
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On-surface synthesis of polyazulene with 2,6-connectivity†
Qiang
Sun‡
 a,
Ian Cheng-Yi
Hou‡
b,
Kristjan
Eimre
a,
Carlo A.
Pignedoli
a,
Pascal
Ruffieux
a,
Akimitsu
Narita
*bc and
Roman
Fasel
*ad
a,
Ian Cheng-Yi
Hou‡
b,
Kristjan
Eimre
a,
Carlo A.
Pignedoli
a,
Pascal
Ruffieux
a,
Akimitsu
Narita
*bc and
Roman
Fasel
*ad
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland. E-mail: Roman.Fasel@empa.ch
bMax Planck Institute for Polymer Research, 55128 Mainz, Germany. E-mail: narita@mpip-mainz.mpg.de
cOrganic and Carbon Nanomaterials Unit, Okinawa Institute of Science and Technology Graduate University, 1919-1 Tancha, Onna-son, Kunigami, Okinawa 904-0495, Japan
dDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland
First published on 17th October 2019
Abstract
Azulene, the smallest neutral nonalternant aromatic hydrocarbon, serves not only as a prototype for fundamental studies but also as a versatile building block for functional materials because of its unique opto(electronic) properties. Here, we report the on-surface synthesis and characterization of the homopolymer of azulene connected exclusively at the 2,6-positions using 2,6-diiodoazulene as the monomer precursor. As an intermediate to the formation of polyazulene, a gold-(2,6-azulenylene) chain is observed.
Properties of carbon-based aromatic systems are sensitively determined by their bond topologies.1 So far, much attention has been paid to the design and synthesis of aromatic materials like conjugated polymers and nanographenes constituted by alternant hydrocarbons, which do not possess odd-membered rings. In contrast, incorporation of non-alternant hydrocarbons has only rarely been explored. The electronic and optical properties of alternant and non-alternant hydrocarbons differ profoundly. Azulene (Scheme 1), for example, an aromatic hydrocarbon containing 10 π-electrons, has several characteristics that differ from its isomer naphthalene.2 Azulene has an intrinsic dipole moment of 1.08 D,3 while naphthalene is non-polar. The dipole moment of azulene arises from an unequal distribution of electron density between its electron-deficient 7-membered ring and electron-rich 5-membered ring due to an aromatic stabilization according to Hückel's rule. In addition, azulene exhibits “anomalous” fluorescence in its second excited singlet state in violation of Kasha's rule,4 which makes it a promising candidate in optoelectronics.5
 | ||
| Scheme 1 (a) The chemical structure of azulene with carbon atom numbering. (b) On-surface synthetic route toward 2,6-polyazulene. | ||
Because of its unique electronic and optical properties, azulene has been employed as a core structure of functional materials for different applications such as stimuli-responsive materials,6 organic field-effect transistors,7 solar cells8 and others.9 The connectivity of azulenylene units (Scheme 1a) in the derived structures has a substantial influence on their optical and electronic properties.10 One remarkable example is that the lowest unoccupied molecular orbital (LUMO) of 2,6′:2′,6′′-terazulene is fully delocalized over the whole molecule, showing strong bonding between the azulene moieties, resulting in a good n-type semiconductor performance.11 This motivates the study of 2,6-polyazulene. However, despite a number of theoretical studies on 2,6-polyazulenes, reports on the synthesis of azulene-based polymers predominantly focus on the incorporation of 1,3-azulenylenes.12 Recent studies showed that integration of 2,6-azulenylenes into copolymers has great potential for field-effect transistors and proton-conducting materials.7b,13 Nevertheless, the synthesis of azulene homopolymers with 2,6-connectivity remains elusive.
On-surface synthesis has recently developed as a complementary strategy for chemical synthesis. A significant number of structures/materials which are challenging by conventional solution chemistry have been obtained by this method.14 Due to its potentially interesting properties, 2,6-polyazulene is starting to gain attention from the community of on-surface synthesis.15 Here, we report the on-surface synthesis of polyazulene with exclusive 2,6-connectivity using 2,6-diiodoazulene as the molecular precursor (Scheme 1b). The electronic properties of the synthesized polyazulene are investigated by scanning tunneling spectroscopy (STS) and density functional theory (DFT) calculations. The electronic gap of the polyazulene is determined to be 1.8 eV on Au(111), which is further supported by differential conductance dI/dV mapping. This work demonstrates not only the synthesis but also the first detailed electronic characterization of azulene homopolymers with pure 2,6-connectivity.
2,6-Diiodoazulene was synthesized following a previous work.16 To access the pristine 2,6-diiodoazulene molecules, we first deposit the molecule under ultrahigh vacuum conditions onto an Au(111) surface maintained at 160 K to avoid possible deiodination that would occur at room temperature.17 As shown in the scanning tunneling microscopy (STM) images (Fig. 1a and b), 2,6-diiodoazulene forms porous structures on Au(111). A zoom-in image reveals the monomers appearing as rod-shaped structures with brighter iodine protrusions at both ends according to the chemical model displayed in Fig. 1b. Note that it is not possible to differentiate the 7- and 5-membered rings and the exact orientation of azulenes solely based on the STM images. After annealing the sample to 350 K, a chain structure is observed on the surface (Fig. 1c and d). The molecular chains are composed of two alternating subunits (Fig. 1d), assigned to 2,6-azulenylene moieties and to gold adatoms, respectively. We superimpose an equally scaled chemical structure of a chain segment onto a small-scale STM image in Fig. 1d, which shows a good match. It is not surprising that gold atoms are involved in nanostructures obtained upon dehalogenation of molecules on Au substrates, and there are many reports on organo-metallic species on Au(111).18 The detached iodine atoms are detected as round protrusions staying aside the organo-metallic chains (indicated by a blue arrow in Fig. 1d), forming some honeycomb-type structure as shown on the left side of Fig. 1c. This is consistent with the fact that iodine desorption from Au(111) starts only at around 540 K.17 Closer inspection of the structures formed at 350 K reveals that a few 2,6-connected oligoazulenes without the involvement of Au atoms have already been formed (one of them is indicated by a green arrow in Fig. 1d). The one-dimensional organometallic chains are straight due to the formation of the C–Au–C organo-metallic motifs as aryl radicals on Au(111).18 Further annealing the sample to 580 K triggers the release of all Au atoms from the organo-metallic chains and the formation of the carbon–carbon bonded polymer, as well as partial desorption of iodines (Fig. 1e and f). A high-resolution STM image of the polymer reveals that the azulene units are connected along their long axis, implying the formation of 2,6-connected polyazulene.
 | ||
| Fig. 1 (a and b) STM images after deposition of 2,6-diiodoazulene on Au(111) held at 160 K. Chemical structures of four molecules are overlaid on the corresponding STM image in (b). (c and d) STM images of the sample after annealing to 350 K. The inset in (d) highlights the organo-metallic chain. The blue arrow indicates a detached iodine atom. The green arrow indicates oligomeric 2,6-connected azulene segments. (e and f) STM images after annealing the sample to 580 K. A scaled chemical model of a 2,6-oligoazulene is overlaid on a chain in (f). Scanning parameters: (a) Vs = −1 V, It = 0.09 nA; (b) Vs = −0.02 V, It = 0.25 nA; (c) Vs = −1 V, It = 0.06 nA; (d) Vs = −0.005 V, It = 0.1 nA; (e) Vs = −1 V, It = 0.08 nA; (f) Vs = −0.01 V, It = 1 nA. | ||
To further support the formation of polyazulene and determine the connectivity between azulene units, we resort to bond-resolved non-contact atomic force microscopy (nc-AFM).19 As shown in Fig. 2a, the structure of the polymer can be clearly resolved, with the 5- and 7-membered rings being imaged with different sizes. Typical defects in the straight 2,6-connected polyazulene are kinks (cf. Fig. S1, ESI†) arising from 1,6-connected azulene units, which are attributed to a small amount of impurity from the precursor16 or to a 2,1-sigmatropic rearrangement. Within a 2,6-polyazulene chain, the neighboring 2,6-azulenylenes can be connected in three different ways, namely pentagon vs. heptagon (p–h), pentagon vs. pentagon (p–p), or heptagon vs. heptagon (h–h), which correspond to 2,6-, 2,2- and 6,6-connectivity, respectively (see the arrows in Fig. 2a). The h–h-connectivity is the most easily recognized, since it results in a non-zero dihedral angle between the neighboring azulenylene units due to the steric hindrance between the hydrogen atoms at 5,7-positions of azulenylene, which is clearly reflected in the nc-AFM images. High resolution images however also allow identification of p–h- and p–p-connectivity. An interesting aspect about the reaction between azulenylenes is whether there is any preference for the p–h-connectivity, since azulene has an intrinsic dipole moment of 1.08 D which might direct the polymerization process via dipole–dipole interactions. To this end, we have performed a statistical analysis of the linkages between the neighboring azulenylenes. It turns out that the ratio between p–h-, p–p- and h–h-linkages is 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (Fig. 2b), which indicates a non-selective, random connection between the 5- and 7-membered rings (see discussion in Fig. S2, ESI†). Thus, the intrinsic dipole moment of pristine azulene does not play a role in orienting the azulenylenes during on-surface polymerization. We attribute this to the screening effect of the electrons of the metal which produces an image dipole, with the resulting dipolar interaction being inconsequentially small for molecular dipoles oriented parallel to the surface.20 A possible way to align the azulenylenes and to obtain only p–h linkages may thus be through the use of an insulating substrate.
:1:1 (Fig. 2b), which indicates a non-selective, random connection between the 5- and 7-membered rings (see discussion in Fig. S2, ESI†). Thus, the intrinsic dipole moment of pristine azulene does not play a role in orienting the azulenylenes during on-surface polymerization. We attribute this to the screening effect of the electrons of the metal which produces an image dipole, with the resulting dipolar interaction being inconsequentially small for molecular dipoles oriented parallel to the surface.20 A possible way to align the azulenylenes and to obtain only p–h linkages may thus be through the use of an insulating substrate.
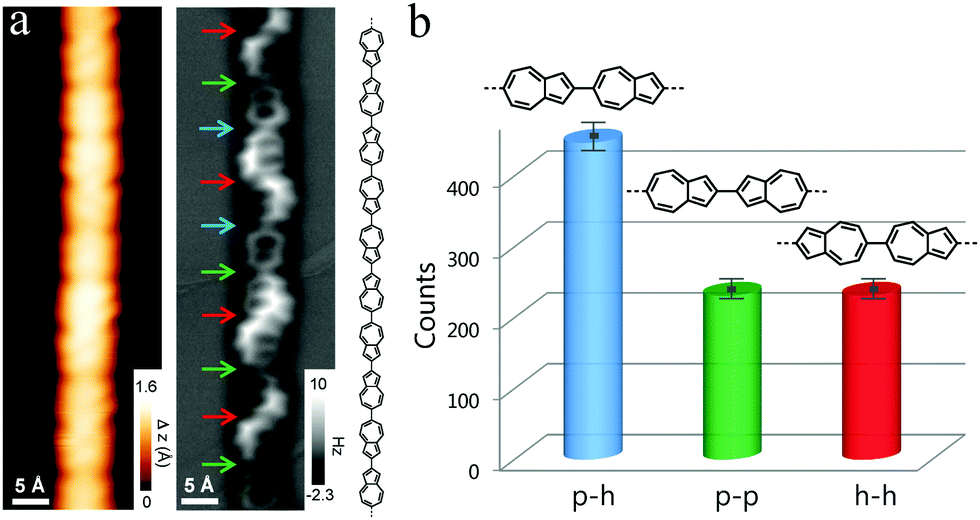 | ||
| Fig. 2 (a) High-resolution STM image of a 2,6-polyazulene chain (Vs = −0.1 V, It = 0.3 nA) and the corresponding nc-AFM image (Vs = −5 mV, Oscillation amplitude: ∼80 pm). The pentagon–heptagon (p–h), pentagon–pentagon (p–p) and heptagon–heptagon (h–h) linkages are indicated by blue, green and red arrows, respectively. The corresponding chemical model of the chain is also shown. (b) Statistical analysis of the different linkages within the polymers. | ||
To study the electronic properties of 2,6-polyazulene, we first carry out DFT calculations of an azulene molecule and polyazulene. Fig. 3a shows the frontier orbitals from HOMO−2 to LUMO+1 of azulene, and Fig. 3b shows the band structure of the 2,6-polyazulene originating from these orbitals. The bands originating from the highest occupied molecular orbital (HOMO), HOMO−2 and LUMO+1 are relatively flat due to the orbitals having low amplitudes at the 2,6-positions where azulene units are connected. In contrast, the bands originating from HOMO−1 and LUMO show significant dispersion due to a considerable orbital overlap between the neighboring azulenylenes. To further investigate how the random distribution of p–h-, p–p- and h–h-linkages affects the electronic structure, we perform band structure calculations for three different structures consisting of a supercell with four azulene units. The first structure with only the p–h-linkage between all the azulenylenes reproduces the band structure of 2,6-polyazulene (Fig. 3c), but folded to a four times smaller Brillouin zone. The second structure (star) in Fig. 3c has alternating p–p- and h–h-linkages, and the third structure (diamond) has every third unit flipped compared to the others. Besides a splitting of the bands due to lifting of degeneracy near k-vectors corresponding to the modified periodicity, the band structures near the valence band (VB) and conduction band (CB) onsets do not differ much from each other. All of the three structures have an electronic band gap of 0.94 ± 0.03 eV, and their frontier bands display similar dispersions. This is in line with the fact that the molecular orbitals of azulene possess no weight at 2,6-positions for HOMO−2 and HOMO but considerable weight for HOMO−1 and LUMO (Fig. 3a). Although three different linkages are randomly distributed within our experimentally obtained polymers, the electronic properties of the synthesized polyazulene shall remain very similar to the ones of the perfectly regular poly(2,6-azulenylene).
 | ||
| Fig. 3 Electronic structure of azulene and polyazulenes with 2,6-connectivity. (a) The shape of frontier orbitals of an azulene molecule. (b) Band structure of 2,6-polyazulene and the orbital shapes of the frontier bands which evolve from the corresponding molecular orbital of azulene shown in (a). (c) Band structures of three polyazulenes with different connectivity patterns in a supercell containing four 2,6-azulenylenes with all units oriented in the same way (triangle); first and third unit flipped (star); and only third unit flipped (rhombus). The green arrows show two examples of degeneracy liftings caused by the flipped units. In all band structure plots, the top of the valence band is taken as zero energy, the orange lines are occupied bands while the blue lines correspond to the unoccupied bands. In all orbital plots wavefunction isosurfaces at ±0.03 Å−3/2 are shown. a is the length of one azulene unit within the homopolymer. | ||
We have used dI/dV spectroscopy to experimentally characterize the electronic properties. The point spectra of the polymer reveal two peaks at around −1.25 V and 0.6 V, which correspond to its VB and CB onsets, respectively (Fig. 4a and b). To verify the assignment of the molecular orbitals, we have performed dI/dV mapping at two corresponding bias voltages (Fig. 4c). The dI/dV maps show clear patterns confirming their origin from molecular orbitals. Moreover, the position of the h–h-linkage has a higher contrast in dI/dV mapping of the CB onset, which is highlighted by the white rectangle in Fig. 4. This feature could also be seen in the constant-current STM image at a bias voltage of 0.6 V (Fig. S3, ESI†), and is consistent with the fact that the LUMO of an azulene monomer is mainly located at the 7-membered ring (see also Fig. 3a).12a To further support our experimental findings, we determine the electronic properties of the experimentally observed polyazulene segment shown in Fig. 4a by DFT calculations (Fig. S4, ESI†). The DFT calculated local density of states (LDOS) maps at the VB and CB onsets (i.e., HOMO and LUMO) are in good agreement with the experimental dI/dV maps at both negative and positive biases (Fig. 4c), and thus confirm that the peaks at −1.25 V and 0.6 V in Fig. 4b originate from the VB and CB onsets. Note that the energy positions of the DFT computed LDOS are determined from the calculated DOS shown in Fig. S4 (ESI†). Notably, the h–h-linkage displays characteristic contrast in both DFT calculated maps, and appears particularly bright in the experimental map (highlighted by dashed rectangles).
 | ||
| Fig. 4 (a) STM image of a polyazulene segment (Vs = −1.5 V, It = 0.9 nA) and (b) its differential conductance dI/dV spectra. The spectra were taken at the locations marked by crosses with corresponding colors in (a). The grey spectrum has been obtained on a clean Au(111) area nearby. Spectra are vertically offset for clarity. (c) Constant-current dI/dV maps of the HOMO and LUMO, and the corresponding DFT-calculated LDOS maps. The h–h-linkage which shows a higher contrast is highlighted by dashed rectangles in both dI/dV and LDOS maps. | ||
The frontier states (i.e., the first occupied and unoccupied states) of the polymer are positioned symmetrically with respect to a negative bias voltage of around −0.3 V (Fig. 4b). In contrast, the frontier states of most carbon nanowires/ribbons composed of alternant (poly)benzenoids are positioned symmetrically to positive bias voltages on Au(111).21 This is due to the systems having different valence and conduction band alignments with respect to the vacuum level, which determines the corresponding positioning with respect to the Au Fermi level upon adsorption for weakly interacting systems. This is reflected in our DFT calculations, which demonstrate that for polyazulene the VB and CB onsets are respectively at −5.0 eV and −4.1 eV with respect to the vacuum level. In contrast, the band onsets for armchair graphene nanoribbons of width 7 (7AGNR) are found at −4.7 eV and −3.2 eV (Fig. S5, ESI†).
In conclusion, the homopolymer of azulene with exclusive 2,6-connectivity has been synthesized by dehalogenative coupling of 2,6-diiodoazulene on Au(111). Although azulene has an intrinsic dipole moment, dipole–dipole interactions do not yield a preferential pentagon–heptagon-linkage along the polymer. However, the electronic properties of 2,6-polyazulene do not significantly depend on the ratio of the p–p, p–h and h–h linkages. STS yields an electronic gap of 1.8 eV for polyazulene on Au(111), confirmed by STS mapping and DFT-based LDOS simulations. The reported synthesis route may enable future applications of azulene-based functional materials.
This work was supported by the Swiss National Science Foundation under Grant No. 200020_182015, the NCCR MARVEL funded by the Swiss National Science Foundation (51NF40-182892), the European Union's Horizon 2020 research and innovation programme under grant agreement number 785219 (Graphene Flagship Core 2), the Office of Naval Research (N00014-18-1-2708), and a grant from the Swiss National Supercomputing Centre (CSCS) under project ID s904. Q. S. acknowledges the EMPAPOSTDOCS-II programme under the Marie Sklodowska-Curie grant agreement no. 754364. Open Access funding provided by the Max Planck Society. R. F. thanks Oliver Gröning for insightful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. P. Klein, N. J. van der Heijden, S. R. Kachel, M. Franke, C. K. Krug, K. K. Greulich, L. Ruppenthal, P. Müller, P. Rosenow, S. Parhizkar, F. C. Bocquet, M. Schmid, W. Hieringer, R. J. Maurer, R. Tonner, C. Kumpf, I. Swart and J. M. Gottfried, Phys. Rev. X, 2019, 9, 011030 CAS; (b) S. Mishra, T. G. Lohr, C. A. Pignedoli, J. Liu, R. Berger, J. I. Urgel, K. Müllen, X. Feng, P. Ruffieux and R. Fasel, ACS Nano, 2018, 12, 11917–11927 CrossRef CAS PubMed.
- (a) J. Michl and E. W. Thulstrup, Tetrahedron, 1976, 32, 205–209 CrossRef CAS; (b) J. W. Sidman and D. S. McClure, J. Chem. Phys., 1956, 24, 757–763 CrossRef CAS.
- A. G. Anderson Jr and B. M. Steckler, J. Am. Chem. Soc., 1959, 81, 4941–4946 CrossRef.
- Y. Zhou, G. Baryshnikov, X. Li, M. Zhu, H. AAgren and L. Zhu, Chem. Mater., 2018, 30, 8008–8016 CrossRef CAS.
- (a) N. Tétreault, R. S. Muthyala, R. S. H. Liu and R. P. Steer, J. Phys. Chem. A, 1999, 103, 2524–2531 CrossRef; (b) D. R. Mitchell and G. D. Gillispie, J. Phys. Chem., 1989, 93, 4390–4393 CrossRef CAS.
- M. Murai, E. Amir, R. J. Amir and C. J. Hawker, Chem. Sci., 2012, 3, 2721–2725 RSC.
- (a) Y. Yamaguchi, K. Ogawa, K. Nakayama, Y. Ohba and H. Katagiri, J. Am. Chem. Soc., 2013, 135, 19095–19098 CrossRef CAS PubMed; (b) H. Xin, C. Ge, X. Jiao, X. Yang, K. Rundel, C. R. McNeill and X. Gao, Angew. Chem., 2018, 130, 1336–1340 CrossRef.
- H. Nishimura, N. Ishida, A. Shimazaki, A. Wakamiya, A. Saeki, L. T. Scott and Y. Murata, J. Am. Chem. Soc., 2015, 137, 15656–15659 CrossRef CAS PubMed.
- (a) J.-X. Dong and H.-L. Zhang, Chin. Chem. Lett., 2016, 27, 1097–1104 CrossRef CAS; (b) H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945–956 CrossRef CAS.
- E. Amir, M. Murai, R. J. Amir, J. S. Cowart, M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 5, 4483–4489 RSC.
- Y. Yamaguchi, M. Takubo, K. Ogawa, K. Nakayama, T. Koganezawa and H. Katagiri, J. Am. Chem. Soc., 2016, 138, 11335–11343 CrossRef CAS PubMed.
- (a) G. Nöll, C. Lambert, M. Lynch, M. Porsch and J. Daub, J. Phys. Chem. C, 2008, 112, 2156–2164 CrossRef; (b) R. P. Steer, J. Photochem. Photobiol., C, 2019, 40, 68–80 CrossRef CAS.
- I. C.-Y. Hou, V. Shetti, S.-L. Huang, K.-L. Liu, C.-Y. Chao, S.-C. Lin, Y.-J. Lin, L.-Y. Chen and T.-Y. Luh, Org. Chem. Front., 2017, 4, 773–778 RSC.
- (a) J. Björk and F. Hanke, Chem. – Eur. J., 2014, 20, 928–934 CrossRef PubMed; (b) P. A. Held, H. Fuchs and A. Studer, Chem. – Eur. J., 2017, 23, 5874–5892 CrossRef CAS PubMed; (c) Q. Sun, R. Zhang, J. Qiu, R. Liu and W. Xu, Adv. Mater., 2018, 30, 1705630 CrossRef PubMed.
- Q. Fan, D. Martin-Jimenez, D. Ebeling, C. K. Krug, L. Brechmann, C. Kohlmeyer, G. Hilt, W. Hieringer, A. Schirmeisen and M. Gottfried, J. Am. Chem. Soc., 2019 DOI:10.1021/jacs.9b08060.
- M. Narita, T. Murafuji, S. Yamashita, M. Fujinaga, K. Hiyama, Y. Oka, F. Tani, S. Kamijo and K. Ishiguro, J. Org. Chem., 2018, 83, 1298–1303 CrossRef CAS PubMed.
- M. Di Giovannantonio, O. Deniz, J. I. Urgel, R. Widmer, T. Dienel, S. Stolz, C. Sánchez-Sánchez, M. Muntwiler, T. Dumslaff, R. Berger, A. Narita, X. Feng, K. Müllen, P. Ruffieux and R. Fasel, ACS Nano, 2018, 12, 74–81 CrossRef CAS PubMed.
- (a) J. I. Urgel, H. Hayashi, M. Di Giovannantonio, C. A. Pignedoli, S. Mishra, O. Deniz, M. Yamashita, T. Dienel, P. Ruffieux, H. Yamada and R. Fasel, J. Am. Chem. Soc., 2017, 139, 11658–11661 CrossRef CAS PubMed; (b) H. Zhang, H. Lin, K. Sun, L. Chen, Y. Zagranyarski, N. Aghdassi, S. Duhm, Q. Li, D. Zhong, Y. Li, K. Müllen, H. Fuchs and L. Chi, J. Am. Chem. Soc., 2015, 137, 4022–4025 CrossRef CAS PubMed; (c) Q. Sun, L. Cai, H. Ma, C. Yuan and W. Xu, ACS Nano, 2016, 10, 7023–7030 CrossRef CAS PubMed.
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed.
- D. A. Kunkel, J. Hooper, S. Simpson, D. P. Miller, L. Routaboul, P. Braunstein, B. Doudin, S. Beniwal, P. Dowben, R. Skomski, E. Zurek and A. Enders, J. Chem. Phys., 2015, 142, 101921 CrossRef PubMed.
- P. Ruffieux, J. Cai, N. C. Plumb, L. Patthey, D. Prezzi, A. Ferretti, E. Molinari, X. Feng, K. Müllen, C. A. Pignedoli and R. Fasel, ACS Nano, 2012, 6, 6930–6935 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07168g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |