
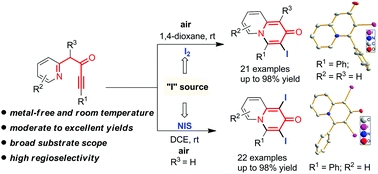
Controllable synthesis of 3-iodo-2H-quinolizin-2-ones and 1,3-diiodo-2H-quinolizin-2-ones via electrophilic cyclization of azacyclic ynones†
Yan-Bo
Wang  *a
*a
*a
Abstract
An effective electrophilic annulation reaction of azacyclic ynones was reported, divergently affording various functionalized 3-iodo-2H-quinolizin-2-ones and 1,3-diiodo-2H-quinolizin-2-ones in moderate to excellent yields with different iodide reagents. This reaction shows high regioselectivity and broad substrate scope under metal-free, room temperature conditions in air. In addition, the products with highly active C–I bonds have an opportunity for further functionalization.
 Please wait while we load your content...
Please wait while we load your content...

