteworthy, activation of 13 or 14 with the strong Lewis acid TMSOTf (trimethylsilyl trifluoromethanesulfonate) resulted in lower yields (as shown for 14, Table 1, entry 6). The imidate donor 18 could successfully be used for the synthesis of 24 in 93% yield, but in contrast to most reactions with the thioglucosides 11–14 we have observed formation of the cyclic carbonate 17, even though less than 10% (Table 1, entry 7). Glucosylation of the benzyl-protected glycosyl acceptor 20 gave results similar to the model compound 19 (Table 1, entries 8–12). The SEt donor 11 (activated with NIS/TfOH) and the SPym donor 14 (activated with AgOTf) performed best throughout this study. Both methods are compatible with base-labile and potentially migrating acetyl-groups, e.g. as shown for the glycosylation of the acetyl-protected glycosyl acceptor 21 using the SEt donor 11 (Table 1, entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Acceptor | Activation | Temp. (°C) | Yielda (productb/17) |
| a Determined by HPLC after 3 h, or * after 24 h;. b β/α > 99/1 (no α detected); NIS = N-iodosuccinimide, TfOH = trifluoromethanesulfonic acid, n.d. = not detected. | |||||
| 1 | 11 (SEt) | 19 (1.5) | NIS/TfOH | −10 | >99% (24)/n.d. |
| 2 | 11 (SEt) | 19 (1.5) | I2 | 20 | 22%* (24)/46% (17) |
| 3 | 12 (STol) | 19 (1.5) | NIS/TfOH | −10 | 98% (24)/n.d. |
| 4 | 13 (STaz) | 19 (1.5) | AgOTf | 20 | 71% (24)/n.d. |
| 5 | 14 (SPym) | 19 (1.5) | AgOTf | 0 | 97% (24)/n.d. |
| 6 | 14 (SPym) | 19 (1.5) | TMSOTf | −10 | 61% (24)/n.d. |
| 7 | 18 (imidate) | 19 (1.5) | TMSOTf | −10 | 93% (24)/6% (17) |
| 8 | 11 (SEt) | 20 (1.0) | NIS/TfOH | −10 | >99% (25)/n.d. |
| 9 | 12 (STol) | 20 (1.5) | NIS/TfOH | −10 | 97% (25)/n.d. |
| 10 | 13 (STaz) | 20 (1.5) | AgOTf | 20 | 59% (25)/n.d. |
| 11 | 14 (SPym) | 20 (1.0) | AgOTf | 0 | 98% (25)/n.d. |
| 12 | 18 (imidate) | 20 (1.5) | TMSOTf | −10 | 95% (25)/4% (17) |
| 13 | 11 (SEt) | 21 (1.5) | NIS/TfOH | −10 | >99% (26)/n.d. |
| 14 | 14 (SPym) | 22 (1.5) | AgOTf | 0 | 68% (27)/32% (17) |
| 15 | 11 (SEt) | 23 (1.5) | NIS/TfOH | −10 | 88% (28)/12% (17) |
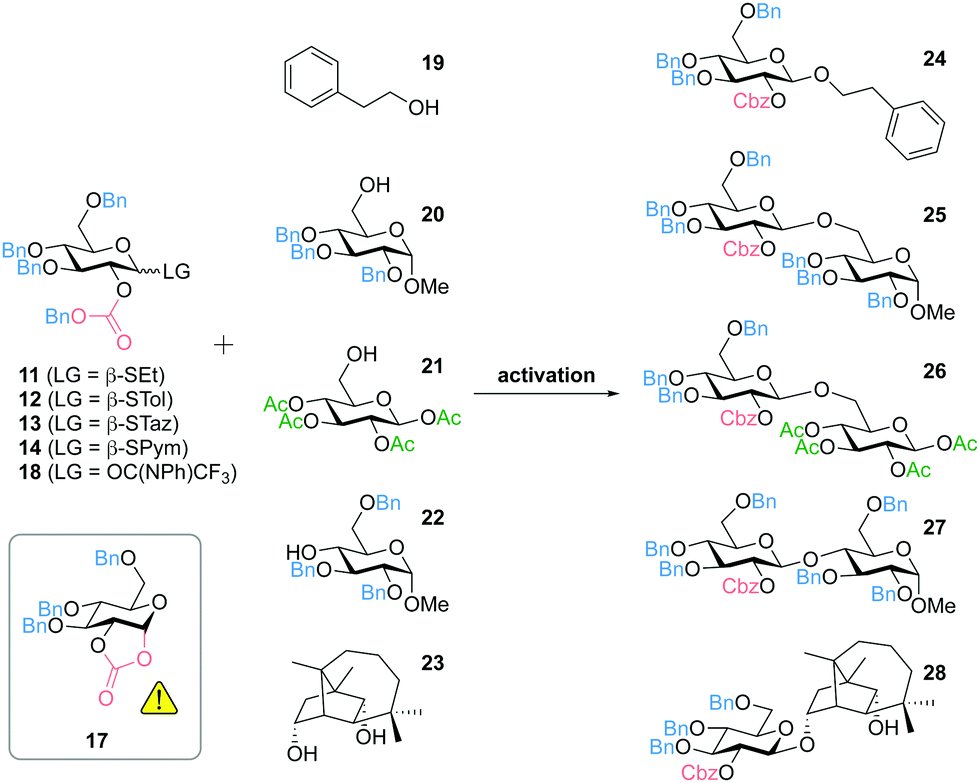
When using sterically more hindered secondary alcohols as acceptors we have observed increased formation of the cyclic carbonate 17 indicating the scope of the use of Cbz for diastereoselective glycosylation. Nevertheless, we have been able to prepare the 1,4-linked disaccharide 27 by glycosylation of the 2,3,6-tri-O-benzyl-protected glycosyl acceptor 22 in 68% yield using the SPym donor 14 (Table 1, entry 14). Moreover, we have recently reported the first application of our 2-O-Cbz protected donors for the diastereoselective glycosylation of natural products. The mycotoxin culmorin (23) was reacted with the SEt donor 11 (activated with NIS/TfOH) regioselectively affording the desired product 28 (88% by HPLC, 80% isolated yield) with a ratio to the cyclic carbonate 17 of approximately 7/1 (Table 1, entry 15). Noteworthy, this glycosylation could not be achieved by using acetyl-protected donors due to acyl transfer.39
Moreover, the used combination of Cbz and Bn protective groups enables single-step deprotection compatible with base-labile compounds. For example, simultaneous cleavage of Cbz and Bn groups of glucoside 24 and disaccharide 26 applying palladium-catalyzed hydrogenation was carried out under mild reaction conditions to obtain 2-phenylethyl β-D-glucoside (29) and 1,2,3,4-tetra-O-acetyl-gentiobioside (30), respectively, showing that acetyl groups are not affected (Fig. 4). Complete orthogonality was shown by selective cleavage of the acetyl groups of the disaccharide 26 under mild basic conditions using potassium cyanide in methanol to obtain 31 (Fig. 4b).
 | ||
| Fig. 4 (a) Simultaneous removal of Cbz and Bn groups affording β-D-glucoside 29; (b) selective single-step deprotection in the presence of ester functionalities, and orthogonal cleavage of acetyl groups. | ||
In addition to the glycosylation of alcohol functionalities, we focused on the diastereoselective synthesis of glycosyl esters, in particular C-terminal glycosylated peptides, as an even more labile class of compounds not compatible with deprotection under basic conditions. Such peptide-carbohydrate adducts play an important role for the improvement of the physicochemical properties of peptide pharmaceuticals.40 To mimic the glycosyl ester bond formation between a peptide and a sugar moiety, we used trans-N-(tert-butoxycarbonyl)-4-acetoxy-L-proline (32) as a model compound. Glycosylation with glucosyl donor 11 afforded the benzyl-protected glycosyl ester 33 with complete β-diastereoselectivity. Deprotection was achieved by palladium-catalyzed hydrogenation to obtain glycosyl ester 34 without observing any cleavage or migration of the acyl group (Fig. 5a). Furthermore, the β-glucosyl ester 37, a precursor of an aspirin prodrug,41 was prepared using donor 11 for glycosylation of acetylsalicylic acid (35) (Fig. 5b), showing compatibility with even more labile phenolic ester functionalities (e.g. no transesterification was observed during catalytic hydrogenation in ethanol).
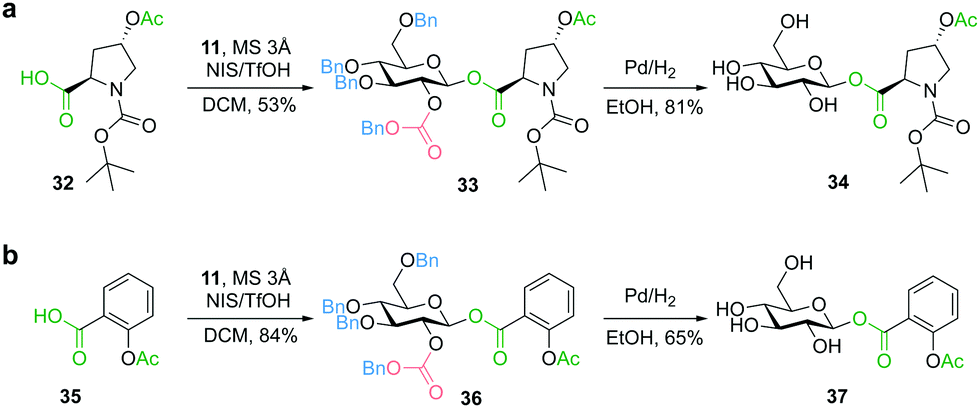 | ||
| Fig. 5 Preparation of glycosyl esters: (a) diastereoselective synthesis of an amino acid glucosyl ester as a model for C-terminal glycosylation of peptides; (b) compatibility with phenolic ester functionalities. | ||
In summary, the synthesis and application of several 3,4,6-tri-O-benzyl-2-O-Cbz protected thioglucosides and glucosyl imidates as diastereoselective glycosyl donors is presented. We show that Cbz can be efficiently used for the 1,2-trans glycosylation of several acceptors followed by deprotection via catalytic hydrogenation not affecting ester functionalities. Even though we have observed formation of cyclic carbonates as a side reaction when using secondary alcohols, the developed strategy is particularly suitable for the glycosylation of base-labile compounds and the synthesis of glycosyl esters. Hence, we are convinced that the presented concept will be useful for the development of several strategies expanding the glycosylation tool box.
The authors thank the Austrian Research Promotion Agency (FFG) for financial support (BRIDGE-Project 843488).
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. van der Vorm, T. Hansen, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Sci., 2017, 8, 1867–1875 RSC.
- A. M. Downey, R. Pohl, J. Roithová and M. Hocek, Chem. – Eur. J., 2017, 23, 3910–3917 CrossRef CAS PubMed.
- T. Fang, Y. Gu, W. Huang and G.-J. Boons, J. Am. Chem. Soc., 2016, 138, 3002–3011 CrossRef CAS PubMed.
- H. Yao, S. Zhang, W.-L. Leng, M.-L. Leow, S. Xiang, J. He, H. Liao, K. L. M. Hoang and X.-W. Liu, ACS Catal., 2017, 7, 5456–5460 CrossRef CAS.
- J.-H. Kim, H. Yang, J. Park and G.-J. Boons, J. Am. Chem. Soc., 2005, 127, 12090–12097 CrossRef CAS PubMed.
- A. V. Demchenko, Handbook of Chemical Glycosylation, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- T. Nukada, A. Berces, M. Z. Zgierski and D. M. Whitfield, J. Am. Chem. Soc., 1998, 120, 13291–13295 CrossRef CAS.
- P. Fruhmann, B. Warth, C. Hametner, F. Berthiller, E. Horkel, G. Adam, M. Sulyok, R. Krska and J. Fröhlich, World Mycotoxin J., 2012, 5, 127–132 CrossRef CAS.
- N. W. McGill and S. J. Williams, J. Org. Chem., 2009, 74, 9388–9398 CrossRef CAS PubMed.
- T. Nukada, A. Berces and D. M. Whitfield, J. Org. Chem., 1999, 64, 9030–9045 CrossRef CAS.
- G. Gu, M. Fang and Y. Du, Carbohydr. Res., 2011, 346, 2801–2804 CrossRef CAS PubMed.
- J. Xiong, Z. Lu, N. Ding, S. Ren and Y. Li, Eur. J. Org. Chem., 2013, 6158–6166 CrossRef CAS.
- K. Daragics and P. Fügedi, Org. Lett., 2010, 12, 2076–2079 CrossRef CAS PubMed.
- E. Arranz and G.-J. Boons, Tetrahedron Lett., 2001, 42, 6469–6471 CrossRef CAS.
- K. R. Love, R. B. Andrade and P. H. Seeberger, J. Org. Chem., 2001, 66, 8165–8176 CrossRef CAS PubMed.
- J.-M. Vatèle, Tetrahedron Lett., 2005, 46, 2299–2301 CrossRef.
- T. Wada, A. Ohkubo, A. Mochizuki and M. Sekine, Tetrahedron Lett., 2001, 42, 1069–1072 CrossRef CAS.
- S. Buda, P. Gołębiowska and J. Mlynarski, Eur. J. Org. Chem., 2013, 3988–3991 CrossRef CAS.
- J. T. Smoot, P. Pornsuriyasak and A. V. Demchenko, Angew. Chem., Int. Ed., 2005, 44, 7123–7126 CrossRef CAS PubMed.
- D. Crich and F. Cai, Org. Lett., 2007, 9, 1613–1615 CrossRef CAS PubMed.
- A. Geert Volbeda, N. R. M. Reintjens, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Eur. J. Org. Chem., 2016, 5282–5293 CrossRef.
- P. Pornsuriyasak, X. G. Jia, S. Kaeothip and A. V. Demchenko, Org. Lett., 2016, 18, 2316–2319 CrossRef CAS PubMed.
- J. P. Yasomanee and A. V. Demchenko, J. Am. Chem. Soc., 2012, 134, 20097–20102 CrossRef CAS PubMed.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162 CrossRef CAS PubMed.
- C. M. Pedersen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2007, 129, 9222–9235 CrossRef CAS PubMed.
- E. Gentil, M. Potier, P. Boullanger and G. Descotes, Carbohydr. Res., 1990, 197, 75–91 CrossRef CAS.
- A. Morère, F. Mouffouk, S. Leiris, A. Leydet and J. L. Montero, J. Carbohydr. Chem., 2006, 25, 451–459 CrossRef.
- M. Calosso, D. Charpentier, M. Vaillancourt, M. Bencheqroun, G. St-Pierre, B. C. Wilkes and Y. Guindon, ACS Med. Chem. Lett., 2012, 3, 1045–1049 CrossRef CAS PubMed.
- L. J. Ingram, The Synthesis of Sulfated Carbohydrates Using a Sulfate Protecting Group Strategy, University of Waterloo, Canada, 2010, http://hdl.handle.net/10012/5437 Search PubMed.
- A. Desoky, Synthesis of Sulfated Carbohydrates Using Sulfuryl Imidazolium Salts, University of Waterloo, Canada, 2010, http://hdl.handle.net/10012/5635 Search PubMed.
- A. Ali, R. J. B. H. N. van den Berg, H. S. Overkleeft, D. V. Filippov, G. A. van der Marel and J. D. C. Codée, Tetrahedron Lett., 2009, 50, 2185–2188 CrossRef CAS.
- M. Delbianco, A. Kononov, A. Poveda, Y. Yu, T. Diercks, J. Jiménez-Barbero and P. H. Seeberger, J. Am. Chem. Soc., 2018, 140, 5421–5426 CrossRef CAS PubMed.
- I. A. Gagarinov, T. Li, J. S. Toraño, T. Caval, A. D. Srivastava, J. A. W. Kruijtzer, A. J. R. Heck and G.-J. Boons, J. Am. Chem. Soc., 2017, 139, 1011–1018 CrossRef CAS PubMed.
- K. Plé, M. Chwalek and L. Voutquenne-Nazabadioko, Tetrahedron, 2005, 61, 4347–4362 CrossRef.
- S. J. Danishefsky, S. Hu, P. F. Cirillo, M. Eckhardt and P. H. Seeberger, Chem. – Eur. J., 1997, 3, 1617–1628 CrossRef CAS.
- M. Adinolfi, G. Barone, L. Guariniello and A. Iadonisi, Tetrahedron Lett., 2000, 41, 9305–9309 CrossRef CAS.
- P. I. Dosa, T. Ward, R. E. Castro, C. M. P. Rodrigues and C. J. Steer, ChemMedChem, 2013, 8, 1002–1011 CrossRef CAS PubMed.
- J. Weber, M. Vaclavikova, G. Wiesenberger, M. Haider, C. Hametner, J. Fröhlich, F. Berthiller, G. Adam, H. Mikula and P. Fruhmann, Org. Biomol. Chem., 2018, 16, 2043–2048 RSC.
- I. Jerić and Š. Horvat, Eur. J. Org. Chem., 2001, 1533–1539 CrossRef.
- A. A. Hussain, H. B. Kostenbauder and J. E. Truelove, J. Pharm. Sci., 1980, 69, 231–232 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthetic procedures, compound characterization. See DOI: 10.1039/c9cc07194f |
| This journal is © The Royal Society of Chemistry 2019 |