
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUV photostability of three 2-aminoazoles with key roles in prebiotic chemistry on the early earth†
Zoe R.
Todd
 *ab,
Rafał
Szabla
c,
Jack W.
Szostak
b and
Dimitar D.
Sasselov
a
*ab,
Rafał
Szabla
c,
Jack W.
Szostak
b and
Dimitar D.
Sasselov
a
aDepartment of Astronomy, Harvard-Smithsonian Center for Astrophysics, 60 Garden Street, Cambridge, MA 02138, USA. E-mail: zoe.todd@cfa.harvard.edu
bHoward Hughes Medical Institute, Department of Molecular Biology and Center for Computational and Integrative Biology, Massachusetts General Hospital, 185 Cambridge Street, Boston, MA 02114, USA
cInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, PL-02668, Warsaw, Poland
First published on 5th August 2019
Abstract
Three related molecules in the 2-aminoazole family are potentially important for prebiotic chemistry: 2-aminooxazole, 2-aminoimidazole, and 2-aminothiazole, which can provide critical functions as an intermediate in nucleotide synthesis, a nucleotide activating agent, and a selective agent, respectively. Here, we examine the wavelength-dependent photodegradation of these three molecules under mid-range UV light (210–290 nm). We then assess the implications of the observed degradation rates for the proposed prebiotic roles of these compounds. We find that all three 2-aminoazoles degrade under UV light, with half lives ranging from ≈7–100 hours under a solar-like spectrum. 2-Aminooxazole is the least photostable, while 2-aminoimidazole is the most photostable. The relative photostabilities are consistent with the order in which these molecules would be used prebiotically: AO is used first to build nucleotides and AI is used last to activate them.
Recent advances in prebiotic cyanosulfidic chemistry informed by the environmental constraints on the Hadean–Archaean Earth suggest that a robust chemical network may have generated all four major types of building blocks of life: sugars, amino acids, ribonucleotides, and lipid precursors.1 In this network, critical roles are played by three molecules in the 2-aminoazole family: 2-aminooxazole (AO), 2-aminoimidazole (AI), and 2-aminothiazole (AT) (Fig. 1). 2-Aminooxazole is a key intermediate in the pathway towards synthesizing pyrimidine ribonucleotides2 and a complete set of arabino nucleotides.3 2-Aminoimidazole is capable of activating ribonucleotides so that they are able to copy RNA templates non-enzymatically with greater efficiency than previously observed with other leaving groups.4 Finally, 2-aminothiazole has been shown to be a potentially important selecting agent, allowing for stabilization and purification of simple sugars as their crystalline aminal derivatives. These derivatives enable nucleotide synthesis from a complex mixture of starting products as well as a pathway to amino acids without formation of non-canonical α–α-disubstituted derivatives.5 All three of these 2-aminoazoles could have potentially been synthesized prebiotically in the same or similar environments. Recent results show a divergent synthesis of 2-aminooxazole and 2-aminoimidazole from cyanamide and glycolaldehyde in the presence or absence of ammonia.6 2-Aminothiazole can similarly be synthesized from cyanamide and β-mercaptoacetaldehyde.5
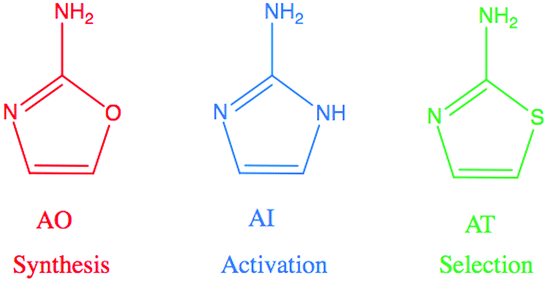 | ||
| Fig. 1 2-Aminooxazole (AO), 2-aminoimidazole (AI), and 2-aminothiazole (AT) are related molecules in the 2-aminoazole family with distinct potential roles in prebiotic chemistry. AO is a key intermediate in the synthesis of pyrimidine ribonucleotides. AI can activate RNA monomers to allow for efficient non-enzymatic replication, and AT can help to sequester, stabilize, and crystallize molecules. | ||
UV light has been suggested to be a potentially important source of energy for driving prebiotic chemistry.7–9 Indeed, UV light is required in key steps of the chemistry yielding pyrimidine nucleosides and nucleotides under prebiotically plausible conditions. For instance, the UV irradiation of ferrocyanide and sulfite generates the solvated electrons that drive the cyanosulfidic redox chemistry that could generate simple sugars and amino acids from one-carbon feedstock molecules.10,11 Additionally, the selective conversion of C to U ribonucleotides and the degradation of non-canonical nucleotide stereoisomers is also driven by UV.2 Alternative nucleobase analogs such as 2-aminopurine or 5-hydroxyuracil exhibit lower photostability than canonical nucleobases, potentially indicating why they were not incorporated into DNA.12 Consequently, UV light may have been both an important source of energy and a powerful selection factor in the prebiotic era.
The UV light used in laboratory experiments simulating prebiotic chemistry has typically been limited to 254 nm UV from a mercury lamp,1,2,10,13 but the wavelength range of UV light available on the surface of the early Earth is not well represented by monochromatic emission at 254 nm and instead would have reached down to roughly 200 nm.14 The fact that photochemical reactions typically display a dependence on the irradiation wavelength (e.g. Matsunga et al.15) has inspired us to study these reactions as a function of wavelength.16
Furthermore, UV-induced damage (and in some cases, self-repair) of RNA and/or DNA strands has been extensively studied,17 however, much less effort has been put into studying the photostability of prebiotically credible precursors of nucleotides such as the three 2-aminoazoles. Here, following the recent investigation of the photostability of oxazoline precursors of RNA,18 we investigate the wavelength-dependent UV photostability of the three 2-aminoazoles. We then discuss whether these rates allow for a self-consistent network of reactions, in order to place constraints on the relevant prebiotic environment.
To perform these studies, a xenon lamp was coupled with a diffraction grating to allow tunable wavelength selection, as in Todd et al.16 We used experimental irradiation wavelengths of 215–285 nm, in 10 nm intervals with a 10 nm bandwidth, to irradiate 0.1 mM solutions of each of the 2-aminoazoles for 1–8 hours (depending on the rate of the reaction, see ESI,† Section S9). We observed decreases in the UV-vis absorption spectra of each species as a function of irradiation time (Fig. 2) and converted the absorbances into concentrations of each molecule (see ESI,† Section S2). The photodegradation reactions are first order (see ESI,† Section S4), so the logarithm of the concentration vs. time gives a linear trend, with the rate constant given by the slope (Fig. 3). Destruction rates were measured for irradiation wavelengths from 215–285 nm. These rates were then normalized by the incident photon flux (determined by measuring the power at each wavelength) so that rates could be compared across wavelength for a constant photon flux expected on the surface of the early Earth (see ESI,† Section S3 for details). The wavelength dependencies and comparative photostability of the three molecules are shown in Fig. 4. AO and AI are less photostable at shorter irradiation wavelengths compared to longer wavelengths, as would be expected from the maxima in their absorption spectra. It is also worth noting that we have recorded the highest photodestruction rate for AO at 215 nm, which indicates that short irradiation wavelengths (high energies) are necessary to efficiently trigger destructive photorelaxation mechanisms in this molecule. AT has an approximately constant rate of photodestruction at wavelengths below 255 nm and is more photostable at longer wavelengths. Comparing the three species to each other shows that AO is much less photostable than either AI or AT. The comparative photostability of AI and AT depends on the wavelength of light considered (AI is less stable at the shortest wavelengths, while AT is less stable at wavelengths >230 nm). Co-irradiation of AO and AI (0.05 mM) at 215 nm is shown in Fig. 3 (see ESI,† Section S7 for details). The rate of degradation of the AO + AI mixture is less than that of AO alone but more than that of AI alone (see Fig. 4, purple points), indicating a potential protection of AO when irradiated in the presence of another more UV-photostable molecule.
 | ||
| Fig. 2 Absorption spectra of AI, AO, and AT as a function of irradiation time at irradiation wavelengths of 215, 215, and 255 nm, respectively. These wavelengths were chosen to be near the absorption maxima for each molecule, which will have larger changes in absorption. The decreasing absorption intensities with irradiation time allow for the destruction rate to be calculated. | ||
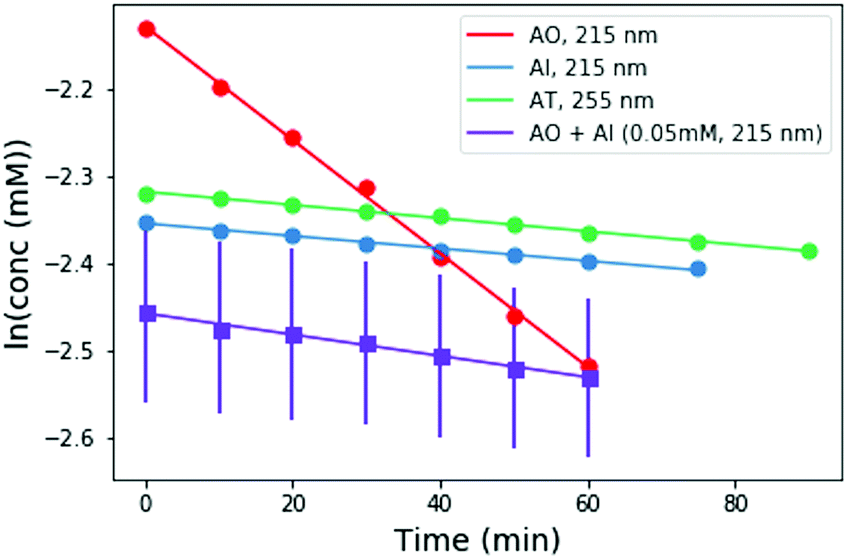 | ||
| Fig. 3 Concentration as a function of irradiation time for AI, and AO at 215 nm, and AT at 255 nm. Concentrations were calculated from the absorption spectra shown in Fig. 2. A mixture of 0.05 mM AO + 0.05 mM AI was also irradiated at 215 nm for comparison (purple points). The rate of degradation of the mixture is less than that of AO alone, indicating that AO can perhaps be protected by co-irradiation with a more UV-photostable molecule. | ||
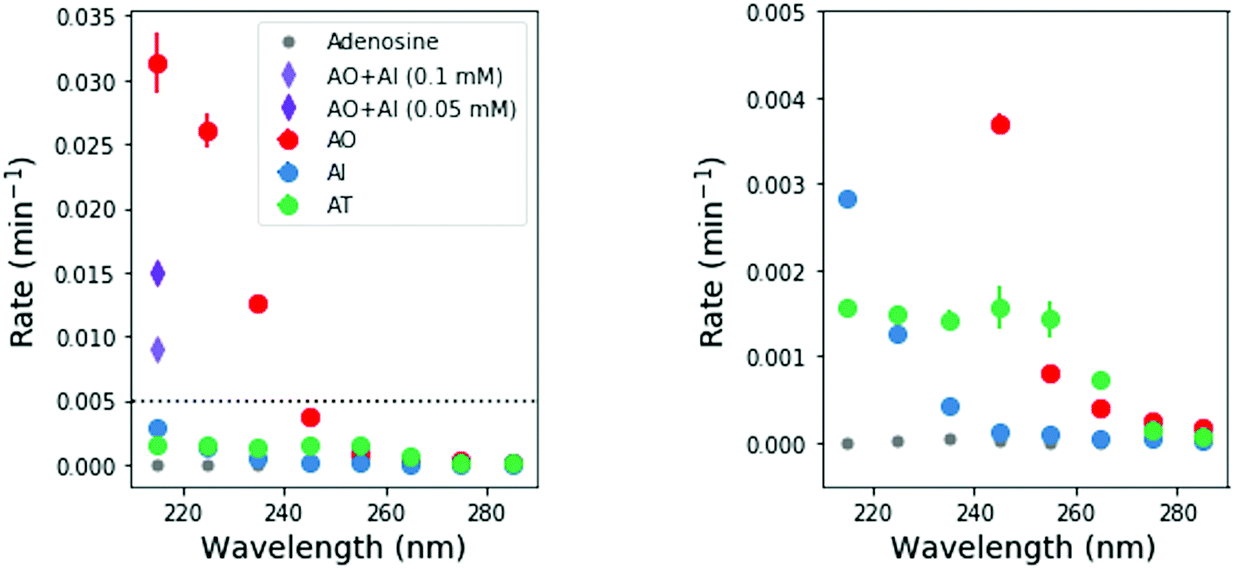 | ||
| Fig. 4 Destruction reaction rate normalized by incident photon flux as a function of irradiation wavelength. The right hand panel shows a zoomed in view of the left hand panel, to show the wavelength dependence of AI and AT. AO has the highest relative rate of photodegradation, especially at short wavelengths. AI and AT are more photostable, but AI is susceptible only to short wavelengths, while AT is most affected by short-to-mid-range wavelengths. We repeated each irradiation wavelength for each molecule in duplicate. Points show the average rates, while error bars represent the 1σ standard deviation. | ||
Quantum chemical simulations and pump–probe spectroscopic techniques revealed important details of the excited state dynamics of model azole chromophores, including AO and AI.19 These studies demonstrated a significant contribution of repulsive πσ* states to their overall photochemistry.19 In particular, πσNH* excitations were shown to promote an ultrafast two stage electron-driven proton transfer (EDPT) photorelaxation mechanism which is initiated with photoinduced electron transfer to neighbouring water molecules. The subsequent proton transfer from the amino group in the direction of the hydrated electron can be treated as an example of photoacidity and enables efficient photorelaxation through the πσ*/S0 state crossing. It was suggested that after the repopulation of the S0 state the hydrogen atom could be readily returned to the chromophore and EDPT could be a photostabilizing deactivation mechanism. The very high photostability of AI reported here corroborates this computational prediction, since EDPT was found as the dominant, if not the only photodeactivation mechanism in this microhydrated chromophore.19 In contrast, excited-state dynamics simulations revealed a considerable contribution from photoinduced ring-opening mechanism (C–O bond breaking) to the photochemistry of AO, which was observed in one third of photoexcitation events.19 The repulsive πσ* state responsible for the ring-opening mechanism is present in the higher energy range of the spectrum, which is also consistent with our observation that AO is most efficiently decomposed at shorter irradiation wavelengths.19 Consequently, we anticipate that the relatively high rate of photodestruction observed for AO could be the result of photochemical opening of its heteroaromatic ring. Photochemical opening and destruction of thiazole rings has also been observed in past work.20,21
We note that we did not observe the appearance of new absorption bands in the monitored UV ranges throughout the irradiation period that could correspond to any photoproducts and could affect our estimated photodegradation rates. The open-ring photoproducts of AO and AI are not expected to absorb in similar regions. If any of the possible photoproducts of AT contains a thiocarbonyl group it might absorb between 210 and 290 nm. Nevertheless, we see a linear decrease in absorbance of the AT sample and no new spectral features (see Fig. 2 and 3), which indicates that the initial photoproducts formed from AT are also prone to photodegradation or do not absorb within the studied wavelength range either.
Based on our measured wavelength-dependent rates of degradation, we can then ask how stable these molecules would have been in the environment of the early Earth. We used the two-stream radiative transfer model described in Ranjan and Sasselov22 to calculate the surface radiance of the Sun on the surface of the early Earth through a model prebiotic atmosphere. The yellow dashed line in Fig. 5 shows the spectral surface radiance for this model, which is consistent within an order of magnitude with the flux from our experimental apparatus. In the absence of oxygen and ozone in the atmosphere, UV light all the way down to 200 nm reaches the surface of the planet; however, the shorter wavelengths have a much lower intensity than the longer wavelengths. We then integrated the spectral surface intensity in the same 10 nm intervals as the experimental conditions and calculated the relative rate of the reaction on the surface of the planet as the product of the integrated surface radiance and the experimental photon-flux normalized rate (Fig. 5 circles, see ESI,† Section S8 for details). Under the UV environment on the surface of the early Earth, AO (red) has the highest rate of photodestruction, peaking at a wavelength of 225 nm. AI has a maximum rate at 215 nm, even though there is much less light coming at this wavelength, indicating that AI is especially susceptible to short-wavelength UV irradiation. The relative rate for AT destruction is maximum at 265 nm.
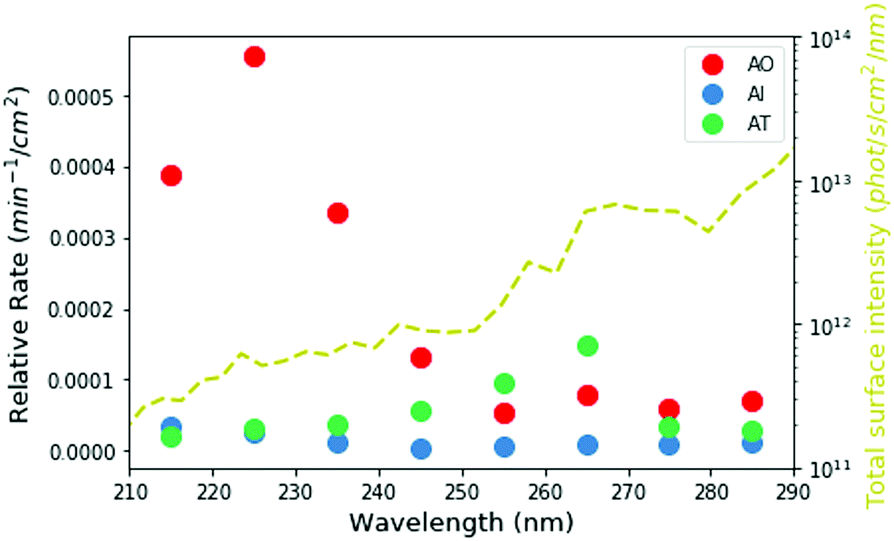 | ||
| Fig. 5 Calculated relative rates of UV degradation on the surface of the early Earth, derived from the product of experimentally determined rates and integrated surface intensity in the given wavelength ranges. The yellow dashed line shows the total surface intensity from the young sun through a 1 bar N2/CO2-dominated atmosphere. The lower fluxes at shorter wavelengths help mitigate somewhat the higher experimental rates of destruction of AI and AO at these wavelengths. | ||
We then integrated the relative rates of photodestruction over wavelengths from 210–290 nm to obtain an estimate of the total rate of destruction, assuming a solar like flux. Fig. 6 shows the total destruction rates and half lives for the three molecules. AO has the shortest half life, of approximately 6.9 hours. AT has an intermediate half life of ≈26 h, and AI has the longest half life (≈99 h). Interestingly, AI is less photostable than other imidazoles under select irradiation conditions (e.g. 2-methylimidazole and 2-ethylimidazole, see ESI† Section S6). From these estimates, this family of prebiotically relevant molecules would only be stable if exposed to the surface UV flux expected on the early Earth for on the order of 7–100 hours. Further exposure to irradiation would significantly deplete the reservoir of these materials. These estimates for the photodestruction rates can help to constrain the relevant concentrations, timescales, and environments necessary for making and using these molecules in a prebiotically relevant manner.
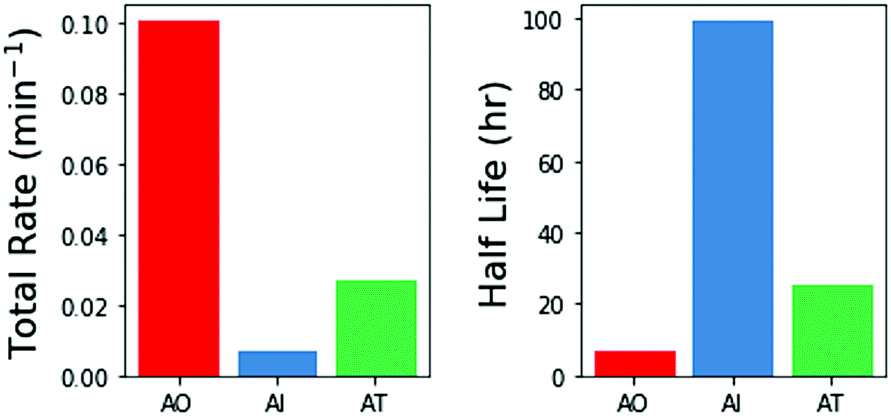 | ||
| Fig. 6 Total rate of degradation when integrating over all wavelengths from 210–290 nm for each molecule, and half-life of each molecule to photodegradation under a solar-like spectrum passing through a N2/CO2-dominated atmosphere. | ||
The relative stabilities of the three aminoazoles are of interest in the context of their potential roles in nucleotide synthesis. As described above, AO, the key intermediate in the prebiotic synthesis of the pyrimidine ribonucleotides is the least photostable. This may not be an issue if the key chemical step involving AO, namely its reaction with glyceraldehyde to form pentose aminooxazolines, which then form anhydronucleosides and ultimately the pyrimidine ribonucleotides,2 proceeds rapidly and efficiently. In the original synthesis proposed by Powner et al.,2 the reaction of AO and glyceraldehyde gives a 70% yield over 16 hours at 40 °C at concentrations that are significantly higher than the concentrations we used for photostability measurements (1 M vs. 0.1 mM). We measured the rate of reaction of AO and glyceraldehyde at reduced concentrations (10, 1, and 0.1 mM each), and saw the reaction proceed over tens of hours for concentrations of 10 mM and 1 mM (see ESI,† Section S5). At 0.1 mM AO, 0.1 mM glyceraldehyde, AO has a half life of >1200 h for reaction with glyceraldehyde, which is significantly longer than the photodestruction half life of 6.9 hours for AO. This potentially places a constraint on the environmental scenario in order for the UV degradation of AO not to hinder the overall pathway. At higher concentrations, AO is consumed faster by reaction with glyceraldehyde while the rate of UV degradation could be slower as the optical depth is decreased, allowing for self-shielding from UV photons. Furthermore, the pentose aminooxazoline products of this reaction were demonstrated to be much more photostable than AO.18
In summary, we have constrained the UV photostability of three key molecules for prebiotic chemistry under conditions in which they might form and be used. AI is the most photostable, while AO is the least. The comparatively low UV photostability of AO may or may not be an issue for this prebiotic chemical network, depending on the concentrations and timescales under which it is produced and used and the presence of other more UV-photostable molecules that could act as sunscreens, such as AI. It is also encouraging that AI, which would be needed to activate already synthesized nucleotides23 and facilitate nonenzymatic RNA template copying4 has the longest lifetime under UV irradiation.
We thank F. Ng for laboratory assistance, C. Giurgiu and C. Kufner for helpful comments and discussions. This work was supported in part by grants from the Simons Foundation (290363 to J. W. S. and 290360 to D. D. S. and 494188 to R. S.) and by the Foundation for Polish Science (FNP, START 2019 Fellowship to R. S.). Z. R. T. and D. D. S. acknowledge support from the Harvard Origins of Life Initiative. J. W. S. is an investigator of the Howard Hughes Medical Institute.
Conflicts of interest
There are no conflicts to declare.References
- B. H. Patel, C. Percivalle, D. J. Ritson, C. D. Duffy and J. D. Sutherland, Nat. Chem., 2015, 7, 301–307 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- S. J. Roberts, R. Szabla, Z. R. Todd, S. Stairs, D.-K. Bučar, J. Šponer, D. D. Sasselov and M. W. Powner, Nat. Commun., 2018, 9, 4073 CrossRef PubMed.
- L. Li, N. Prywes, C. P. Tam, D. K. O'Flaherty, V. S. Lelyveld, E. C. Izgu, A. Pal and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 1810–1813 CrossRef CAS PubMed.
- S. Islam, D.-K. Bučar and M. W. Powner, Nat. Chem., 2017, 9, 584–589 CrossRef CAS.
- A. C. Fahrenbach, C. Giurgiu, C. P. Tam, L. Li, Y. Hongo, M. Aono and J. W. Szostak, J. Am. Chem. Soc., 2017, 139, 8780–8783 CrossRef CAS PubMed.
- C. Sagan and B. N. Khare, Science, 1971, 173, 417–420 CrossRef CAS PubMed.
- C. Chyba and C. Sagan, Nature, 1992, 355, 125–132 CrossRef CAS PubMed.
- O. Pestunova, A. Simonov, V. Snytnikov, V. Stoyanovsky and V. Parmon, Adv. Space Res., 2005, 36, 214–219 CrossRef CAS.
- J. Xu, D. J. Ritson, S. Ranjan, Z. R. Todd, D. D. Sasselov and J. D. Sutherland, Chem. Commun., 2018, 54, 5566–5569 RSC.
- P. B. Rimmer, J. Xu, S. J. Thompson, E. Gillen, J. D. Sutherland and D. Queloz, Sci. Adv., 2018, 4, eaar3302 CrossRef PubMed.
- (a) A. A. Beckstead, Y. Zhang, M. S. de Vries and B. Kohler, Phys. Chem. Chem. Phys., 2016, 18, 24228–24238 RSC; (b) K. Kleinermanns, D. Nachtigallová and M. S. de Vries, Int. Rev. Phys. Chem., 2013, 32, 308–342 Search PubMed; (c) C. T. Middleton, K. D. L. Harpe, C. Su, Y. K. Law, C. E. Crespo-Hernández and B. Kohler, Annu. Rev. Phys. Chem., 2009, 60, 217–239 CrossRef CAS PubMed.
- D. Ritson and J. D. Sutherland, Nat. Chem., 2012, 4, 895–899 CrossRef CAS PubMed.
- S. Ranjan and D. D. Sasselov, Astrobiology, 2016, 16, 68–88 CrossRef CAS PubMed.
- T. Matsunga, K. Hieda and O. Nikaido, Photochem. Photobiol., 1991, 54, 403–410 CrossRef.
- Z. R. Todd, A. C. Fahrenbach, C. J. Magnani, S. Ranjan, A. Björkbom, J. W. Szostak and D. D. Sasselov, Chem. Commun., 2018, 54, 1121–1124 RSC.
- (a) A. Barlev and D. Sen, Acc. Chem. Res., 2018, 51, 526–533 CrossRef CAS PubMed; (b) S. Reiter, D. Keefer and R. di Vivie-Riedle, J. Am. Chem. Soc., 2018, 140, 8714–8720 CrossRef CAS PubMed; (c) D. B. Bucher, C. L. Kufner, A. Schlueter, T. Carell and W. Zinth, J. Am. Chem. Soc., 2016, 138, 186–190 CrossRef CAS PubMed; (d) B. Durbeej and L. A. Eriksson, Photochem. Photobiol., 2003, 78, 159–167 CrossRef CAS PubMed; (e) R. B. Zhang and L. A. Ericsson, J. Phys. Chem. B, 2006, 110, 7556–7562 CrossRef CAS PubMed.
- M. J. Janicki, S. J. Roberts, J. Šponer, M. W. Powner, R. W. Góra and R. Szabla, Chem. Commun., 2018, 54, 13407–13410 RSC.
- (a) R. Szabla, D. Tuna, R. W. Góra, J. Šponer, A. L. Sobolewski and W. Domcke, J. Phys. Chem. Lett., 2013, 4, 2785–2788 CrossRef CAS; (b) R. Szabla, J. Šponer and R. W. Góra, J. Phys. Chem. Lett., 2015, 6, 1467–1471 CrossRef CAS PubMed; (c) M. J. Janicki, R. Szabla, J. Sponer and R. W. Góra, Faraday Discuss., 2018, 212, 345–358 RSC; (d) G. M. Roberts, C. A. Williams, M. J. Paterson, S. Ullrich and V. G. Stavros, Chem. Sci., 2012, 3, 1192–1199 RSC; (e) J. Cao, Z.-Z. Xie and X. Yu, Chem. Phys., 2016, 474, 25–35 CrossRef CAS.
- F. M. Uber and F. Verbrugge, J. Biol. Chem., 1940, 136, 81–86 CAS.
- J. Miyazaki, H. Takiyama and M. Nakata, RSC Adv., 2017, 7, 4960–4974 RSC.
- S. Ranjan and D. D. Sasselov, Astrobiology, 2017, 17, 169–204 CrossRef CAS PubMed.
- A. Mariani, D. A. Russell, T. Javelle and J. D. Sutherland, J. Am. Chem. Soc., 2018, 140, 8657–8661 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc05265h |
| This journal is © The Royal Society of Chemistry 2019 |