
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective modification of tryptophan and protein tryptophan residues through PdNP bionanohybrid-catalysed C–H activation in aqueous media†
Carlos
Perez-Rizquez
 a,
Olga
Abian
bc and
Jose M.
Palomo
*a
a,
Olga
Abian
bc and
Jose M.
Palomo
*a
aDepartment of Biocatalysis, Institute of Catalysis (ICP-CSIC), Marie Curie 2, Cantoblanco, Campus UAM, 28049 Madrid, Spain. E-mail: josempalomo@icp.csic.es
bInstituto Aragonés de Ciencias de la Salud (IACS), Zaragoza, 50009, Spain
cInstitute of Biocomputation and Physics of Complex Systems (BIFI), Joint Units IQFR-CSIC-BIFI, and GBsC-CSIC-BIFI, Universidad de Zaragoza, Zaragoza, 50018, Spain
First published on 3rd October 2019
Abstract
Herein we report for the first time the site-selective C–H bond arylation of tryptophan and tryptophan residues in proteins in aqueous media at room temperature by using a PdNP bionanohybrid as a heterogeneous catalyst. The reaction proceeds using a stable aryldiazonium salt without a base.
Tryptophan (Trp) is one of the rarest amino acids present in proteins with a natural abundance of only 1.09%.1
However, in most cases Trp has a very important role in the biological activity of proteins and especially enzymes, affecting not only activity but also in many cases the selectivity.2–6
Strategies for selective chemical modification of proteins have been well developed in recent years.7–15 The selective functionalization of traditional groups such as N-terminal, Lys amino residues, Asp or Glu carboxylic acid residues or side chain cysteines has been successfully employed.7–10 However, the established location and number of functionalizable groups on the protein makes this strategy limited, being in most cases a non-specific modification. Thus, new elegant procedures – combining chemical and biological techniques – through C–C strategies (cycloadditions, Staudinger ligation, Michael additions, etc.)10–15 have been developed for protein site-selective modification. This has allowed modified proteins with enhanced or altered biological properties to be obtained.12,13
Recently, the C(sp2)–H arylation reactions of α-amino acids and peptide-like compounds have attracted attention, especially in the indole unit of tryptophan.16–20 Over the years, several protocols with different coupling partners and oxidation systems were developed for functionalization at the indole C2-position using transition metal catalysis, in which palladium acetate has been the most used catalyst.17–22 However, this protocol showed some limitations in the extension to protein modification. The conditions normally require the use of organic solvents, while protein modifications must be performed in water to preserve protein structure, and catalytic performance in enzymes.
Therefore, to overcome all these drawbacks the application of a heterogeneous catalyst that works well in aqueous media would be mandatory. An interesting alternative could be the recently described Pd nanobiohybrids,23 a heterogeneous metal biohybrid formed by very small dispersed nanoparticles of Pd(0) in a protein network. This catalyst has been successfully applied in C–C bonding or reduction processes.23
In this work we present for the first time the site-selective modification of tryptophan residues by C–H activation under mild conditions (aqueous media and room temperature) catalyzed by the heterogeneous PdNP biohybrid. The concept was first tested using a protected tryptophan and finally used successfully in the modification of tryptophan residues of a particular enzyme (Fig. 1).
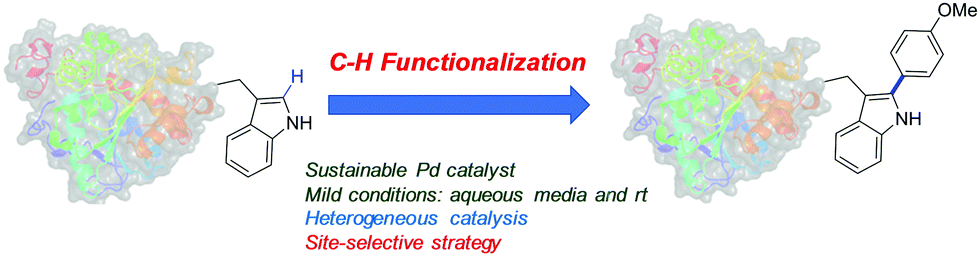 | ||
| Fig. 1 Pd biohybrid-catalysed C–H activation of tryptophan in proteins. | ||
First, the Pd nanoparticle biohybrid (PdNPs-E) was synthesized using the strategy previously described,23a scaling up the synthesis five times. The characterization of the heterogeneous biohybrid was performed, and Pd(0) nanoparticles of around 5 nm diameter size were obtained (Fig. S1, ESI†).
Our studies started by exploring the capacity of this Pd biohybrid as a catalyst in the C–H activation reaction by selective arylation of N-acetyl-L-tryptophan methyl ester (1) (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Time [h] | Conversione [%] |
a Conditions: 1 (0.192 mmol), 2 (0.192 mmol), solvent (5 mL), catalyst (2 mg), r.t (ca. 20 °C).
b (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 v/v).
c (50:50 v/v).
d (40:60 v/v).
e Conversion of the product 3 was quantified by HPLC. :30 v/v).
c (50:50 v/v).
d (40:60 v/v).
e Conversion of the product 3 was quantified by HPLC.
|
||||
| 1 | Pd(OAc)2 | EtOAc | 16 | >99 |
| 2 | PdNPs-E | EtOAc | 16 | >99 |
| 3 | PdNPs-E | Dioxane | 16 | 87 |
| 4 | PdNPs-E | THF | 16 | 36 |
| 5 | PdNPs-E | MeOH | 2 | >99 |
| 6 | PdNPs-E | MeOH:H2Ob |
2 | >99 |
| 7 | PdNPs-E | MeOH:H2Oc |
2 | >99 |
| 8 | PdNPs-E | EtOH | 2 | 37 |
| 9 | PdNPs-E | EtOH:H2Oc |
2 | 70 |
| 10 | PdNPs-E | EtOH:H2Od |
2 | 97 |
| 11 | PdNPs-E | i-PrOH:H2Oc |
2 | 75 |
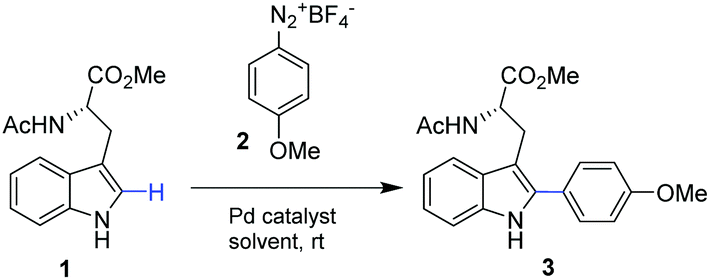
The reaction was performed using 4-methoxybenzenediazonium tetrafluoroborate (2), as an electrophilic arylating coupling partner, firstly in ethyl acetate using Pd(OAc)2 or the PdNPs-E biohybrid as a catalyst. In both cases, >99% of arylated tryptophan 3 was obtained after 16 h incubation. In particular, this result using Pd acetate (5 mol%) is in concordance with reported results.19
However, our PdNPs-E catalyst showed better performance under these conditions, being at least two times more active.
Among a variety of solvents (Table 1, entries 3–5), an excellent result was obtained in methanol, with full conversion of 1 after 2 h (Table 1, entry 5).
Based on the previous results with PdNPs-E catalysing C–C bonding reaction in aqueous/solvent mixtures,23 the C–H activation of 1 was performed in a mixture of MeOH/water. The PdNPs-E catalyst showed the same catalytic performance in the mixtures 70/30 or 50/50 (v/v) as in pure methanol (Table 1, entries 6 and 7). Although the full conversion was completed at the same time in pure methanol or aqueous methanol, evaluating the reaction profile we can observe great differences (Fig. S2, ESI†). The conversion of 3 was very low (<20%) within 40 min using pure methanol as a reaction medium, while 90% conversion was obtained at that time using the 50/50 mixture (Fig. S2, ESI†).
Following these results, other pure or water–mixture alcohols were tested as solvents (Table 1, entries 8–11, Fig. S2b and c, ESI†). More hydrophobic alcohols showed lower conversions, although in the case of ethanol, 97% conversion of 3 was achieved in 2 hours using a mixture of 40/60 (v/v) of ethanol/water as a reaction medium (Table 1, entry 10). Isopropanol showed solubility problems of substrates, but the 50/50 mixture with water gave the best results.
Some studies were conducted to evaluate the catalytic results by decreasing the amount of Trp 1 with respect to compound 2 (Fig. S3, ESI†). Performing the reaction in MeOH:water (1:1), similar conversion value of 3 as before was obtained using five times less amount of 1, while more than 40% decrease in catalytic efficiency of PdNPs-E was obtained using twenty times less of 1.
These results demonstrate the ability of this Pd nanobiohybrid to perform the C–H arylation reaction under selective and mild conditions.
This heterogeneous catalyst exhibited high stability while maintaining complete activity without loss of Pd after several recycling processes (Fig S4, ESI†).
Therefore, after these results we tried to modify tryptophan through this strategy in a more complex environment, such as in a protein structure. Taking into account that the problem of the solubility of 1 did not allow us to reduce the amount of MeOH in the reaction solution, in this case Trp residues are included in the structure of the completely water soluble protein. This could allow us to perform the C–H activation reaction in pure water solution.
As a proof of concept, we selected the lipase from Candida antarctica B (renamed as Pseudozyma antarctica) (Cal-B) as a model protein. This is a well-known hydrolase, where its crystal structure is solved.24 The protein sequence and tridimensional structure of Cal-B show the presence of five tryptophan groups (Trp52, Trp65, Trp104, Trp113, Trp155) (Fig. S5, ESI†). However, only three of them are superficially accessible, two of them on the protein surface (Trp65, Trp155) and another in the oxyanion hole (Trp104) (Fig. S5b, ESI†).
The modification of Cal-B with 2 was performed in distilled water (pH∼6) at room temperature (Fig. 2). Two different reaction protocols were used for C–H activation of Cal-B Trp residues, modifying the amount of Pd catalyst added, 2 mg or 10 mg of PdNPs-E biohybrid, respectively. After purification, two different modified proteins, Cal-B-T1 and Cal-B-T2 were obtained. Control experiments were also performed, where Cal-B was incubated only in the presence of 2 without a catalyst (protein called Cal-B*) or incubated only in the presence of PdNPs biohybrid without 2 (protein called Cal-B**). Analysis of the different proteins by MALDI-TOF mass spectrometry revealed one Trp modification in the Cal-B-T1 whereas two modifications were obtained in the Cal-B-T2 protein (Fig. 3a).
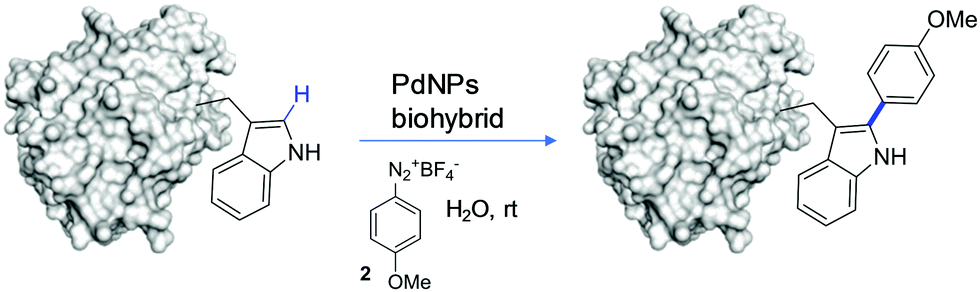 | ||
| Fig. 2 Site-selective modification of Trp in Cal-B by 2 catalysed by the Pd biohybrid. | ||
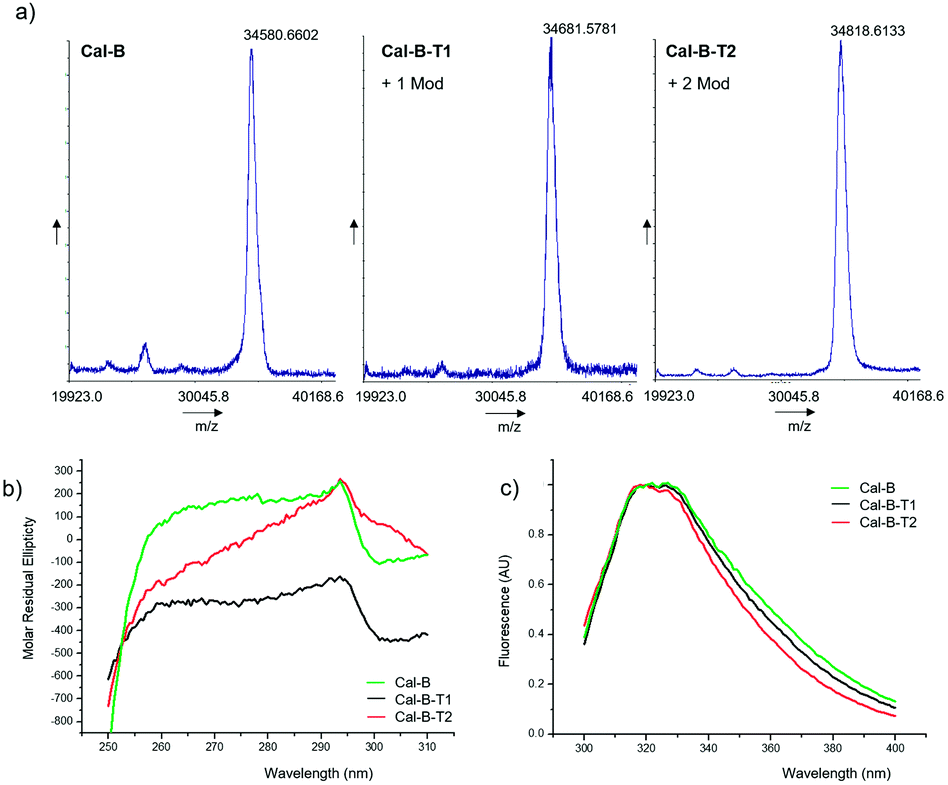 | ||
| Fig. 3 Characterization of the different conjugates Cal-B-T1 and Cal-B-T2. (a) MALDI-TOF spectra, (b) near-UV CD spectra, and (c) fluorescence spectra. | ||
No modification was observed by MALDI in the Cal-B* and Cal-B** control proteins (Fig. S6, ESI†).
Near-circular dichroism (CD) and fluorescence assays were performed on the native and the two modified Cal-B proteins to evaluate the effect of the modifications on the tertiary structure of the protein (Fig. 3b and c). Near-CD spectra show signal loss in the Trp area (around 295 nm), stronger in Cal-B-T2 (Fig. 3b). Fluorescence spectra show a shift of the peak in both modified proteins to wavelengths <330 nm (Fig. 3c), which is related to a less polar environment of Trp.25 Therefore, these structural analyses suggested that the Trp modification alters the tertiary structure of the enzyme.
Trypsin hydrolysis of the different modified proteins was performed to determine which Trp was modified (Fig. S7, ESI†). After the hydrolysis, a peptide profile from MALDI-TOF (MS/MS) was obtained for Cal-B, Cal-B-T1 and Cal-B-T2 and clear differences in peptide sequence were found. However, the peptide GTVLAGPLDALAVSAPSV![[W with combining low line]](https://www.rsc.org/images/entities/char_0057_0332.gif) QQTTGSALTTALR was found in both modified proteins, which contain Trp155, indicating that Trp155 was not modified although this is a highly exposed amino acid in the Cal-B structure (Fig. 4).
QQTTGSALTTALR was found in both modified proteins, which contain Trp155, indicating that Trp155 was not modified although this is a highly exposed amino acid in the Cal-B structure (Fig. 4).
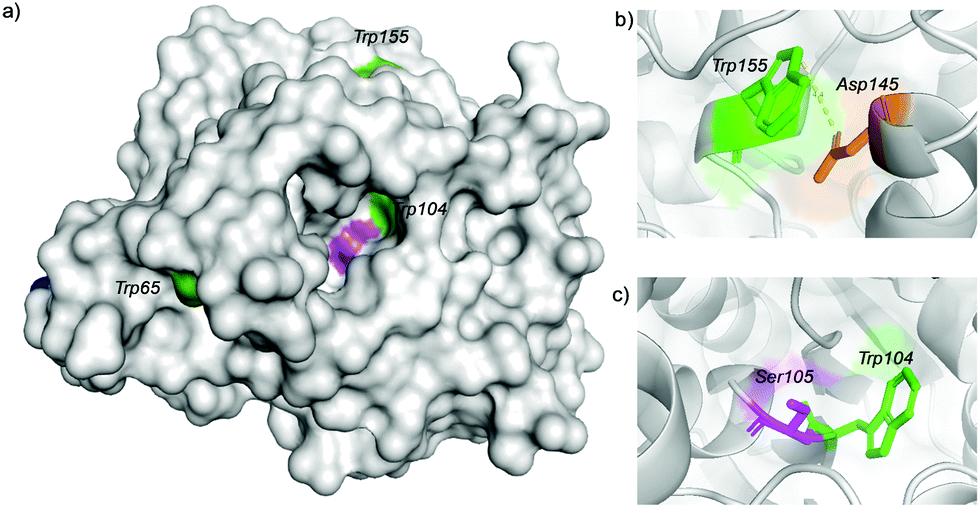 | ||
| Fig. 4 (a) Three-dimensional surface structure of Cal-B marking Trp residues exposed on the surface. (b) Representation of the anion–π interaction between Trp155 and Asp145. (c) Representation of the Trp104 and catalytic Ser105. Tryptophan (green), serine (purple). The 3D structure was obtained from the Protein Data Bank (PDB) using Pymol vs 2.0.5. The pdb code for Cal-B is TCA. | ||
The possible reason why this Trp was not modified could be due to an anion–π interaction26 between Trp155 and Asp145 (Fig. 4b and Fig. S8, ESI†).
From these results, we can suggest that Trp65 was modified in both Cal-B proteins (Cal-B-T1 and Cal-B-T2). The Trp104 residue is the other that could be more accessible for C–H modification. This amino acid shows an important role in the catalytic activity of this enzyme because it is close to the catalytic serine (Fig. 4c and Fig. S9, ESI†); therefore enzymatic hydrolysis of p-nitro-phenyl propionate (pNPP) was evaluated with all Cal-B proteins (Fig. 5). Native, control and Cal-B-T1 enzymes showed a similar activity, while the activity of Cal-B-T2 decreased approximately 40% of the initial enzymatic activity (Fig. 5). Also the trypsin digestion of Cal-B-T2 showed a peptide sequence with observed mass of 2808.2366 [Mmodified CAL-B + Na] corresponding to the peptide sequence LPVLT(104)SQ GGLVAQ(113)GLTFFPSIRS including the modification of one Trp residue (Fig. S7, ESI†). Therefore, considering all results, we suggest that Trp104 is the second Trp modified in Cal-B-T2.
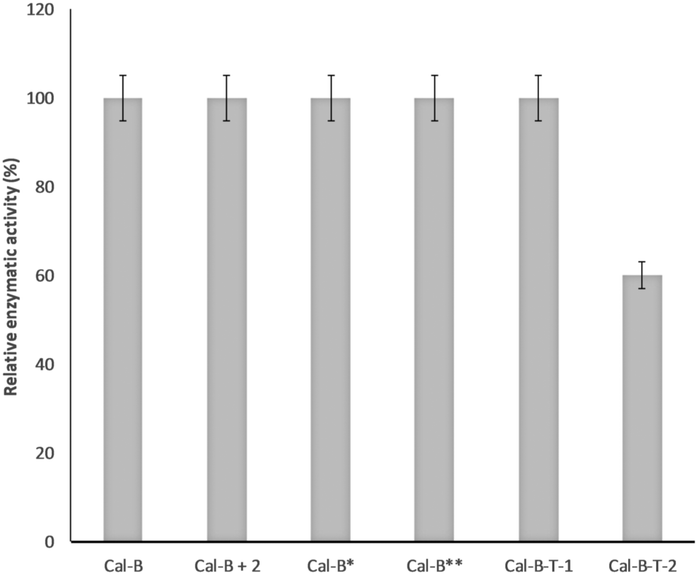 | ||
| Fig. 5 Enzymatic activity of the different conjugate Cal-B proteins against p-nitro-phenyl propionate hydrolysis. Cal-B* refers to Cal-B after the reaction only in the presence of 2, Cal-B** refers to Cal-B after reaction only in the presence of the PdNP biohybrid. | ||
In conclusion, we have described for the first time the site-selective C–H bond arylation of protected-tryptophan, with the best conditions in an aqueous solution containing methanol, and more interestingly, tryptophan residues in proteins in aqueous media at room temperature by the use of a PdNP bionanohybrid as a heterogeneous catalyst. It was possible to selectively modify one or two Trp residues in the Cal-B protein by controlling the amount of Pd catalyst used. The structural characterization allowed the identification of the modified Trp in each case. These results open the door to the application of this strategy for the selective and mild modification of biologically relevant proteins.
This work was supported by the Spanish Government the Spanish National Research Council (CSIC), and the Ministry of Education, Youth and Sports of the Community of Madrid and the European Social Fund (PEJD-2017PRE/SAL-3762). The authors thank the European Cooperation in Science and Technology (COST) program under CA15106 grant (CHAOS: CH Activation in Organic Synthesis). The authors also thank Irene Orera and Giuseppe Lattanzio from Proteomics Unit of Servicios Científico Técnicos del CIBA (IACS-Universidad de Zaragoza), ProteoRed ISCIII member, for mass spectra analysis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- UniProtKB/Swiss-Prot protein knowledgebase release 2019 07 statistics, http://web.expasy.org/docs/relnotes/relstat.html.
- V. P. Jaakola, M. T. Griffith, M. A. Hanson, V. Cherezov, E. Y. T. Chien, J. R. Lane, A. P. Ijzerman and R. C. Stevens, Science, 2008, 322, 1211–1217 CrossRef CAS PubMed.
- Z. Chen, L. C. Trotman, D. Shaffer, H.-K. Lin, Z. A. Dotan, M. Niki, J. A. Koutcher, H. I. Scher, T. Ludwig, W. Gerald, C. Cordon-Cardo and P. P. Pandolfi, Nature, 2005, 436, 725–730 CrossRef CAS PubMed.
- H. Li, S. Ilin, W. Wang, E. M. Duncan, J. Wysocka, C. D. Allis and D. J. Patel, Nature, 2006, 442, 91–95 CrossRef CAS PubMed.
- J. M. Watermeyer, W. L. Kröger, H. G. O'Neill, B. T. Sewell and E. D. Sturrock, Biochem. J., 2010, 428, 67–74 CrossRef CAS PubMed.
- M. Miyanokoshi, T. Yokosawa and K. Wakasugi, J. Biol. Chem., 2018, 293, 8428–8438 CrossRef CAS PubMed.
- B. Wu, H. J. Wijma, L. Song, H. J. Rozeboom, C. Poloni, Y. Tian, M. I. Arif, T. Nuijens, P. J. L. M. Quaedflieg, W. Szymanski, B. L. Feringa and D. B. Janssen, ACS Catal., 2016, 6, 5405–5414 CrossRef CAS.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS PubMed.
- C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- T. H. Wright, B. J. Bower, J. M. Chalker, G. J. L. Bernardes, R. Wiewiora, W.-L. Nag, R. Raj, S. Faulkner, M. R. J. Vallée, A. Phanumartwiwath, O. D. Coleman, M.-L. Thézénas, M. Khan, S. R. G. Galan, L. Lercher, M. W. Schombs, S. Gerstberger, M. E. Palm-Espling, A. J. Baldwin, B. M. Kessler, T. D. W. Claridge, S. Mohammed and B. G. Davis, Science, 2016, 354, 597 CrossRef CAS PubMed.
- A. Díaz-Rodríguez and B. G. Davis, Curr. Opin. Chem. Biol., 2011, 15, 211–219 CrossRef PubMed.
- O. Romero, M. Filice, B. de las Rivas, C. Carrasco-Lopez, J. Klett, A. Morreale, J. A. Hermoso, J. M. Guisan, O. Abian and J. M. Palomo, Chem. Commun., 2012, 72, 9053–9055 RSC.
- B. Bhushan, Y. A. Lin, M. Bak, A. Phanumartwiwath, N. Yang, M. K. Bilyard, T. Tanaka, K. L. Hudson, L. Lercher, M. Stegmann, S. Mohammed and B. G. Davis, J. Am. Chem. Soc., 2018, 140, 14599 CrossRef CAS PubMed.
- M. Filice, O. Romero, J. M. Guisan and J. M. Palomo, Org. Biomol. Chem., 2011, 9, 5535–5540 RSC.
- M. M. Lorion, N. Kaplaneris, J. Son, R. Kuniyil and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 1684–1688 CrossRef CAS PubMed.
- A. J. Reay, T. J. Williams and I. J. S. Fairlamb, Org. Biomol. Chem., 2015, 13, 8298 RSC.
- L. Mendive-Tapia, S. Preciado, J. Garcia, R. Ramon, N. Kielland, F. Albericio and R. Lavilla, Nat. Commun., 2015, 6, 7160 CrossRef PubMed.
- A. J. Reay, L. A. Hammarback, J. T. W. Bray, T. Sheridan, D. Turnbull, A. C. Whitwood and I. J. S. Fairlamb, ACS Catal., 2017, 78, 5174–5179 CrossRef PubMed.
- T. Brandhofer and O. García Mancheño, Eur. J. Org. Chem., 2018, 6050–6067 CrossRef CAS.
- Y. Zhu, M. Bauer and L. Ackermann, Chem. – Eur. J., 2015, 21, 9980 CrossRef CAS PubMed.
- S. Preciado, L. Mendive-Tapia, F. Albericio and R. Lavilla, J. Org. Chem., 2013, 78, 8129 CrossRef CAS PubMed.
- (a) M. Filice, M. Marciello, M. P. Morales and J. M. Palomo, Chem. Commun., 2013, 49, 6876–6878 RSC; (b) J. M. Palomo, Chem. Commun., 2019, 55, 9583–9589 RSC.
- J. Uppenberg, M. T. Hansen, S. Patkar and T. A. Jones, Structure, 1994, 2, 293–308 CrossRef CAS PubMed.
- J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 CrossRef CAS PubMed.
- X. Lucas, A. Bauza, A. Frontera and D. Quiñonero, Chem. Sci., 2016, 7, 1038–1050 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization data and additional tables and figures. See DOI: 10.1039/c9cc06971b |
| This journal is © The Royal Society of Chemistry 2019 |