
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced oxygen reduction activity of platinum subnanocluster catalysts through charge redistribution†
Hironori
Tsunoyama‡
 a,
Akira
Ohnuma‡
b,
Koki
Takahashi
a,
Archana
Velloth
c,
Masahiro
Ehara
c,
Nobuyuki
Ichikuni
d,
Masao
Tabuchi
e and
Atsushi
Nakajima
*a
a,
Akira
Ohnuma‡
b,
Koki
Takahashi
a,
Archana
Velloth
c,
Masahiro
Ehara
c,
Nobuyuki
Ichikuni
d,
Masao
Tabuchi
e and
Atsushi
Nakajima
*a
aDepartment of Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, Kanagawa 223-8522, Japan. E-mail: nakajima@chem.keio.ac.jp
bNew Field Pioneering Division, Toyota Boshoku Corp., 1-1 Toyoda-cho, Kariya, Aichi 448-8651, Japan
cDepartment of Theoretical and Computational Molecular Science, Institute for Molecular Science, Myodaiji, Okazaki 444-8585, Japan
dDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
eSynchrotron Radiation Research Center, Nagoya University, Furo-cho, Chikusa, Nagoya 464-8603, Japan
First published on 26th September 2019
Abstract
Single-size platinum Pt6 subnanoclusters exhibit superior mass-specific and surface-specific activities for the oxygen reduction reaction. The enhanced activity is attributed to polarized electron distributions based on rigorous structure characterization by X-ray absorption fine structure spectroscopy and density functional theory.
The development of durable and active catalysts with high atomic efficiency has been a key issue for a sustainable world, where size-selected, sub-nanoscale clusters (sub-NCs) consisting of a few to tens of atoms have been strong potential candidates attributable to their high surface area and tunability of activity by “atomicity”. A wide variety of catalytic properties of sub-NCs have been extensively reported,1–8 and among the various uses for the catalytic transformations, fuel cell (FC) technology appears to be a promising way to reduce environmental loads based on the potential of a principally carbon-free energy source.9–11 Among the various types of FCs, proton-exchange membrane FCs have attracted much attention due to their low-temperature operation, which makes them particularly suitable for mobile vehicles. Although various technological advancements are needed to upgrade FCs, the development of more active catalysts is required for the cost reduction and the downsizing of FC units. Since the activity for the cathodic reaction, the oxygen reduction reaction (ORR), is lower than that for the anodic hydrogen oxidation reaction, improvements in mass specific activity (MA/A gPt−1) for the ORR are essential. To this end, various active catalysts have been developed so far, using e.g., core–shell, alloy, and unique nanostructures,9,10,12–14 by tuning the reactivity through ligand, strain, and ensemble effects.15,16
For platinum (Pt)-based nanoparticle (NP) catalysts, one of the convincing arguments for catalyst development is to increase the surface-to-volume ratio through downsizing the catalytic NPs; MA increases with the surface ratio, and reaches a constant value above 80 m2 gPt−1.14 In terms of high surface ratio, NCs with less than ten atoms are good candidates to enhance the activity. Although the atomicity is a unique parameter for the activity in monometallic Pt NCs, size specificity is still controversial;2,17–24 the difficulty arises from the complex nature of catalysts, e.g., non-systematic variation of structures with the size and effect of surrounding materials (supports and ligated molecules). For a systematic understanding of NC catalysis, a dry fabrication method which reduces the complexity through the entirely clean nature of the synthetic procedure carried out under high vacuum conditions, facilitates the catalysis preparation of naked NCs;2,3,25–30 high surface specific activity (SA) of Pt46 NCs has been reported at high coverage.2 Despite numerous efforts, the correlation between activity and structure has not been elucidated so far, which is crucial to establish a reliable guiding principle for NC catalysts.
We have demonstrated herein that the ORR activity of Pt6 sub-NCs supported on a glassy carbon (GC) electrode shows superior activity compared to a standard Pt/C catalyst, the origin of which is discussed in terms of the geometric and electronic structures investigated via X-ray absorption fine structure (XAFS) spectroscopy and density functional theory (DFT) calculations.
Pt NC catalysts were prepared by soft-landing of size-selected NCs using a high-flux single-size NC generation system, nanojima®-NAP01,31,32 combined with a quadrupole mass filter (Fig. S2, ESI†), where the collision energy of the NCs was kept under 1 eV per atom to avoid fragmentation during the deposition. In order to suppress agglomerations of NCs on the GC surface, the density of deposited Pt6 sub-NCs was kept under 0.5 ML equivalents based on the density of a Pt(111) surface by assuming that the GC electrode has a flat surface. In addition, prepared catalysts were kept under an oxygen-free argon atmosphere until just before the electrochemical (EC) measurements to avoid oxidation of tiny NCs and contamination by organic substances. Successful immobilization of Pt NCs was confirmed by high-resolution scanning transmission electron microscopy as shown in Fig. S3 (ESI†).
Fig. 1a shows a representative cyclic voltammogram under an argon atmosphere. Current for underpotential adsorption/desorption of hydrogen is clearly observed around 0.05–0.3 VRHE. The electrochemically active surface area (ECSA) was evaluated based on the reported procedure33 by integrating the hydrogen desorption current using a factor of 210 μC cmPt−2. Surprisingly the ECSA for Pt6 (Table 1) is ca. 0.8 times that of a commercial standard Pt/C catalyst (TEC10E50E, TKK) with a crystallite size of 3 nm, which is a large deviation from the hypothetical surface ratio for such tiny Pt6 sub-NCs. The origin of the decrease in the ECSA is discussed based on its structure in a later section. A hydrodynamic voltammogram under oxygen (Fig. 1b) represents clear ORR activity despite the ultra-low loading of NC catalysts using the reported procedure.34 Since high sensitivity measurements for the present ultra-low loading catalyst are susceptible to impurities, it is difficult to evaluate kinetic limiting current density (Jkin) from the Koutecky–Levich plot, and so the ORR Jkin has been estimated using the Koutecky–Levich equation based procedure reported by Yamamoto and co-workers.17,18 Despite the relatively lower ECSA for Pt6, the MA at 0.9 VRHE for Pt6/GC is 74% higher than that of the standard Pt/C catalyst. Furthermore, the Pt6 SA at 0.9 VRHE is 2.3 times higher than that for the Pt/C catalyst. For the typical Pt NP catalyst with a diameter larger than 3 nm, the MA increases with increasing ECSA, while SA increases with decreasing ECSA for larger NPs.14 In this system, both the MA and SA of Pt6 are higher than those for Pt NPs, which is in sharp contrast to conventional NP catalysts.
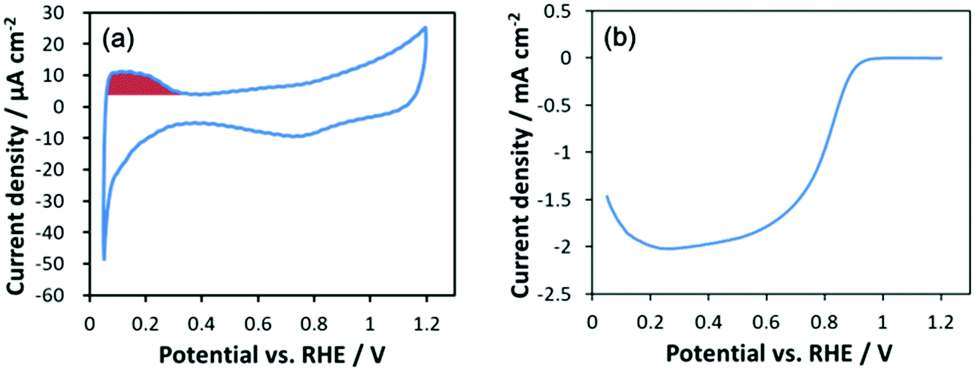 | ||
| Fig. 1 A cyclic voltammogram under argon (a) and a hydrodynamic voltammogram under oxygen (b) for Pt6/GC. The red area in (a) corresponds to the desorption of hydrogen. Experimental conditions: concentration of HClO4; 0.1 M and sweep rate; 50 mV s−1. | ||
| ECSAb (m2 gPt−1) | MAc (A mgPt−1) | SAd (mA cmPt−2) | |
|---|---|---|---|
| a Uncertainties are given in parentheses, referred to the corresponding last digit. b Evaluated from desorption current of hydrogen. c Obtained from kinetic limiting current at 0.9 VRHE. d Obtained from kinetic limiting current at 0.9 VRHE and ECSA. e Standard Pt/C catalyst (TEC10E50E). | |||
| Pt6/GC | 54 (8) | 0.52 (8) | 0.96 (7) |
| Pt/Ce | 71 (2) | 0.30 (0) | 0.42 (1) |
We now discuss the origin of the superior activity of Pt6/GC catalysts in terms of their geometric and electronic structures through rigorous characterizations using extended XAFS (EXAFS) and DFT calculations. The charge state for deposited Pt6 sub-NCs is almost neutral based on analysis of the X-ray absorption near edge structure (XANES) as shown in Fig. 2a, where the white-line for the Pt L3 edge is almost identical with that for bulk Pt foil. Surprisingly the Pt6/GC catalyst is robust against oxidation under ambient conditions (Fig. S4, ESI†), suggesting that the structure of Pt6 sub-NCs remains unchanged in EC experiments under an oxygen atmosphere from the EXAFS measurements under argon. The XAFS χ(R) profile (Fig. 2b) clearly exhibits a radial distribution peak for Pt–Pt bonds; Pt–Pt bond distance and its coordination number (CN) for Pt6/GC are slightly smaller and much smaller than those for the bulk Pt, respectively (Table S2, ESI†). In order to characterize the Pt6 structure in detail, we have compared between (1) experimental χ(k) spectra (χobs(k)) extracted with the Athena program35 and (2) those simulated for the calculated equilibrium geometries by the FEFF code36 (χsim(k)). As a result of DFT calculations using the VASP program,37,38 where the GC surface was modeled with a graphene (Gr) structure, seven isomers were found for Pt6/Gr (Fig. S5, ESI†) within a total energy difference of 0.5 eV. Details of geometry optimization are explained in the ESI.† Based on the matching of the phases between χobs(k) and χsim(k) spectra for Pt6/GC, we can safely exclude planar isomers (Pt6-1–3) due to the deviation at the higher k regime (k > 6 Å−1) (Fig. S6, ESI†). Bent triangular isomers (Pt6-4–6) can also be excluded due to a slight phase deviation at k > 8 Å−1. By considering the structure scaling (−2% to +2%) for the optimized Pt6/Gr geometry, the above conclusion is thoroughly supported (Fig. S7, ESI†). Since both the phase and amplitude are quantitatively matched in Fig. 2c, the most probable structure for Pt6/GC is concluded to be the isomer Pt6-7 (Fig. 2d). Although the isomer Pt6-7 is not the most stable one in the DFT calculations, the discrepancy may be explained by the occurrence of a different structure on the GC surface39 from that on Gr.
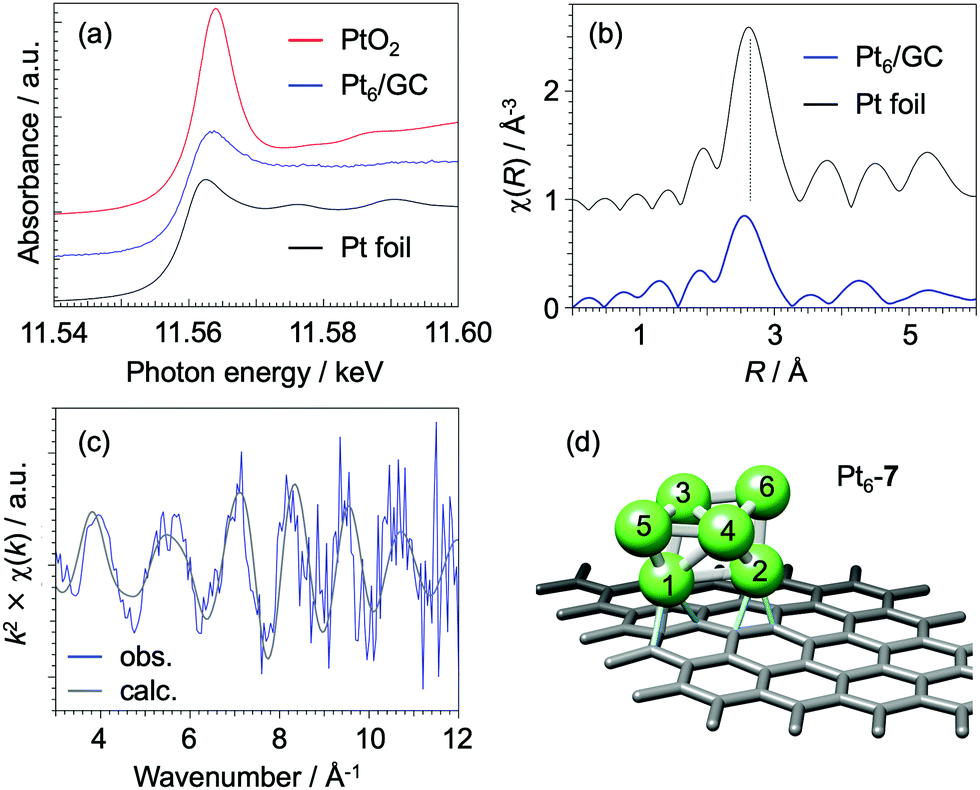 | ||
| Fig. 2 Results of EXAFS spectroscopy for Pt6/GC. (a) XANES spectra that have been offset vertically for clarity of presentation, (b) χ(R) profile obtained by Fourier transform of χ(k) spectra in the range k = 5–12 Å−1, and (c) observed χ(k) spectra along with simulation for the most probable isomer of Pt6-7. Simulation conditions: energy shift: +5.0 eV, distance shift: +0.075 Å, Debye–Waller factor: 0.008. (d) Optimized structure for Pt6-7 on graphene from DFT calculations. | ||
The origin of the size-specific activity is discussed in terms of their electronic structure and the reported reaction mechanism for the ORR.40,41 A Bader charge analysis (Fig. 3a) shows that Pt6 sub-NC is polarized on the Gr surface; two Pt sites with a low CN (CN = 3, Pt(6) and Pt(5)) are negatively charged (q = −0.15) while the other Pt atoms are positively charged; two Pt not coordinated to Gr (q = +0.04) and two Pt coordinated to Gr (q = +0.09, +0.13). Charge redistribution upon adsorption can be also identified from the electron-density-difference isosurface shown in Fig. 3b; charge accumulation regions are spread over the Pt(5) atom and Pt(1)–Pt(5) bond. In the ORR mechanism,40,41 dissociative adsorption of molecular O2 on a Pt site is an important step to 4e− reduction for direct formation of water, which results in higher ORR activity rather than 2e− reduction to form hydrogen peroxide through molecular O2 adsorption. Electron density tends to accumulate at the apex site in NCs and the negatively charged site is reactive.42 In a kinetic picture, the first electron transfer step to an O2 molecule is an important step, where cleaving the O–O bond forms adsorbed atomic oxygen (O*), although the rate determining step for the 4e− reduction pathway in a thermodynamic picture is reduction of adsorbed hydroxy radical species (OH*).40 To activate the adsorbed molecular O2 through electron transfer, the d-band center level for a Pt adsorption site plays a crucial role, because a relatively higher d-band-center with respect to bulk Pt(111) enhances the activity.41 In fact, the d-band-center closely correlates with a partial charge on the site; a partial negative charge pushes the d-band-center up.
 | ||
| Fig. 3 Atomic charge distribution of Pt6/Gr (isomer 7). (a) Bader charge and (b) electron density difference iso-surface; charge depletion and accumulation regions are shown in red and green, respectively. | ||
Two possibilities can be considered as the reasons for the high activity of Pt6/GC; (i) the high surface area of Pt6 and (ii) the site-specific activation of oxygen molecules as described above. The ratio of surface atoms (Psurf) in NPs depends on the diameter; Psurf values are 45% for Pt561 (d = 2.97 nm) and 63% for Pt147 (d = 1.89 nm) based on a spherical geometry of cuboctahedron or icosahedron. On the other hand, the Pt6-7 sub-NC has four outermost Pt atoms and two Pt atoms ligating to carbon, producing a Psurf of 66%, which is almost comparable or at most 20% higher than that of the standard Pt/C catalyst (d = 3 nm). Therefore, the effect of surface atom exposure probably plays a minor role in the enhanced activity. In the electronic structure of larger NPs, such as Pt309 (d = 2.43 nm), the density of states and d-band-center energy look similar to those for a bulk Pt surface based on DFT.43 In addition, atomic charge distribution in Pt309 NC seems less polarized even after adsorption on Gr, excluding the peripheral atoms.43 As a result, the ORR activity of NPs is mainly influenced by Psurf values as summarized in the previous literature,14 which has been believed to be a key principle in the catalyst development. On the other hand, Pt6 is highly polarized (−0.15 to +0.13 e) after adsorption on the Gr (Fig. 3a). As a result of charge redistribution in Pt6, the average of the d-orbital energy, corresponding to the d-band-center in the bulk, for negatively charged Pt sites is −1.47 eV from the Fermi energy (εF), which is higher in energy than other Pt sites (−1.81 to −1.66 eV). The d-orbital-center energy is much higher than the d-band center for larger NPs and the bulk (111) surface.44 Based on our knowledge of the correlation between the d-band-center and ORR activity,41 the enhanced MA for Pt6/GC can be attributed to its electronic structure changed by a large degree of charge separation in the tiny NCs. On the other hand, ECSA determined from hydrogen adsorption/desorption is smaller than the larger NP of Pt/C. This probably originates from site specific adsorption44 of hydrogen on the smaller NCs, which also supports the idea of a large change in the surface chemical nature for Pt6 catalysts; Pt sites charged neutral presumably act as adsorption sites for hydrogen. The nature of site-specificity for sub-NCs, originating from a greater flexibility of charge redistribution, will enable much higher activity by further alloying and strong support interactions through electronic interaction (ligand and strain effects)16 in addition to the geometric ensemble effect.15
In conclusion, we have demonstrated superior mass-specific and surface-specific activities of Pt6 subnanoclusters for oxygen electroreduction, which are 1.7 and 2.3 times higher than the conventional standard Pt/C catalysts with a crystallite diameter of 3 nm. Based on rigorous structure analyses by X-ray absorption fine structure spectroscopy and density functional theory calculations, we have identified the geometric structure of Pt6. The origin of simultaneous enhancements of SA and MA for ORR, which is unusual in pure metallic Pt catalysts, is attributed to larger charge redistribution in the tiny sub-NC, leading to pushing the d-band-center energy up compared to the NP catalysts. The present study provides an important guideline for catalyst developments and opens “precise NC catalysis chemistry” for a future FC technology.
The authors thank Dr Tomoyuki Nagai for the advice on electrochemical measurements, Dr Kouta Iwasaki for discussion and comments on the manuscript, Mr Motoki Kondo for the experimental support, and Prof. Dr Hidehiro Yasuda and Prof. Dr Kazuhisa Sato for STEM measurements. Development on nanojima® apparatus was supported by Ayabo Corp. The computations were partly performed using Research Center for Computational Science, Okazaki, Japan. HAADF-STEM measurements were supported by “Advanced Characterization Nanotechnology Platform, Nanotechnology Platform Program of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan” at the Research Center for Ultra-High Voltage Electron Microscopy (Nanotechnology Open Facilities) in Osaka University. This work is partly supported by JSPS KAKENHI of Grants-in-Aid for Scientific Research (A) Grant No. 15H02002 and 19H00890, of Challenging Research (Pioneering) Grant No. 17H06226, and of Grant-in-Aid for Young Scientists (A) Grant No. 15H05475.
Conflicts of interest
The authors declare the following competing financial interest(s): H. T. and A. N. are inventors on JAPAN patent JP 5493139, submitted by JST agency and Ayabo Corp., which covers the nanocluster generator.Notes and references
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS PubMed.
- M. Nesselberger, M. Roefzaad, R. F. Hamou, P. U. Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919–924 CrossRef CAS PubMed.
- A. von Weber and S. L. Anderson, Acc. Chem. Res., 2016, 49, 2632–2639 CrossRef CAS PubMed.
- S. Yamazoe, K. Koyasu and T. Tsukuda, Acc. Chem. Res., 2014, 47, 816–824 CrossRef CAS PubMed.
- K. Yamamoto and T. Imaoka, Acc. Chem. Res., 2014, 47, 1127–1136 CrossRef CAS PubMed.
- R. M. Anderson, D. F. Yancey, L. Zhang, S. T. Chill, G. Henkelman and R. M. Crooks, Acc. Chem. Res., 2015, 48, 1351–1357 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019 DOI:10.1021/acs.chemrev.8b00726.
- D. Banham and S. Ye, ACS Energy Lett., 2017, 2, 629–638 CrossRef CAS.
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- X. Wang, A. Ruditskiy and Y. Xia, Natl. Sci. Rev., 2016, 3, 520–533 CAS.
- D. Thompsett, in Handbook of Fuel Cells: Fundamentals, Technology, and Applications, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, Wiley, Chichester England, 2003, ch. 37, vol. 3, pp. 467–480 Search PubMed.
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed.
- L. Zhang and G. Henkelman, J. Phys. Chem. C, 2012, 116, 20860–20865 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9810–9815 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS PubMed.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- M. Rück, A. Bandarenka, F. Calle-Vallejo and A. Gagliardi, J. Phys. Chem. Lett., 2018, 9, 4463–4468 CrossRef PubMed.
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- M. Peuckert, T. Yoneda, R. A. Dalla Betta and M. Boudart, J. Electrochem. Soc., 1986, 133, 944–947 CrossRef CAS.
- N. Giordano, E. Passalacqua, L. Pino, A. S. Arico, V. Antonucci, M. Vivaldi and K. Kinoshita, Electrochim. Acta, 1991, 36, 1979–1984 CrossRef CAS.
- P. Brault, A. Caillard and A.-L. Thomann, Chem. Vap. Deposition, 2011, 17, 296–304 CrossRef CAS.
- H. Qayyum, C.-J. Tseng, T.-W. Huang and S.-y. Chen, Catalysts, 2016, 6, 180 CrossRef.
- N. Todoroki, T. Kato, T. Hayashi, S. Takahashi and T. Wadayama, ACS Catal., 2015, 5, 2209–2212 CrossRef CAS.
- Y. Agawa, H. Tanaka, S. Torisu, S. Endo, A. Tsujimoto, N. Gonohe, V. Malgras, A. Aldalbahi, S. M. Alshehri, Y. Kamachi, C. Li and Y. Yamauchi, Sci. Technol. Adv. Mater., 2015, 16, 024804 CrossRef PubMed.
- R. E. Palmer, R. Cai and J. Vernieres, Acc. Chem. Res., 2018, 51, 2296–2304 CrossRef CAS PubMed.
- J. Laskin, G. E. Johnson, J. Warneke and V. Prabhakaran, Angew. Chem., Int. Ed., 2018, 57, 16270–16284 CrossRef CAS PubMed.
- H. Tsunoyama, C. Zhang, H. Akatsuka, H. Sekiya, T. Nagase and A. Nakajima, Chem. Lett., 2013, 42, 857–859 CrossRef CAS.
- H. Tsunoyama, M. Shibuta, M. Nakaya, T. Eguchi and A. Nakajima, Acc. Chem. Res., 2018, 51, 1735–1745 CrossRef CAS PubMed.
- K. J. J. Mayrhofer, D. Strmcnik, B. B. Blizanac, V. Stamenkovic, M. Arenz and N. M. Markovic, Electrochim. Acta, 2008, 53, 3181–3188 CrossRef CAS.
- T. Nagai, H. Murata and Y. Morimoto, ECS Trans., 2012, 50, 1539–1545 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- S. I. Zabinsky, J. J. Rehr, A. Ankudinov, R. C. Albers and M. J. Eller, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 2995–3009 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- Z. Zhao, E. F. Wang, H. Yan, Y. Kono, B. Wen, L. Bai, F. Shi, J. Zhang, C. Kenney-Benson, C. Park, Y. Wang and G. Shen, Nat. Commun., 2015, 6, 6212 CrossRef CAS PubMed.
- H. A. Hansen, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. C, 2014, 118, 6706–6718 CrossRef CAS.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., 2005, 117, 2170–2173 CrossRef.
- K. Bobuatong, S. Karanjit, R. Fukuda, M. Ehara and H. Sakurai, Phys. Chem. Chem. Phys., 2012, 14, 3103–3111 RSC.
- L. G. Verga, J. Aarons, M. Sarwar, D. Thompsett, A. E. Russell and C.-K. Skylaris, Phys. Chem. Chem. Phys., 2016, 18, 32713–32722 RSC.
- T. R. Garrick, T. E. Moylan, M. K. Carpenter and A. Kongkanand, J. Electrochem. Soc., 2017, 164, F55–F59 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Mass spectrum of Pt NC, STEM image, XANES spectra, EXAFS fitting results, optimized structures of free and adsorbed Pt NC by DFT calculations, and simulations for XAFS. See DOI: 10.1039/c9cc06327g |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2019 |