
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glutathione activation of an organometallic half-sandwich anticancer drug candidate by ligand attack†
Xin
Zhang‡
a,
Fortuna
Ponte‡
b,
Elisa
Borfecchia
 c,
Andrea
Martini
c,
Carlos
Sanchez-Cano§
*a,
Emilia
Sicilia
*b and
Peter J.
Sadler
*a
c,
Andrea
Martini
c,
Carlos
Sanchez-Cano§
*a,
Emilia
Sicilia
*b and
Peter J.
Sadler
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: p.j.sadler@warwick.ac.uk
bDipartimento di Chimica e Tecnologie Chimiche, Università della Calabria, Ponte P. Bucci, Cubo 14cI-87030, Arcavacata di Rende, Italy. E-mail: emilia.sicilia@unical.it
cDepartment of Chemistry, NIS Center and INSTM Reference Center, University of Turin, via Giuria 7, 10125, Turin, Italy
First published on 19th November 2019
Abstract
In contrast to the clinical drug cisplatin, the anticancer complex [Os(η6-p-cymene)(4-(2-pyridylazo)-N,N-dimethylaniline)I]+ [1-I] is inert towards hydrolysis and targets cancer cell metabolism rather than DNA. A combination of DFT calculations and X-ray absorption spectroscopy (XAS) suggests that hydrolytic activation of 1-I involves catalytic attack by the intracellular tripeptide glutathione (GSH) on the azo bond of the chelating ligand in the complex.
[Os(η6-p-cymene)(4-(2-pyridylazo)-N,N-dimethylaniline)I]+ [1-I] and its analogues (Chart 1), are inert prodrugs activated by the reducing environment in cancer cells.11-I, is a promising drug candidate capable of overcoming Pt resistance,2 with a novel mechanism of action.31-I increases oxidative stress in cancer cells through mitochondrial and other pathways, and exploits an inherent weakness in cancer cell energy generation.3
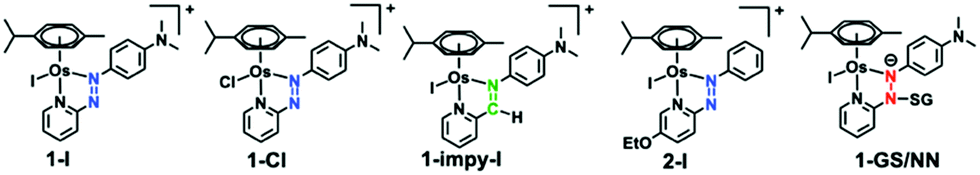 | ||
| Chart 1 Structures of complexes and captured intermediate (1-GS/NN). | ||
Prodrugs hypothesised to be activated by intracellular reduction include PtIV complexes which are reduced to active PtII species.3 A number of RuIII anticancer complexes are also thought to be prodrugs activated in a similar way,4 but recent experiments have not detected reduction of RuIII compounds in tumours.5 However, activation of 1-I and its analogues does not appear to involve changes in the oxidation state of OsII in cells. On the contrary, initial experiments suggest that hydrolysis of the Os–I bond is promoted by the tripeptide glutathione (GSH). This generates the more reactive hydroxido adduct 1-OH, which can react with other molecules in cells, such as H2O2 (generating hydroxyl radicals).1 The aim of the present work was to establish both experimental and theoretical (density functional theory, DFT) evidence for this proposed highly unusual mechanism of activation of a metallo-prodrug.
DFT computational analysis can provide insights into reaction mechanisms for metal coordination compounds.6 Hence we probed the most favourable mechanism, involving deprotonated glutathione (GS−), for the hydrolysis of 1-I using quantum-mechanical DFT calculations. First, the direct replacement of the iodido ligand by water (pathway a), a process observed under physiological conditions for many OsII, RuII, or IrIII half-sandwich complexes [M(arene)(L)X]+,7 was explored. Our calculations suggested an overall energy barrier of 25.4 kcal mol−1 and an endergonic (by 9.8 kcal mol−1) exchange by second-order nucleophilic substitution (SN2) (pathway a; Fig. S1a, ESI†). An alternative, direct replacement of the iodide ligand by GS−, to form an Os-SG adducts, followed by replacement of the thiolate ligand by water was also studied (pathway b). Such a reaction has been reported for other complexes, e.g. [Ru(arene)(en)Cl]+ and its analogues.8 In this case, the energy barrier to generate the stable 1-SG complex was 23.2 kcal mol−1, and the substitution product lies 18.9 kcal mol−1 below the entrance channel. However, hydrolysis of 1-SG to form the experimentally observed 1-OH required surmounting a very high energy barrier of 30.3 kcal mol−1 (pathway b, Fig. S1b, ESI†). Finally, we explored ligand-based attack on the complex by GSH resulting in hydrolysis of the Os–I bond through redox mediation involving the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond (pathway c).
N double bond (pathway c).
Some organic azo compounds are known to catalyse oxidation of GSH in presence of O2,9 reactions also possible for azo-containing ligands in RuII or IrIII complexes.10 The reported mechanism can involve addition of GSH to the diazene double bond and subsequent reduction to an hydrazo group.9 However, the free azo ligand in 1-I does not react with GSH.11
We probed azo bond attack by GSH using DFT, but the most favourable energy barrier for the only viable reaction mechanism was high (40.2 kcal mol−1 Fig. S2, ESI†). Nevertheless, the attack by GS− on the azo N atom bound to the pyridine ring, preluding hydrolysis of 1-I, yielded the lowest barrier (19.0 kcal mol−1). This led to intermediate 1-GS/NN (pathway c, Fig. 1), that lies 11.8 kcal mol−1 above the reactant reference energy, and accumulated electronic density near Os, causing the weakening of the Os–I bond. This facilitates displacement of the I− ligand by water (13.9 kcal mol−1), which occurred simultaneously with detachment of the GS− and regeneration of the azo bond. Further deprotonation of the coordinated water (requiring 5.5 kcal mol−1) yielded 1-OH, which was stabilised by 7.3 kcal mol−1 (with respect to entrance channel). Computational analysis of the activation of 2-I, an analogue with an electron donating ethoxy group on the pyridine ring gave similar results, but with lower energy barriers (Fig. S3, ESI†). This paralleled experimental data showing faster hydrolysis of 2-I compared to 1-I.1
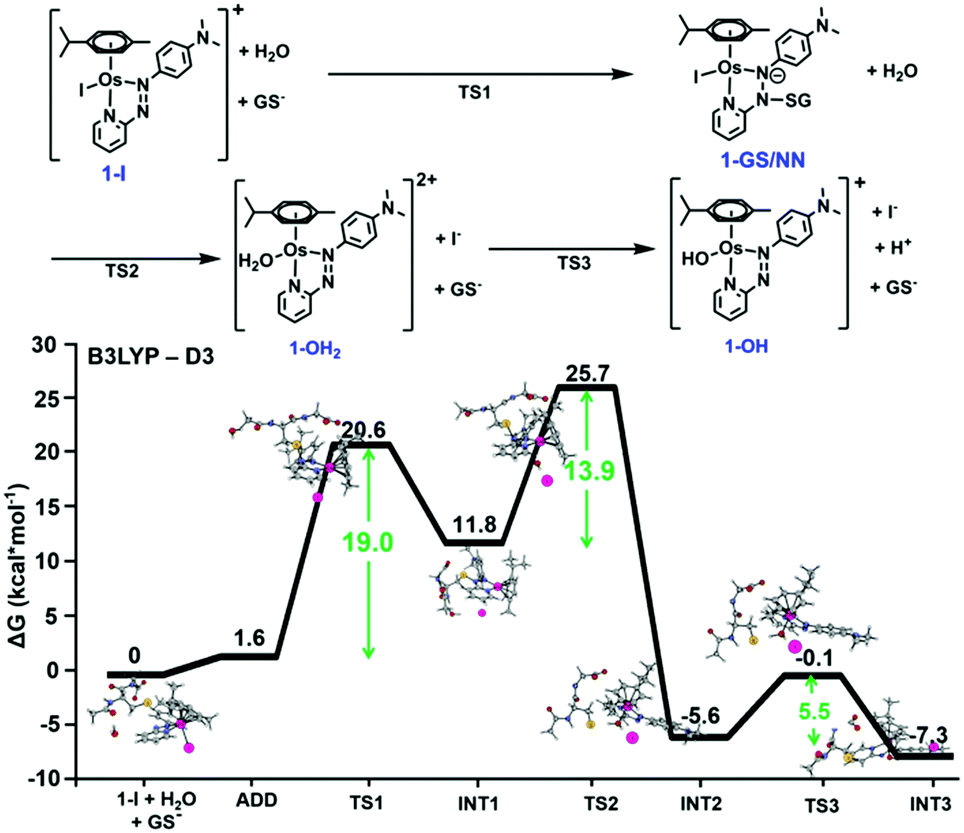 | ||
| Fig. 1 DFT free energy profile describing the most favourable pathway for the reaction of 1-I with GS− (pathway c). Calculated energies are in kcal mol−1 and relative to reference energy of separated reactants. Alternative higher energy pathways a and b are in Fig. S1 (ESI†). | ||
Interestingly, our calculations proposed thermodynamically favoured hydrolysis pathways in the presence of glutathione, and kinetic control favouring the azo-based mechanism. This was confirmed by following the hydrolysis of chlorido [Os(η6-p-cymene)(4-(2-pyridylazo)-N,N-dimethylaniline)Cl]+ [1-Cl] and imino [Os(η6-p-cymene)(4-(2-iminopyridine)-N,N-dimethyl-aniline)I]+ [1-impy-I] analogues in presence or absence of GSH using HPLC (the solvent gradient used is on page S11 of the ESI†). As for 1-I, 1-Cl is stable in aqueous solution, but treatment with GSH promotes faster hydrolysis than 1-I (Fig. S18, ESI†). However, both 1-Cl and 1-I hydrolysed in the presence of GSH following the same mechanism, generating first 1-OH (Fig. S19, S20 and Tables S3, S4, ESI†), which can be attacked by GSH or Cl− to form 1-Cl or 1-SG. 1-SG can be further oxidised to the sulfenate complex 1-SOG (Fig. S21 and S22, ESI†). Notably, similar pathways were observed when different thiols induce hydrolysis of 1-I (Fig. S23, S24 and Table S5, ESI†). In contrast, the iminopyridine complex 1-impy-I readily hydrolysed in the absence of GSH, generating both hydroxido 1-impy-OH and chlorido 1-impy-Cl species. Incubation with GSH did not increase the extent of hydrolysis, but generated the thiolato adduct 1-impy-SG (Fig. S18 and S25, ESI†), even when 1-impy-I was in large excess. This confirmed that the azo bond in the azopyridine complexes is the likely target site for GS− (as it is the only difference between the chemical structure of 1-I and 1-impy-I). Hydrolysis of 1-I was ca. 3× faster at pH 10 compared to pH 7, attributable to the higher proportion of GS− present (pKa(SH): 9.42),12 whereas hydrolysis was inhibited at pH 2.4 (Fig. S26 and Table S6, ESI†). No 1-OH was observed in the absence of thiol at basic pH (Fig. S27, ESI†).
The proposed attack of GSH on the –NN– bond in 1-I during the hydrolysis was further probed using X-ray absorption spectroscopy (XAS), to provide structural information.13 We acquired XAS spectra at the Os L3-edge for solid reference compounds 1-I, 1-Cl, 1-OH and 1-SG (for synthesis and characterisation see ESI†), and an aqueous solution (pH 7 buffer) of 1-I on beamline BM23 at ESRF.14 Discrimination between the different references could not rely on analysis of the XANES region (Fig. 2a and Fig. S28, ESI†).15 However, there were distinguishing differences in the EXAFS oscillations for 1-I, when compared with 1-Cl, 1-OH and 1-SG especially from k = 10 Å−1 upward (Fig. 2b–d and Fig. S29, S30, ESI†). In contrast, the spectra of 1-Cl and 1-SG were similar (due to similar size and atomic number of Cl and S). XAS spectra from the reference compounds were also used to validate structural models generated by our DFT calculations (Fig. S31, ESI†). Based on these data, we simulated an EXAFS signature corresponding to the DFT model of the intermediate 1-GS/NN (Fig. S31, see ESI† for details). This allowed us to probe the presence of 1-GS/NN in reactions between 1-I and GSH followed in situ by EXAFS.
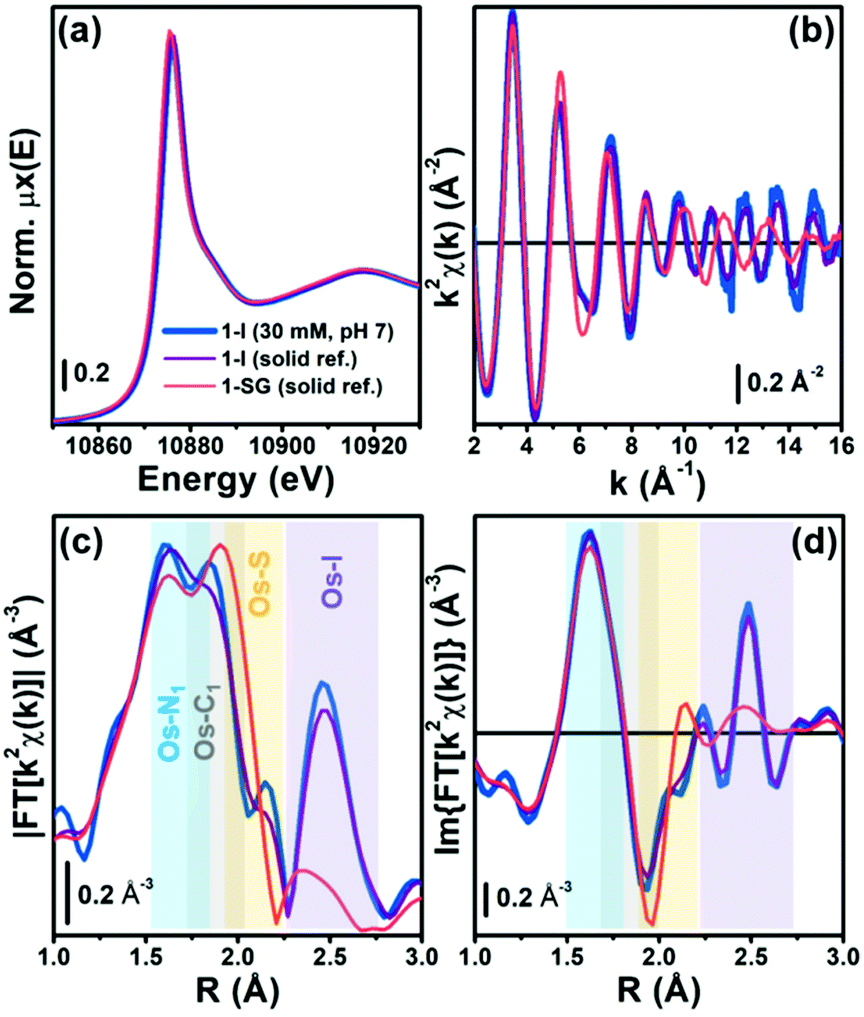 | ||
| Fig. 2 (a) Os L3-edge XANES spectra of aqueous 1-I (30 mM, pH 7; blue), and solid 1-I (purple) and 1-SG (orange). (b) k2χ(k) EXAFS functions for samples in (a). Phase-uncorrected (c) magnitude, and (d) imaginary part of the FT-EXAFS functions for the same samples, obtained by transforming the k2χ(k) functions (b) in the 2.0–15.5 Å−1 range. R-space ranges of major contributions from the different EXAFS components arising from the coordination spheres around Os centres are highlighted by coloured rectangles. | ||
The kinetics for the hydrolysis of 1-I in presence of GSH was further monitored in situ by acquiring consecutive EXAFS spectra (Fig. 3). Data analysis used the structural models obtained from EXAFS spectra of the experimental standards and DFT calculations for the 1-GS/NN intermediate. Initially, an in situ XAS experiment was carried at pH 7, at an Os![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GSH mol ratio of 1:2 (Fig. 3a). Under these conditions (30 mM 1-I, 60 mM GSH; pH 7) Os–I hydrolysis was much faster than in HPLC experiments, and the reaction was complete after ca. 40 min, with only minor further spectral modifications up to 220 min. Linear combination fits (LCF) indicated the dominant presence of 1-SG and 1-Cl after 80 min, accounting for >80% of the Os species in the reaction mixture, together with contributions of <10% from 1-I and 1-OH (Table S9 and Fig. S33, ESI†). LCF provided estimates of the combined % of chlorido and thiolato adducts (Table S9, ESI†) since 1-Cl and 1-SG could not be readily discriminated by EXAFS analysis. In contrast, when the same experiment (30 mM 1-I, 60 mM GSH) was repeated at pH 3, EXAFS spectra were almost identical to 1-I standards, and remained unchanged over the duration of study (Fig. 3b). This suggested that it is possible to capture the dynamics of some steps in these reactions from XAS measurements by careful tuning of the reaction conditions.
:GSH mol ratio of 1:2 (Fig. 3a). Under these conditions (30 mM 1-I, 60 mM GSH; pH 7) Os–I hydrolysis was much faster than in HPLC experiments, and the reaction was complete after ca. 40 min, with only minor further spectral modifications up to 220 min. Linear combination fits (LCF) indicated the dominant presence of 1-SG and 1-Cl after 80 min, accounting for >80% of the Os species in the reaction mixture, together with contributions of <10% from 1-I and 1-OH (Table S9 and Fig. S33, ESI†). LCF provided estimates of the combined % of chlorido and thiolato adducts (Table S9, ESI†) since 1-Cl and 1-SG could not be readily discriminated by EXAFS analysis. In contrast, when the same experiment (30 mM 1-I, 60 mM GSH) was repeated at pH 3, EXAFS spectra were almost identical to 1-I standards, and remained unchanged over the duration of study (Fig. 3b). This suggested that it is possible to capture the dynamics of some steps in these reactions from XAS measurements by careful tuning of the reaction conditions.
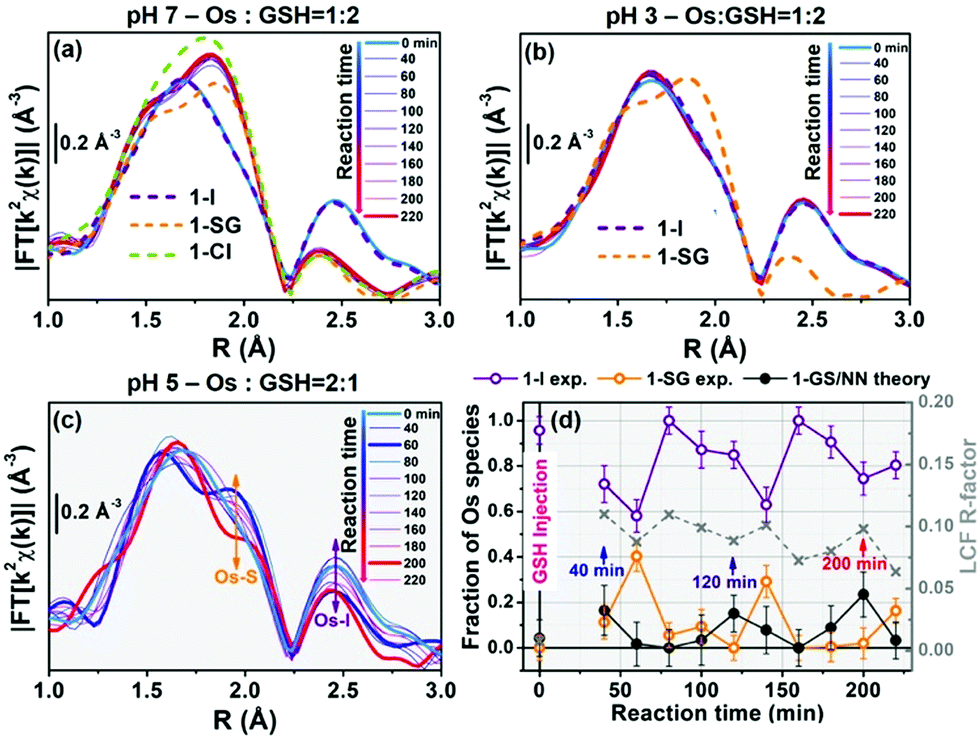 | ||
| Fig. 3 Time-resolved in situ FT-EXAFS during reaction between 1-I (30 mM) and GSH in buffered aqueous solutions at (a) pH 7, Os:GSH = 1:2, (b) pH 3, Os:GSH = 1:2 and (c) pH 5, Os:GSH = 2:1. (d) LCF analysis of the time-resolved k2χ(k) curves resulting in the EXAFS spectra in (c), yielding concentration profiles for reactant 1-I, product 1-SG, and proposed intermediate 1-GS/NN, which all appear to oscillate but with different phases. LCF R-factors (grey x) are also given (right axis). In (a and b) FT-EXAFS spectra of solid-state references are dashed lines, for comparison. All FT-EXAFS spectra have been transformed in the 2.0–12.5 Å−1 range. | ||
The reaction of 1-I (30 mM) with GSH was further studied at pH = 5, at an Os:GSH ratio of 2:1, in an attempt to moderate the reaction rate (as the [GS−] at pH 5 is two orders of magnitude lower than at pH 7, according to the Henderson–Hasselbalch equation;16Fig. 3c and Fig. S34, ESI†). Remarkably, EXAFS spectra in the 40–220 min reaction time range, showed an oscillatory behaviour. Periodic and opposite changes in the intensities of the peaks at ca. 2.5 Å (Os–I single scattering [SS] contribution from 1-I), and the shoulder at ca. 2.0 Å (Os–S SS contribution from 1-SG) in the phase-uncorrected spectra were observed (Fig. 3c). Yet, certain time points showed dampened contributions from Os–I SS without an increase in the characteristic Os–S SS (e.g. after 200 min; red curve in Fig. 3c), suggesting the presence within the reaction of additional unknown Os species. Furthermore, the cyclic behaviour might arise from alternating periods dominated by cleavage or formation of Os–I bonds. To check this, Os-speciation as a function of the reaction time was evaluated by applying EXAFS LCF analysis in k-space (Fig. 3d). Based on our observations, three different components were used to generate the LCF model: the reference compounds 1-I and 1-SG and the simulated EXAFS signature for the 1-GS/NN intermediate predicted by theory. The model adopted reproduced the experimental data well (Fig. S35, ESI†). Inclusion in the LCF model of the 1-GS/NN component resulted in a significant improvement of the fitting quality for the time points with high concentrations of such species (coloured arrows in Fig. 3d) with respect to a two-component model of only 1-I and 1-SG (Fig. S36, ESI†). Crucially, the maxima in the relative abundance of 1-GS/NN (16–24% total Os) systematically preluded increases in the concentration of 1-SG. These data supported the presence of the 1-GS/NN species during the reaction, in line with its intermediate character defined by DFT. LCF results highlighted the oscillating character of the reaction. 1-I was cyclically converted into 1-SG (with a period of 60–80 min), which then was transformed again to 1-I. This may be related to fluctuations in the concentrations of free I− ions and free GSH as a result of the hydrolysis of Os–I bonds, and the formation of new Os–SG species. Indeed, our chromatographic experiments have shown previously that the presence of high concentrations of Cl− ions in the medium can alter the amount of 1-Cl detected during the hydrolysis of 1-I in the presence of GSH. Moreover, a more complex four-component LCF model (with 1-I, 1-SG and 1-OH and simulated 1-GS/NN references) suggested negligible presence of 1-OH in the reaction mixture (Fig. S37, ESI†). This might indicate the presence of high concentrations of free I− ions and free GSH. By calculating the concentrations of free I− ions and free GSH (from amounts of 1-I and 1-SG species indicated by LCF), we observed an alternation between long periods of low I−/GSH ratios and short periods of high I−/GSH ratios (every 60–80 min). Remarkably, the highest concentrations of free I− ions were always just before periods dominated by the formation of Os–I bonds (Fig. S38, ESI†), providing a possible explanation for the regeneration of 1-I. The presence of fluctuations in GSH, together with complete hydrolysis of Os–I bonds (31.2 mM in total, calculated from amounts of 1-I and 1-SG indicated by LCF) by 15 mM of GSH suggested a catalytic reaction, with the thiol being regenerated. This was confirmed by the almost total conversion of 1-Cl to 1-OH after 12 h incubation with sub-stoichiometric quantities of GSH (0.5, 0.25 or 0.125 mol equiv.; Fig. S39, ESI†). Furthermore, significant amounts of 1-OH (525 μM) were generated with little consumption of GSH when 1-I (2 mM) reacted with 0.1 mol equiv. of the thiol (200 μM) for 2 h (Fig. 4) under N2 (protecting GSH from oxidation). 1H NMR spectra provided a turnover frequency (TOF) number of 1.97 h−1 for the reaction under such conditions.
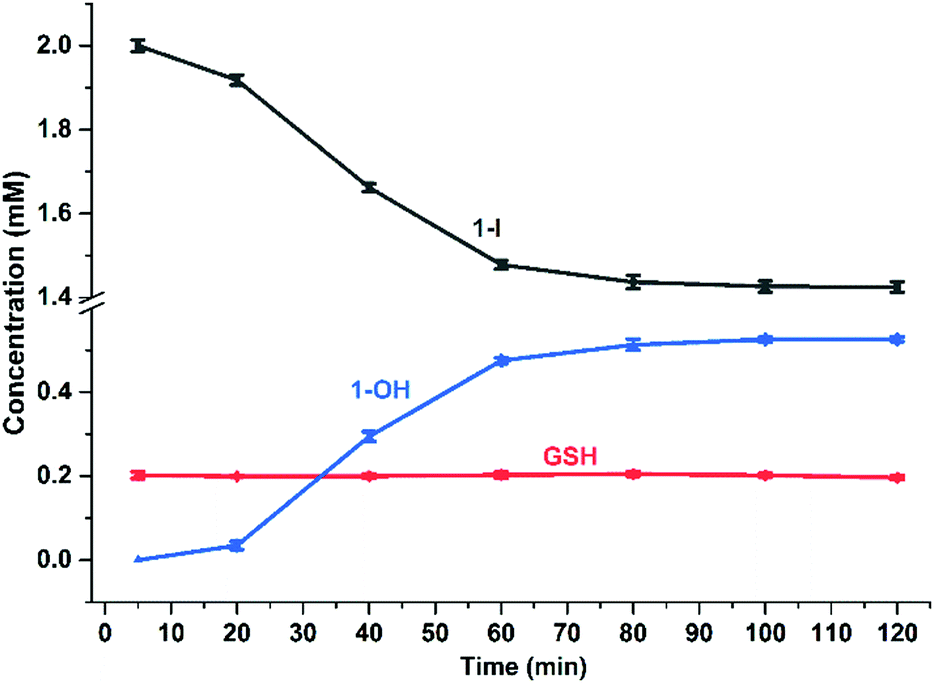 | ||
| Fig. 4 Time-dependent concentration changes for GSH, 1-I and 1-OH in the reaction of 1-I (2 mM) with GSH (200 μM), 75 mM phosphate buffer (pH 7.4), 298 K, for 2 h based on integration of 1H NMR peak areas (Table S10 and Fig. S40, ESI†). | ||
In summary, DFT calculations suggested that substitution of bound iodide by water in the inert organo-osmium anticancer complex 1-I is dependent on high energy kinetic barriers, but revealed a favourable reaction pathway involving attack of GS− on the –NN– azo bond in the chelating ligand, giving a 1-GS/NN intermediate (Fig. S31, ESI†) containing an N–SG bond.
Experimental chromatographic analysis of the GSH-induced hydrolysis then confirmed the importance of the azo ligand in the mechanism of the hydrolysis, and demonstrated enhanced rates at high pH and inhibition at low pH (importance of GS− in the attack). Time resolved XAS data provided evidence for the proposed 1-GS/NN intermediate during the hydrolysis reaction. Our experiments also revealed the possible catalytic nature of the reaction (TOF ca. 2 h−1). Overall, these investigations reveal a novel activation pathway for a metallodrug in which an inert metal–ligand bond is activated towards hydrolysis, and subsequent interactions with biomolecules, by the attack of the abundant intracellular thiol glutathione on a chelating ligand. Such findings have implications for the design of new generations of anticancer metallodrugs.
XAS spectra were acquired in the frame of the ESRF proposal CH-5212. We thank M. Monte Caballero and E. M. Bolitho for the support during experiments at BM23. We thank CRUK/EPSRC (Grant No. C53561/A19933), EPSRC (Grant No. EP/F034210/1) and Wellcome Trust (Grant No. 107691/Z/15/Z) for financial support.
Conflicts of interest
There are no conflicts to declare.References
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelón, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 1017 CrossRef CAS.
- J. M. Hearn, I. Romero-Canelon, A. F. Munro, Y. Fu, A. M. Pizarro, M. J. Garnett, U. McDermott, N. O. Carragher and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3800 CrossRef CAS PubMed.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993 CrossRef CAS; R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 1596 CrossRef; Z. Wang, Z. Deng and G. Zhu, Dalton Trans., 2019, 48, 2536 RSC.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458 RSC; R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925 RSC.
- A. Blazevic, A. A. Hummer, P. Heffeter, W. Berger, M. Filipits, G. Cibin, B. K. Keppler and A. Rompel, Sci. Rep., 2017, 7, 40966 CrossRef CAS; E. Alessio, Eur. J. Inorg. Chem., 2017, 1549 CrossRef; E. Alessio and L. Messori, Molecules, 2019, 24, 1995 CrossRef.
- I. Ritacco, N. Russo and E. Sicilia, Inorg. Chem., 2015, 54, 10801 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161 CrossRef CAS PubMed; W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643 CrossRef; H. Zhang, L. Guo, Z. Tian, M. Tian, S. Zhang, Z. Xu, P. Gong, X. Zheng, J. Zhao and Z. Liu, Chem. Commun., 2018, 54, 4421 RSC.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734 CrossRef CAS PubMed.
- E. M. Kosower and T. Miyadera, J. Med. Chem., 1972, 15, 307 CrossRef CAS PubMed; E. M. Kosower and H. Kanety-Londner, J. Am. Chem. Soc., 1976, 98, 3001 CrossRef PubMed; C. Boulègue, M. Löweneck, C. Renner and L. Moroder, ChemBioChem, 2007, 8, 591 CrossRef PubMed.
- G. Li, Y. Chen, J. Wu, L. Ji and H. Chao, Chem. Commun., 2013, 49, 2040 RSC; Q.-X. Zhou, Y. Zheng, T.-J. Wang, Y.-J. Chen, K. Li, Y.-Y. Zhang, C. Li, Y.-J. Hou and X.-S. Wang, Chem. Commun., 2015, 51, 10684 RSC.
- S. J. Dougan, A. Habtemariam, S. E. McHale, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11628 CrossRef CAS PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508 CrossRef CAS PubMed.
- S. Bordiga, E. Groppo, G. Agostini, J. A. van Bokhoven and C. Lamberti, Chem. Rev., 2013, 113, 1736 CrossRef CAS PubMed; C. Garino, E. Borfecchia, R. Gobetto, J. A. van Bokhoven and C. Lamberti, Coord. Chem. Rev., 2014, 277–278, 130 CrossRef; M. L. Baker, M. W. Mara, J. J. Yan, K. O. Hodgson, B. Hedman and E. I. Solomon, Coord. Chem. Rev., 2017, 345, 182 CrossRef PubMed; S. N. MacMillan and K. M. Lancaster, ACS Catal., 2017, 7, 1773 Search PubMed.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548 CrossRef CAS PubMed.
- C. Sanchez-Cano, D. Gianolio, I. Romero-Canelon, R. Tucoulou and P. J. Sadler, Chem. Commun., 2019, 55, 7065 RSC.
- L. B. Poole, Free Radical Biol. Med., 2015, 80, 148 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc06725f |
| ‡ These authors contributed equally to the manuscript. |
| § Current address: CIC biomaGUNE, Parque Científico y Tecnológico de Gipuzkoa, Paseo Miramón 182, 20014 San Sebastian, Spain. E-mail: csanchez@cicbiomagune.es |
| This journal is © The Royal Society of Chemistry 2019 |